Back to Journals » International Medical Case Reports Journal » Volume 13
Case Report: Klippel-Trenaunay Syndrome – Recurrent Venous Thromboembolism and Vascular Malformation
Authors AlSheef M ![]() , Alotaibi H
, Alotaibi H ![]() , Zaidi ARZ
, Zaidi ARZ ![]() , Bauones S, Kullab GJ
, Bauones S, Kullab GJ ![]() , AlShaikh M
, AlShaikh M
Received 29 October 2019
Accepted for publication 5 May 2020
Published 21 May 2020 Volume 2020:13 Pages 195—200
DOI https://doi.org/10.2147/IMCRJ.S236027
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ronald Prineas
Mohammed AlSheef,1 Hind Alotaibi,1 Abdul Rehman Z Zaidi,1 Salem Bauones,2 Ghaydaa J Kullab,3 Mohammed AlShaikh2
1Department of Medical Specialties, King Fahad Medical City, Riyadh, Saudi Arabia; 2Department of Radiology, King Fahad Medical City, Riyadh, Saudi Arabia; 3Department of Medicine, Dr. Sulaiman Al-Habib Medical Group, Riyadh, Saudi Arabia
Correspondence: Mohammed AlSheef
Department of Medical Specialties, King Fahad Medical City, P.O. Box. 59046, Riyadh 11525, Saudi Arabia
Tel +966 544014035
Email [email protected]
Abstract: Klippel-Trenaunay Syndrome (KTS) is a rare genetic vascular disorder characterized by a limb affected by varicose veins, port wine stains, and hypertrophy of bone and soft tissue. It can also present with vascular malformations in the gastrointestinal tract, liver, spleen, genitourinary tract, and heart. We present a 27-year-old case of KTS diagnosed in adulthood associated with recurrent venous thromboembolism and gastrointestinal bleeding.
Keywords: KTS, VTE, vascular malformation, bleeding
Introduction
Klippel-Trenaunay Syndrome (KTS) is a rare congenital disorder characterized by a triad of varicosities, cutaneous capillary malformations, and soft tissue or bone hypertrophy of one or more limbs. Other clinical manifestations were reported, such as gastrointestinal bleeding due to gastrointestinal vascular malformations,1,2 superficial thrombophlebitis, lymphatic malformation, deep vein thrombosis, pulmonary embolism.1,3 The other differential diagnosis in such cases may be Park Weber syndrome (PKWS), which is a result of a germline mutation of the RASA 1 gene. In PKWS, there are high flow vascular malformations, along with vascular skin nevi (thought to be caused by micro AV shunts in the dermis) and overgrowth. In contrast to KTS, PKWS cases may progress to high output heart failure secondary to a high-flow vascular malformation. Another important differential diagnosis may be CLOVES syndrome, which is a congenital lipomatous disorder that presents with asymmetric overgrowth, vascular malformations, epidermal nevi, and skeletal/spinal anomalies.4 We herein present a case of KTS diagnosed in adulthood associated with recurrent venous thromboembolism and gastrointestinal bleeding.
Case Report
A 27-year-old male labeled by an outside institution as a case of Ehlers-Danlos syndrome since the age of 10 years was referred to the thrombosis clinic at our tertiary care center. He presented with right lower extremity varicose veins, multiple lipomas and hemihypertrophy, recurrent unprovoked (ie, not associated with an environmental risk factor) pulmonary embolism and deep vein thrombosis. Upon further questioning, the mother stated that she started to notice asymmetry of the limbs at the age of 4.
On physical examination, the cardiac and respiratory exams were normal. The patient had a high arched palate, small fused tongue, and hyperelastic skin and joints all over. In addition, there was an anterior abdominal mass (CT abdomen finding was “right-sided hypertrophy of the subcutaneous fat”). During the musculoskeletal exam, hypertrophy of left-hand middle finger was noted. The lower limb showed length discrepancy with bilateral genu valgum deformity, right leg hemihypertrophy and prominent varicose veins with multiple lipomas (Figure 1).
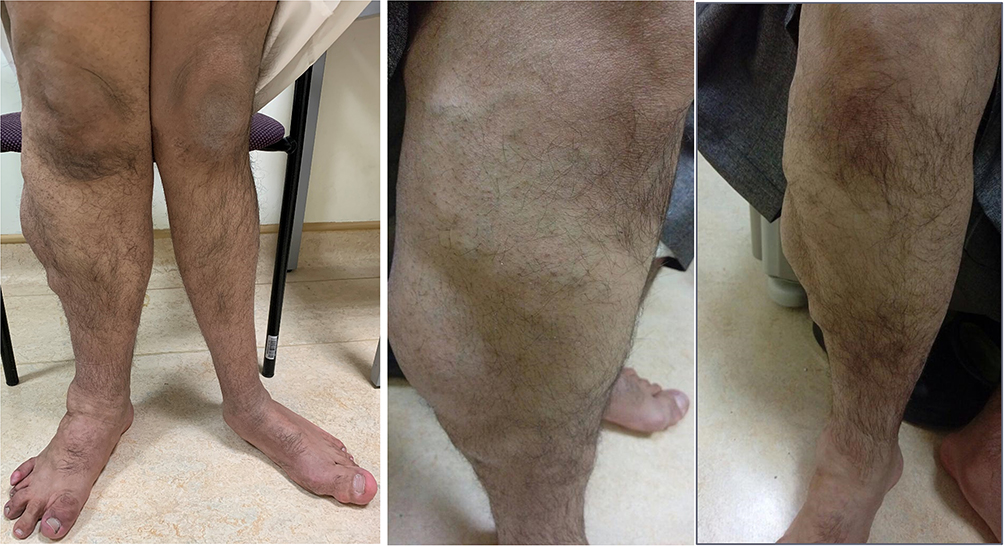 |
Figure 1 Composite figure depicting hemihypertrophy of the right lower limb and mega varicose veins. |
Laboratory examinations were negative for hereditary and acquired thrombophilia and autoimmune investigations (including Anticardiolipin screen IgG, IgM, IgA, B2 - Glycoprotein I IgG, IgM, IgA, Protein C and Protein S, Antithrombin III, Lupus anticoagulation - La1, Factor V Leiden (APCR), Prothrombin gene mutation G20210A). Doppler Ultrasound of right lower limb showed dilatation of the main great saphenous vein measuring 1.7 cm at leg level, the femoral and popliteal veins were aneurysmal but patent; there was detectable reflux in distal popliteal and tibial veins secondary to incompetent valves, and there was no evidence of arteriovenous malformation or deep vein thrombosis. In the skeletal survey, the patient was found to have a gentle levoscoliosis of the lumbar spine, and pelvis was tilted and rotated left. There was bowing of the femoral shaft with bilateral coxa valga deformity, and asymmetrical bowing of tibia and fibula bilaterally with limb length discrepancy. There was focal gigantism involved with the left-hand middle finger phalanges and left big toe. MRI brain showed small T2 abnormal signal in the right frontal white matter superolateral to the head of caudate nucleus suggested of cavernous venous malformation (Figures 2 and 3). There was no hemi-megalocephaly on MRI and there was no evidence of arteriovenous malformation (AVM).
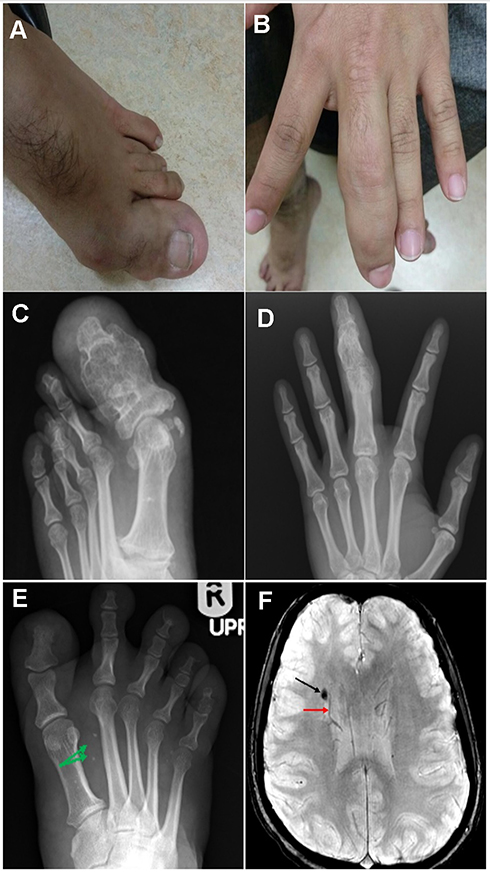 |
Figure 2 (A-D) The composite figure illustrates clinical and radiological findings, showing focal gigantism involving the left-hand middle finger phalanges and phalanges of the left big toe with their expansion and remodeling. (E) Axial susceptibility-weighted image showing right paraventricular cavernous venous malformation (black arrow) accompanied by an adjacent developmental venous anomaly (red arrow). (F) AP radiograph of the right foot exhibiting diffuse soft tissue hypertrophy with punctuate dystrophic calcifications (green arrows) that reveal slow flow venous malformation. |
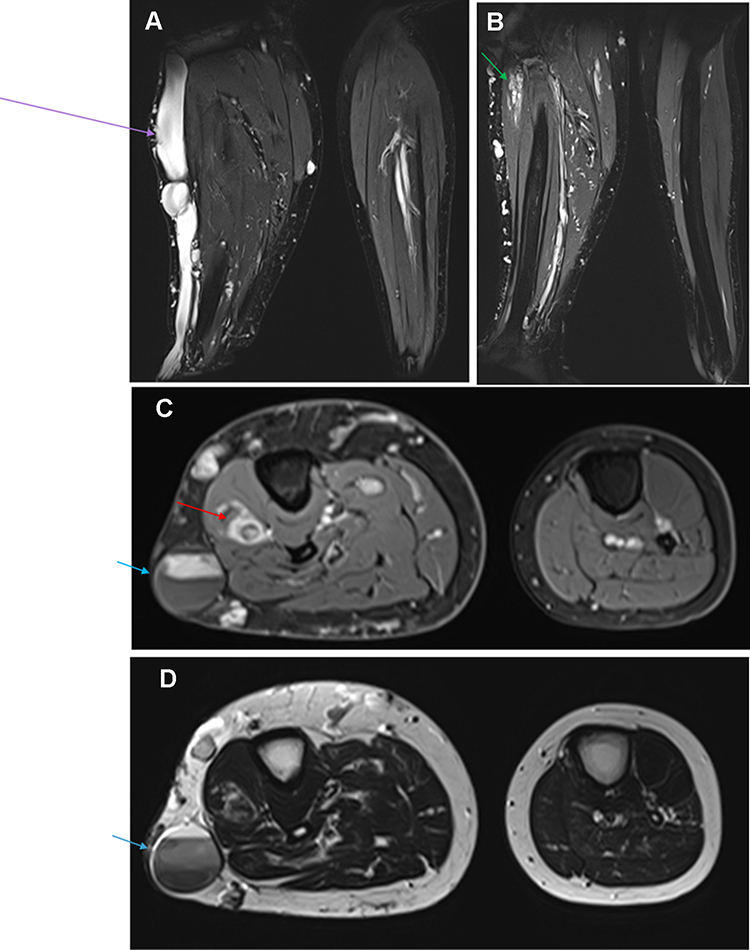 |
Figure 3 Composite figure illustrating: (A and B) Coronal T2 weighted-images with fat saturation showing severely dysplastic and ectatic subcutaneous vein (purple arrow) situated on the lateral aspect of the right leg, which is draining a complex circumferential network of subcutaneous varices and is associated with venostasis. Additionally, intramuscular ectatic deep veins with slow flow venous malformation seated in the proximal part of the deep posterior compartment (green arrow). (C) Axial contrast-enhanced T1 weighted image with fat saturation and (D) Axial T2 weighted-image demonstrate asymmetric hypertrophy of the right leg subcutaneous soft tissue and muscles as well comparing to the normal contralateral side. Furthermore, an intramuscular cavernous hemangioma (red arrow) is seen seated in the proximal part of the extensor digitorum muscle of the right calf. We can note a slow and delayed opacification of the large subcutaneous vein on the post-contrast image. Venostasis is demonstrated by a multilayer of spontaneous contrast (blue arrow). |
During outpatient follow-up and therapeutic anticoagulant for unprovoked pulmonary embolism, the patient was complaining of per rectum bleeding. Colonoscopy showed a single solitary ulcer in the rectum with sigmoid and rectal polyps which were resected. Biopsy revealed hyperplastic polyps and mucosal prolapse syndrome with no dysplasia (Figure 4).
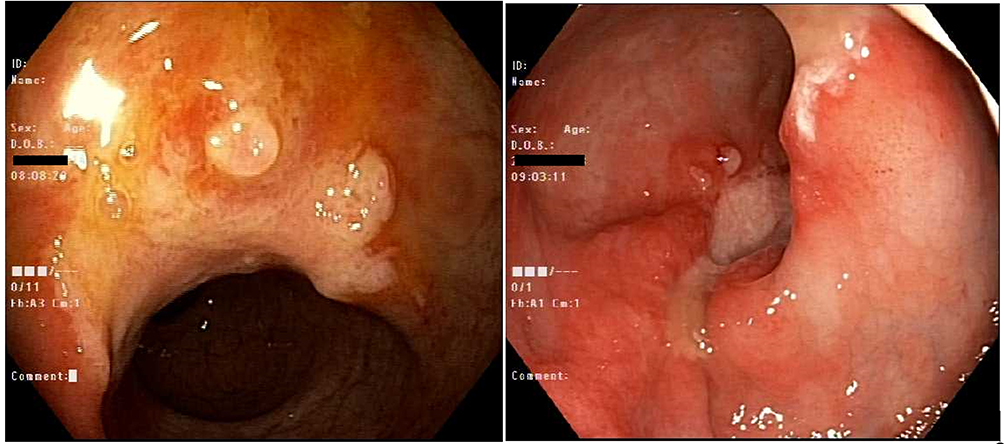 |
Figure 4 Colonoscopy: A single (solitary) 16 mm ulcer in the mid rectum around 10 cm from anal verge. A hyperplastic polyp in the sigmoid colon around 4 mm in size. |
A genetic investigation was performed in Mayo Clinic laboratories in Rochester Main Campus. The interpretation revealed several large regions of homozygosity encompassing at least 6% of the genome with no clinical significant copy number changes were identified.
The most likely diagnosis is Klippel-Trenaunay syndrome based on the presence of extensive lower limb varicosities, soft tissue hypertrophy with bone deformities, central nervous system arteriovenous malformation, and recurrent venous thromboembolism.
Discussion
Klippel-Trenaunay Syndrome (KTS) is a term used for the triad of varicose veins, cutaneous capillary malformation, which are called port-wine stains, and soft tissue or bone hypertrophy. In 1900, KTS was first described as a single disease entity by two French physicians Klippel and Trenaunay.5 In 1907, Parks Weber described a distinct syndrome which includes a similar triad with the presence of arteriovenous malformation.6 In literature, some authors use the term Klippel-Trenaunay-Weber syndrome to describe such cases who have the clinical triad of Klippel-Trenaunay syndrome and arteriovenous malformations.
In a KTS case series study, 252 patients were reviewed, 159 (63%) had all the clinical triad findings, while 93 (37%) had two of the three features.7 The clinical presentation can vary from classical clinical triad to developing potentially life-threatening complications, such as pulmonary embolism, deep vein thrombosis, and massive gastrointestinal bleeding.1–3 The mechanism is unknown, but some studies have investigated groups of KTS patients. In 1985, Baskerville identified that during embryogenesis, a mesodermal defect causes microscopic arteriovenous communications resulting in KTWS.8 The association of thromboembolic events in patients with KTS has been reported.3,9,10 In relation to the prevalence of venous thromboembolism in KTS patients, there was a large cross-sectional cohort study, in which 8% of patients have venous thromboembolism either a deep venous thrombosis or a pulmonary embolism.10 In the same study, the prothrombotic state was identified in the form of high D-dimer levels, higher plasmin-antiplasmin complexes, and lower protein C levels in the adult KTS patients compared to the control group.10
There was a case report from Nigeria where a 15-year-old boy was diagnosed with KTS using X-rays and Doppler studies of the lower limb vessels.11 A study from Michigan from 1965 recounts 18 cases that appeared to correspond to the original description of the syndrome by Klippel and Trenaunay in 1900 in which there was no evidence of an arteriovenous fistula in any of the cases.12 There was also a case reported in 1999, where there was a case of KTS without limb hypertrophy.13
In our case, the differential consideration may include Parkes Weber syndrome (PWS). PWS is characterized by overgrowth of the affected limb combined with arteriovenous fistula or shunt. Both syndromes are hardly to be distinguished in routine clinical practice, and they are easily misdiagnosed. Therefore, Doppler-Ultrasound (Doppler-US) and Dynamic Contrast-Enhanced Magnetic Resonance Angiography (MRA) studies aid to reach the proper diagnosis and are helpful to radiologically differentiate between both conditions.14 PWS is associated with numerous small peri-articular high flow arteriovenous malformations, particularly arteriovenous fistulas that can be obviously depicted on MRA or Doppler-US. In contrast, KTS is associated with slow flow vascular malformation, including capillary, venous, or rarely lymphatic malformations. Those malformations would be demonstrated on MRA as slow flow, dysplastic or ectatic, superficial, or deep venous structures with gradual opacification on the delayed phase. Other differential diagnoses of KTS include cutis marmorata telangiectatica congenita, hemihyperplasia and multiple lipomas, Beckwith-Wiedemann syndrome, and Servelle-Martorell syndrome.15,16 Our patient was also misdiagnosed as a case of Ehler Danlos Syndrome at the age of 10 years. However, our patient did not have skin hyperextensibility or widened atrophic papyraceous scars, neither did he have easy bruising. Usually, Ehler Danlos Syndrome is a clinical diagnosis with the combination of multiple features (skin hyperextensibility, widened atrophic cigarette paper-like scars with poor healing and joint hypermobility, soft doughy skin, easy bruising, fragile skin, molluscoid pseudotumors, subcutaneous spheroids, complications of joint hypermobility, epicanthal folds, hernias, and a positive family history).17 Another differential diagnosis might be juvenile polyposis combined with hemorrhagic telangiectasia (JPHT) syndrome. The JPHT syndrome has findings of arteriovenous malformations of the brain, lungs, liver, and gastrointestinal tract, aneurysms, hamartomatous polyps, hemorrhagic events, and cartilage/bone involvement of connective tissue.18 Lastly, CLOVES syndrome is a mimicker of KTS, that presents with overgrowth features and cutaneous lesions that mostly present at birth and are asymmetric, disproportionate, and less progressive. Both CLOVES and KTS are segmental overgrowth syndromes that share the same gene mutation (PIK3CA).4 However, KTS is a clinical diagnosis and absence of PIK3CA gene mutation does not rule it out.19 Although there are many overlapping similarities in presentation, KTS patients (such as our patient) do not have epidermal nevi, hemi-megalocephaly on MRI, and the overgrowth features of KTS patients may be appreciated at birth (like in CLOVES syndrome), but usually present later in early infancy or childhood and the hypertrophy is more progressive in KTS patients.
Conclusion
In this case, we are reporting a common presentation of an uncommon syndrome that has variable clinical presentations from the classical clinical triad to potentially life-threatening complications, such as pulmonary embolism, deep vein thrombosis, and massive gastrointestinal bleeding. Increasing awareness of KTS as a diagnosis is vital in the Middle East.
Ethics and Consent
Written informed consent to have the case details and images published was obtained from the patient and the patient’s family and an IRB exemption was obtained from from our institution.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Cha SH, Romeo MA, Nentze JA. Visceral manifestations of Kippel-Trenaunay syndrome. Radiographs. 2005;25(6):1694–1697. doi:10.1148/rg.256055042
2. Wang Z-K, Wang F-Y, Zhu R-M, Liu J. Klippel-Trenaunay syndrome with gastrointestinal bleeding, splenic hemangiomas and left inferior vena cava. World J Gastroenterol. 2010;16(12):1548–1552. doi:10.3748/wjg.v16.i12.1548
3. Huiras EE, Barnes CJ, Eichenfield LF, Pelech AN, Drolet BA. Pulmonary thromboembolism associated with Klippel-Trenaunay syndrome. Pediatric. 2005;116:596–600. doi:10.1542/peds.2004-1607
4. Mahajan VK, Gupta M, Chauhan P, Mehta KS. Cloves syndrome: A rare disorder of overgrowth with unusual features – an uncommon phenotype? Indian Dermatol Online J. 2019;10:447–452. doi:10.4103/idoj.IDOJ_418_18
5. Klippel M, Trenaunay P. Du noevus variqueux osteo-hypertrophique. Arch Gen Med. 1900;185:641–672.
6. Weber PF. Angioma: formation in connection with hypertrophy of limbs and hemi hypertrophy. Br J Dermatol. 1907;19:231–235.
7. Jacob AG, Driscoll DJ, Shaughnessy WJ, Stanson AW, Clay RP, Gloviczki P. Klippel-Trenaunay syndrome: spectrum and management. Mayo Clin Proc. 1998;73(1):28–36. doi:10.1016/S0025-6196(11)63615-X
8. Baskerville PA, Ackroyd JS, Browse NL. The etiology of the Klippel-Trenaunay syndrome. Ann Surg. 1985;202:624–627. doi:10.1097/00000658-198511000-00015
9. Reis J, Alomari AI, Trenor CC, et al. Pulmonary thromboembolic events in patients with congenital lipomatous overgrowth, vascular malformations, epidermal nevi, and spinal/skeletal abnormalities and Klippel-Trénaunay syndrome. J Vasc Surg Venous Lymphat Disord. 2018;5(4):511–516.
10. Oduber CE, van Beers EJ, Bresser P, van der Horst CM, Meijers JC, Gerdes VE. Venous thromboembolism and prothrombotic parameters in Klippel-Trenaunay syndrome. Neth J Med. 2013;71(5):246–252.
11. Ikpeme AA, Usang UE, Inyang AW, Ani N. Klippel Trenaunay syndrome: a case report in an adolescent nigerian boy. Maced J Med Sci. 2015;3(2):322–325. doi:10.3889/oamjms.2015.036
12. Lindenauer SM. The Klippel-Trenaunay syndrome: varicosity, hypertrophy and haemangioma with no arteriovenous fistula. Ann Surg. 1965;162:303–314. doi:10.1097/00000658-196508000-00023
13. Kutsal A, Lampros TD, Cobanoglu A. Right iliac vein agenesis, varicosities, and widespread hemangiomas: report of a rare case. Tex Heart Inst J. 1999;26(2):149–151.
14. Elsays KM, Menias CO, Dillman JR, et al. Vascular malformations and hemangiomatosis syndromes: spectrum of imaging manifestations. Am J Roentgenol. 2008;190:1291–1299. doi:10.2214/AJR.07.2779
15. Nozaki T, Nosaka S, Miyazaki O, et al. Syndromes associated with vascular tumors and malformations: a pictorial review. Radiographics. 2013;33(1):175–195. doi:10.1148/rg.331125052
16. Nozaki T, Matsusako M, Mimura H, et al. Imaging of vascular tumors with an emphasis on ISSVA classification. Jpn J Radiol. 2013;31(12):775–785. doi:10.1007/s11604-013-0249-x
17. Malfait F, Wenstrup R, De Paepe A. Classic ehlers-danlos syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; May 29, 2007 [Updated 2018 Jul 26]:1993–2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1244/.
18. Williams JC, Hamilton JK, Shiller M, et al. Combined juvenile polyposis and hereditary hemorrhagic telangiectasia. Proc (Bayl Univ Med Cent). 2012;25(4):360–364. doi:10.1080/08998280.2012.11928877
19. Klippel-trenaunay syndrome - NORD (National Organization For Rare Disorders). NORD (National Organization For Rare Disorders); 2020. Available from: https://rarediseases.org/rare-diseases/klippel-trenaunay-syndrome/.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.