Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 10 » Issue 1
Cardiovascular and inflammatory effects of simvastatin therapy in patients with COPD: a randomized controlled trial
Authors John M, Cockcroft J, McKeever T ![]() , Coward W, Shale D, Johnson S, Thornton J, Harrison T, Knox A, Bolton C
, Coward W, Shale D, Johnson S, Thornton J, Harrison T, Knox A, Bolton C ![]()
Received 19 October 2014
Accepted for publication 27 November 2014
Published 29 January 2015 Volume 2015:10(1) Pages 211—221
DOI https://doi.org/10.2147/COPD.S76061
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Michelle E John,1 John R Cockcroft,2 Tricia M McKeever,3 William R Coward,1 Dennis J Shale,2 Simon R Johnson,1 Jim G Thornton,4 Timothy W Harrison,1 Alan J Knox,1 Charlotte E Bolton1
1Nottingham Respiratory Research Unit, School of Medicine, University of Nottingham, Nottingham, UK; 2Wales Heart Research Institute, Cardiff University, Cardiff, UK; 3Division of Epidemiology and Public Health, 4Clinical Trials Unit and Department of Obstetrics and Gynaecology, School of Medicine, University of Nottingham, Nottingham, UK
Background: There is excess cardiovascular mortality in patients with chronic obstructive pulmonary disease. Aortic stiffness, an independent predictor of cardiovascular risk, and systemic and airway inflammation are increased in patients with the disease. Statins modulate aortic stiffness and have anti-inflammatory properties. A proof-of-principle, double-blind, randomized trial determined if 6 weeks of simvastatin 20 mg once daily reduced aortic stiffness and systemic and airway inflammation in patients with chronic obstructive pulmonary disease.
Methods: Stable patients (n=70) were randomized to simvastatin (active) or placebo. Pre-treatment and post-treatment aortic stiffness, blood pressure, spirometry, and circulating and airway inflammatory mediators and lipids were measured. A predefined subgroup analysis was performed where baseline aortic pulse wave velocity (PWV) was >10 m/sec.
Results: Total cholesterol dropped in the active group. There was no significant change in aortic PWV between the active group and the placebo group (-0.7 m/sec, P=0.24). In those with aortic stiffness >10 m/sec (n=22), aortic PWV improved in the active group compared with the placebo group (-2.8 m/sec, P=0.03). Neither systemic nor airway inflammatory markers changed.
Conclusion: There was a nonsignificant improvement in aortic PWV in those taking simvastatin 20 mg compared with placebo, but in those with higher baseline aortic stiffness (a higher risk group) a significant and clinically relevant reduction in PWV was shown.
Keywords: chronic obstructive pulmonary disease, arterial stiffness, statins
Introduction
Cardiovascular (CV) disease is a common comorbidity in COPD.1 Yet other than smoking cessation, routine COPD management is not currently focused on preventing CV disease. The risk of CV disease in COPD is two to three-fold greater than the risk generated by smoking,2 and CV disease accounts for more than a quarter of deaths in COPD patients.3 In other chronic diseases with an increased CV risk, statins have a role in CV prevention.4 There is limited but supportive retrospective and observational evidence for a cardioprotective role of statins in COPD.5,6 In healthy people with normal lipid levels but increased C-reactive protein (CRP) levels, atorvastatin in a large, randomized controlled trial (RCT) significantly reduced CRP and importantly the incidence of CV events.7
Aortic stiffness is an independent predictor of CV risk,8,9 and is increased in subjects with COPD compared with smokers without COPD.10,11 The ability of statins to modulate aortic stiffness has been demonstrated in people with coronary artery disease independent of their lipid-lowering effects and in those with isolated systolic hypertension and normal cholesterol levels.12,13 Further, statins improve aortic stiffness and systemic inflammation in other chronic inflammatory conditions, such as rheumatoid arthritis.14
COPD is an inflammatory disease reflected in increased airway and circulating inflammatory markers. The persisting systemic inflammatory state may well be central to many of the comorbidities of COPD, including CV disease, and decreasing inflammation may alter disease course.15,16 Statins confer pleiotropic benefits in reducing inflammatory mediators, including the COPD-relevant markers of CRP and matrix metalloproteinase (MMP)-9.14,17 Whilst a recent large RCT of simvastatin in COPD reported no change in the primary outcome of exacerbation rate or time to first exacerbation,18 no RCT has examined CV disease as a primary outcome measure in COPD.
Statins are currently prescribed to patients with COPD needing secondary CV prevention. The role in primary prevention is not known. We hypothesized that treatment with simvastatin 20 mg once daily would reduce aortic stiffness compared with placebo in a group of well characterized patients with COPD without coexistent ischemic heart disease, diabetes, or hypercholesterolemia. A 20 mg dose was selected as previous work indicates benefit at lower doses without the incremental musculoskeletal side effects.19 Our secondary hypothesis was that the heightened systemic and airway inflammation would be lowered in the active treatment group compared with the placebo group.
Materials and methods
Subjects
Clinically stable patients (n=70) with confirmed COPD, ie, forced expiratory volume in one second (FEV1) 30%–80% predicted, FEV1 to forced vital capacity (FVC) ratio (FEV1/FVC) <0.7, salbutamol reversibility <12% and 200 mL, and a supportive smoking history were recruited into this double-blind, randomized, parallel-group, placebo-controlled study. Clinical stability was defined as no change in regular therapy in the preceding 4 weeks or change in symptoms beyond day-to-day variation. Exclusion criteria included prescribed statin or fibrate, hypercholesterolemia (total cholesterol >6.5 mmol/L), documented ischemic heart disease, and diabetes mellitus. The full inclusion/exclusion criteria are provided in the Supplementary materials.
Subjects were recruited from departmental databases of volunteers, advertisements, and outpatient clinics, and with assistance from the Primary Care Research Network. Ethical (REC 10/H0408/10) and governance (including Medicines and Healthcare Products Regulatory Agency) approvals were granted (clinical trials identifier NCT01151306). The study was performed at a single center, ie, Nottingham Respiratory Research Unit, University of Nottingham, City Hospital Campus, Nottingham, from July 2010 to March 2013. Subjects gave written informed consent and the study was performed according to the Declaration of Helsinki. Measurements were performed pre-treatment and after 6 weeks (±3 days). Subjects were randomized 1:1 in a double-blind fashion, stratified by age (45–62 years and 63–80 years), to either simvastatin (active) or placebo using a computer-generated code of random permuted blocks of randomly varying size by the clinical trials unit. The sequence was stored in the pharmacy and concealed from investigators and participants. Treatment allocation was performed by an independent pharmacist. The active treatment and placebo had an identical capsule appearance, the simvastatin being overencapsulated.
Cardiovascular measurements
Having refrained from short-acting and long-acting bronchodilators for 4 and 12 hours respectively, heart rate (HR) and peripheral blood pressure were measured (705-IT, Omron, Milton Keynes, UK). Mean arterial pressure (MAP) was calculated. Aortic pulse wave velocity (PWV) was performed after resting supine for 10 minutes using a Sphygmocor device (Atcor Medical, West Ryde, Australia) with a single experienced operator.10,20
Anthropometry, lung function, and functional tests
Height and weight (Seca GmbH & Co. KG, Hamburg, Germany) were measured and body mass index was calculated. Post-bronchodilator spirometry was performed using a Microlab MK6 spirometer (Micro Medical Ltd, Rochester, UK). The six-minute walking test was performed following a standardized protocol using a 10 m course.21 Handgrip strength was determined from the mean of three attempts using the dominant arm.
Biochemistry
Fasting lipids, liver function tests, and creatine phosphokinase were measured on an Olympus AU2700 platform (Beckman Coulter, Brea, CA, USA). Low-density lipoprotein cholesterol levels were estimated using the Friedewald equation.22 Serum was centrifuged, aliquoted, and stored at -80°C for later determination of MMP-9 and high sensitivity C-reactive protein (hs-CRP). Total MMP-9 was measured in duplicate and in batches using an enzyme-linked immunosorbent assay kit (R&D Systems, Abingdon, UK). Hs-CRP was measured using an immunoturbidimetric assay (Beckman Coulter) on an Olympus AU5400 analyzer.
Airway inflammatory measurements
Five-flow exhaled nitric oxide (NiOX flex, Aerocrine, Solna, Sweden) was used to determine FeNO50, bronchial intercept, and alveolar gradient. Induced sputum was performed by nebulizing hypertonic saline solution and a differential cell count was obtained, both as described in the Supplementary material.
Compliance
The tablets remaining at visit two were counted and compared with the expected number remaining. Good compliance was defined as taking at least 90% of the medication.
Sample size
A power calculation based on a 1 m/sec reduction in aortic PWV post-treatment, a standard deviation of 1.5 m/sec, a significance of 0.05, and a power of 0.8, gave a sample size of 29 patients per group.23 Given likely attrition, we aimed to recruit 35 patients per arm.
Statistical analysis
The data were analyzed using Statistical Package for the Social Sciences version 21.0 software (SPSS, Chicago, IL, USA). Main analyses compared the difference between the two groups in the mean change over 6 weeks. The primary analysis was to compare the difference in the mean change in the active and placebo arms for aortic PWV using an unpaired t-test. A predefined subgroup analysis was performed in patients with a baseline aortic PWV measurement >10 m/sec.24–26 The difference between arms for the change in aortic PWV was calculated. A secondary stratified analysis was performed using baseline characteristics thought to influence the outcome, including age, sex, and change in MAP, using multiple regression analysis. Unless otherwise specified, the arithmetic mean and standard deviation was used to show central tendency. Positively skewed data, including hs-CRP, exhaled nitric oxide measures, and sputum counts were log10 transformed.
Results
Demographics
The sample size was met: of the 70 subjects randomized and prescribed treatment, 33 were randomized to the active treatment arm (31 completed) and 37 to the placebo arm (33 completed), as shown in Figure 1. Baseline demographics were well matched between groups (see Table 1). Baseline lung function and aortic PWV between those who dropped out (n=6) and those who completed the study (n=64) were similar.
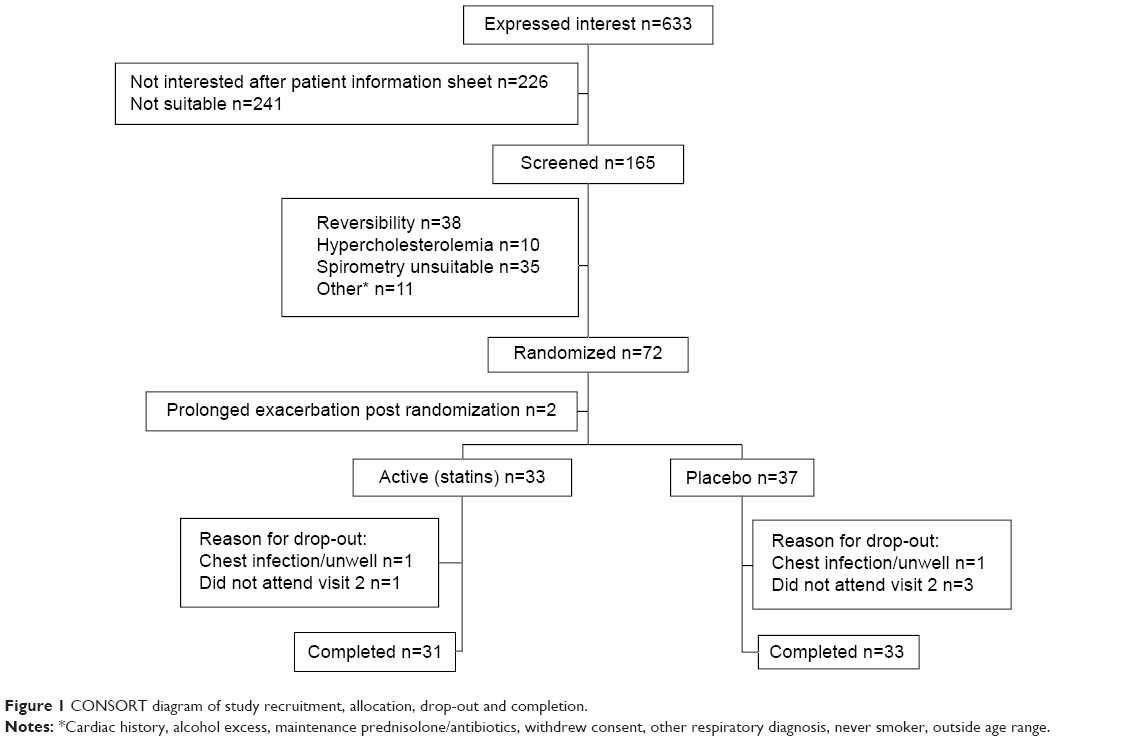 | Figure 1 CONSORT diagram of study recruitment, allocation, drop-out and completion. |
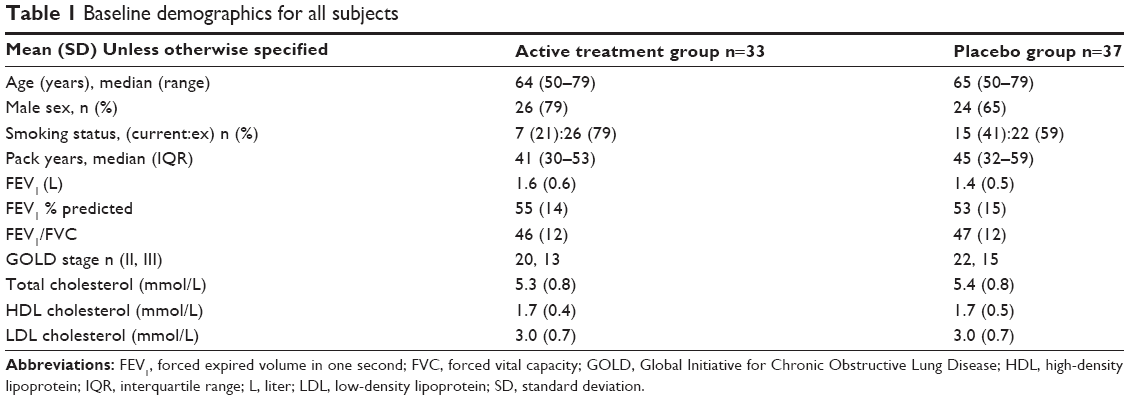 | Table 1 Baseline demographics for all subjects |
Aortic PWV and other hemodynamic measures
There was a fall in aortic PWV in the active group of −1 m/sec; however, the difference in change between the active and placebo arms was not significant (−0.7 [−1.8, 0.5] m/sec, see Table 2 and Figure 2). Adjusting for potential confounders of age, sex, and change in MAP did not alter this.
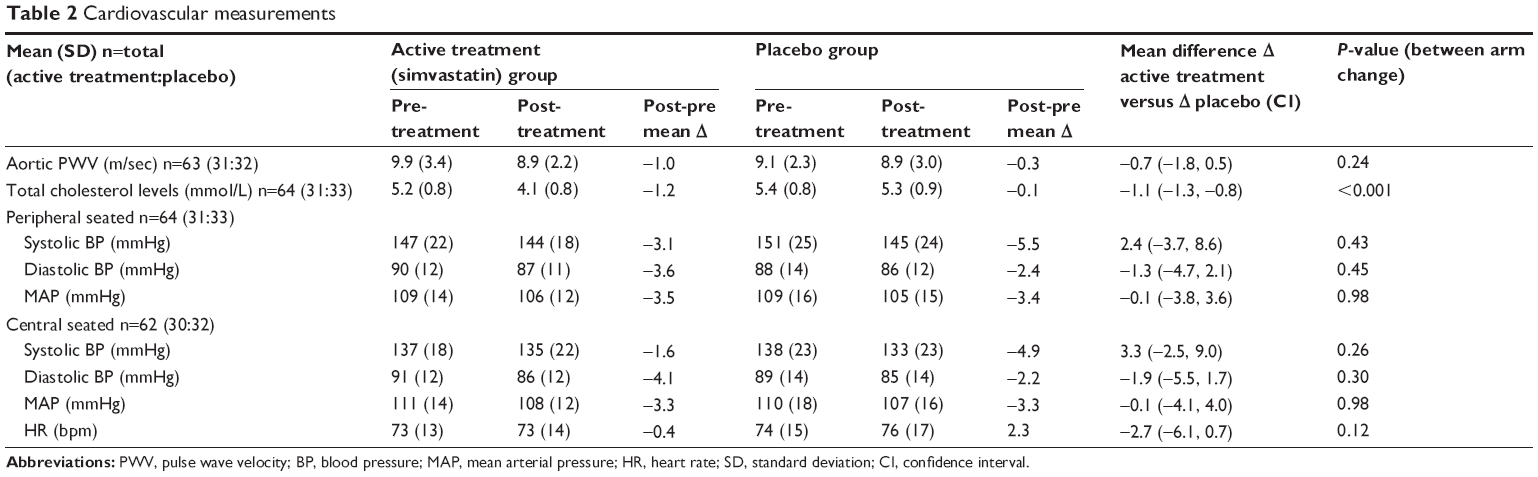 | Table 2 Cardiovascular measurements |
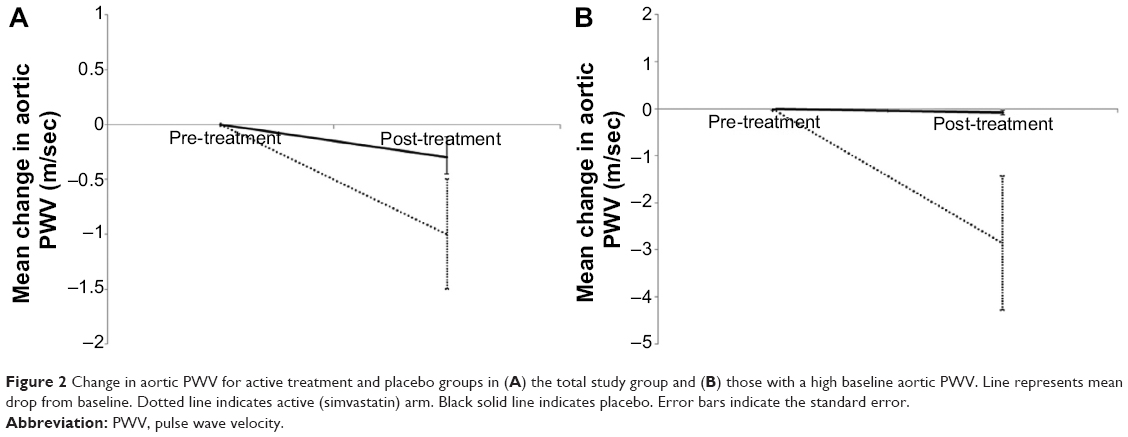 | Figure 2 Change in aortic PWV for active treatment and placebo groups in (A) the total study group and (B) those with a high baseline aortic PWV. Line represents mean drop from baseline. Dotted line indicates active (simvastatin) arm. Black solid line indicates placebo. Error bars indicate the standard error. |
There were no significant changes in central or peripheral blood pressure indices between the active treatment and placebo arms (Table 2).
Multiple linear regression confirmed both MAP and age to be independent predictors of baseline aortic PWV. There was no relationship between baseline total cholesterol, low-density lipoprotein cholesterol level, and baseline aortic PWV. Aortic PWV was greater in males (9.9 [3.2] m/sec) than in females (8.7 [1.8] m/sec, P=0.051).
Hemodynamics in patients with baseline aortic PWV >10 m/sec
In the predefined subgroup analysis of subjects with a baseline aortic PWV >10 m/sec,25,26 n=25 there were n=12 on active treatment and n=13 in the placebo group. Of these n=22 completed the study – active n=11, placebo n=11. The baseline demographics and physiological measures are presented in Table S1. There was a significant difference in the change in aortic PWV between the active and placebo groups of −2.8 m/sec (P=0.03, Table 3 and Figure 2). This significant difference remained after adjusting for age, sex, and change in MAP (P=0.009).
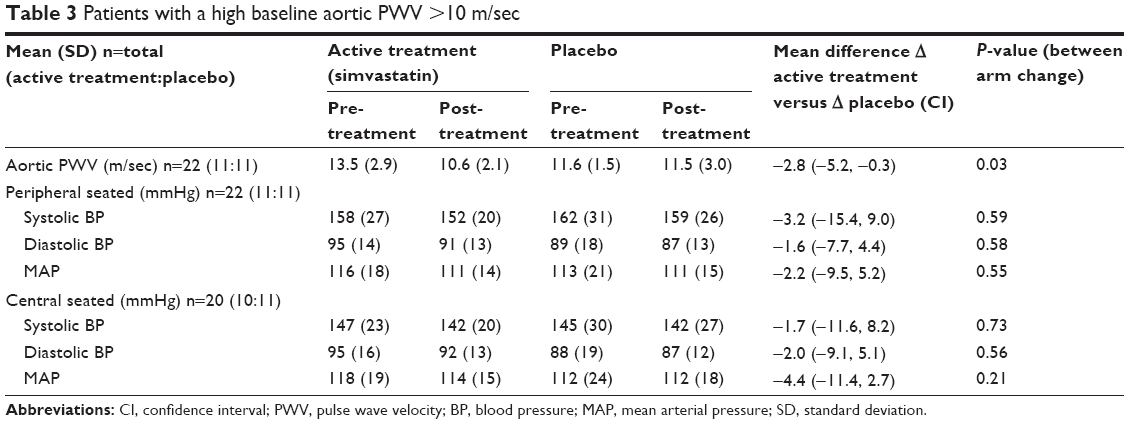 | Table 3 Patients with a high baseline aortic PWV >10 m/sec |
Lipids and inflammatory markers
Total and low-density lipoprotein cholesterol levels dropped in the active treatment arm compared with the placebo arm (both P<0.001, Table 2). There were no significant differences in the mean change of total MMP-9 or hs-CRP, sputum neutrophil cell counts (n=20), or FeNO measurements (n=36) in the active treatment group compared with the placebo group (see Table S2).
Lung function and functional tests
There was no difference in the spirometric and functional tests between the active treatment and placebo groups (Table S2).
Compliance and adverse events
Of the 64 subjects who completed, 15 had poor compliance (placebo n=10, active n=5). However, all 64 subjects took a minimum of 35 tablets. There were no serious adverse events. Patient-reported side effects in both groups were musculoskeletal (muscle and joint aches, pain and cramps) and respiratory (cough, shortness of breath, and chest infections) in nature (see Table S3).
Discussion
This is the first reported double-blind, randomized, placebo-controlled trial of statin therapy examining CV endpoints as the primary outcome in patients with COPD. Further, it focuses on the role of statins as primary prevention in COPD, excluding those with known ischemic heart disease, diabetes, or hypercholesterolemia. Overall, there was a nonsignificant reduction in the primary endpoint of aortic stiffness following 6 weeks of simvastatin 20 mg once daily compared with placebo. In those with a high baseline aortic PWV, statin therapy reduced aortic PWV when compared with placebo. This trial supports the concept that the increased aortic stiffness in COPD is modifiable and that statins may have a primary preventative role in those patients with COPD particularly those at greater CV risk.
Aortic PWV measurements were comparable with those in recently published studies of patients with COPD,27,28 but lower than initial reports of around 11 m/sec despite these original COPD studies using similar methodology and devices.10,11 Age, lung function, and cholesterol levels in our patients were similar to other studies in COPD, and our exclusion of comorbidities was also comparable.10,27,28 As expected, multiple linear regression demonstrated that seated MAP and age were predictors of baseline aortic PWV, which is reassuring given that age and MAP are major contributors to aortic PWV,20 but interestingly, aortic PWV was not associated with baseline cholesterol.
The drop of −1 m/sec in aortic PWV with simvastatin 20 mg over 6 weeks from a baseline of 9.9 m/sec is consistent with a previous study in rheumatoid arthritis,14 and is potentially clinically meaningful given that an increase of 1 m/sec is associated with clinically relevant increases in CV events, CV mortality, and all-cause mortality.23
The large and significant reduction in aortic PWV with simvastatin in the subjects with a baseline aortic PWV >10 m/sec, a value recently adopted by the European Society of Hypertension/European Society of Cardiology as indicating increased aortic stiffness,24–26 suggests that such therapy may be better directed at those with the greater aortic stiffness and CV risk and provides encouragement to consider further investigation in such patients. The magnitude of change seen over the 6 weeks is of clinical relevance.
Low-grade systemic inflammation has been associated with a risk of cardiac injury in COPD, and attenuating the increased inflammation might reduce CV risk.29 Circulating inflammatory markers such as hs-CRP and MMP-9 are elevated in COPD, and patients with COPD and a CRP >3 mg/L are at high risk of future CV events.30–32 Just over half of our study population was identified as being in this high-risk group.
Potential mechanisms of how simvastatin may modulate aortic stiffness involve an anti-inflammatory effect and a subsequent effect on vascular structure,33 a direct cholesterol-lowering effect, or improving endothelial function, and thereby improving endothelial nitric oxide formation.14,34,35 It is likely that these mechanisms are further intertwined with each other. In this proof of principle study, 6 weeks of simvastatin treatment did not affect circulating CRP, although a previous RCT in patients with COPD reported a reduction in CRP levels following 6 months of treatment with pravastatin.36 Circulating MMP-9 levels are associated with aortic stiffness in patients with isolated systolic hypertension and also in healthy subjects,37 and have previously been modulated by statins, but again, were not affected by statins in our study.17 We opted not to study a vast array of inflammatory mediators, selecting two that were relevant to COPD and CV state and had been previously modulated with statins. This aside, the effect of statins on vascular wall inflammation, as opposed to circulating inflammatory biomarkers, is likely to be more relevant mechanistically.17,38
We did not expect nor did we find that simvastatin would alter lung function over such a short period. In the general population, retrospective observational studies suggest that statins may slow the annual rate of decline in lung function.39 It was encouraging that no decline in the six-minute walking distance occurred in the active treatment group compared with placebo, as muscular side effects remain a concern with statin therapy.
Although the lack of significant change in hemodynamic markers on simvastatin compared with placebo might suggest that statins do not alter these parameters, alternative explanations include the short duration of the study, the dose, and the aforementioned lower baseline aortic PWV. The sample size of 70 as per power calculation was reached; however, given the lower baseline results and wider standard deviation than expected, retrospectively it is likely that this study was underpowered. Six weeks was sufficient to lower cholesterol, and a longer duration may have been confounded by the likelihood of exacerbations that could interfere with both CV and inflammatory outcome measures. Furthermore, 6 weeks of statin treatment has previously been sufficient to detect a change in aortic PWV in studies of other chronic inflammatory conditions.14 In clinical practice, simvastatin would be prescribed as long-term therapy. The dose of simvastatin 20 mg once daily was a relatively low one, and there is the possibility that a higher dose may have shown a better response, but this dose was sufficient to significantly lower cholesterol levels and a greater dose may have led to a more side effects. We selected a 20 mg dose for simvastatin because with a higher dose there is a greater risk of musculoskeletal problems and other side effects.19
Until recently, there have been no pharmacological RCTs in COPD with primary CV endpoints, with only observational or retrospective analyses of RCTs that had primary lung endpoints. Since starting this study, a placebo-controlled RCT of fluticasone/salmeterol in subjects with COPD, including those with stable CV disease, with the primary endpoint of aortic PWV, has been reported.28 Overall, there was no beneficial effect on aortic PWV over 12 weeks. However, there was a significant improvement in aortic PWV in patients with the top tertile (>10.9 m/sec) of baseline aortic PWV. The primary outcome of the recently published STATCOPE study of simvastatin 40 mg once daily was exacerbation, with CV events included in the adverse event reporting.18 It should be noted that although the results with simvastatin in patients with COPD and a higher baseline aortic PWV were encouraging, there was some disparity in baseline hemodynamic measures between the two groups.
Conclusion
In patients with COPD without ischemic heart disease, diabetes, or hypercholesterolemia, simvastatin 20 mg once daily for 6 weeks compared with placebo was associated with a nonsignificant improvement in aortic stiffness despite a significant lowering of cholesterol. In those with high baseline aortic PWV, a large and significant reduction in aortic PWV was demonstrated on simvastatin compared with placebo. This may signify a potential benefit in terms of CV mortality in a high-risk group of patients, in whom statins would not previously have been routinely considered.
Acknowledgments
This research was supported by a National Institute for Health Research (NIHR) for Patient Benefit grant (PB-PG-0110-21178). The views expressed are those of the authors and not necessarily those of the National Health Service, the NIHR, or the Department of Health. The Nottingham NIHR Biomedical Research Unit (2008–2012) provided support for the study. The authors thank all the study volunteers; Helen Bailey, Garry Meakin, and research staff, Nottingham Respiratory Research Unit; Sheila Hodgson and Rosie Roberts, Clinical Trials Pharmacy, Nottingham University Hospitals Trust; the Clinical Trials Unit, University of Nottingham; the Trent Clinical Local Research Network; the Primary Care Research Network; and local general practices.
Disclosure
DJS reports having received personal fees from GlaxoSmithKline, Almirall, Novartis, and AstraZeneca. JRC reports having received personal fees from GSK. The other authors have no conflicts of interest to report.
References
Fabbri LM, Luppi F, Beghe B, Rabe KF. Complex chronic comorbidities of COPD. Eur Respir J. 2008;31(1):204–212. | ||
Sin DD, Wu L, Man SF. The relationship between reduced lung function and cardiovascular mortality: a population-based study and a systematic review of the literature. Chest. 2005;127(6):1952–1959. | ||
McGarvey LP, John M, Anderson JA, Zvarich M, Wise RA. Ascertainment of cause-specific mortality in COPD: operations of the TORCH Clinical Endpoint Committee. Thorax. 2007;62(5):411–415. | ||
Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): multicentre randomised placebo-controlled trial. Lancet. 2004;364(9435):685–696. | ||
Soyseth V, Brekke PH, Smith P, Omland T. Statin use is associated with reduced mortality in COPD. Eur Respir J. 2007;29(2):279–283. | ||
Mancini GB, Etminan M, Zhang B, et al. Reduction of morbidity and mortality by statins, angiotensin-converting enzyme inhibitors, and angiotensin receptor blockers in patients with chronic obstructive pulmonary disease. J Am Coll Cardiol. 2006;47(12):2554–2560. | ||
Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359(21):2195–2207. | ||
Laurent S, Boutouyrie P, Asmar R, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37(5):1236–1241. | ||
Ben-Shlomo Y, Spears M, Boustred C, et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol. 2014;63(7):636–646. | ||
Sabit R, Bolton CE, Edwards PH, et al. Arterial stiffness and osteoporosis in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(12):1259–1265. | ||
Maclay JD, McAllister DA, Mills NL, et al. Vascular dysfunction in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180(6):513–520. | ||
Ferrier KE, Muhlmann MH, Baguet JP, et al. Intensive cholesterol reduction lowers blood pressure and large artery stiffness in isolated systolic hypertension. J Am Coll Cardiol. 2002;39(6):1020–1025. | ||
Meng X, Qie L, Wang Y, Zhong M, Li L. Assessment of arterial stiffness affected by atorvastatin in coronary artery disease using pulse wave velocity. Clin Invest Med. 2009;32(6):E238. | ||
Maki-Petaja KM, Booth AD, Hall FC, et al. Ezetimibe and simvastatin reduce inflammation, disease activity, and aortic stiffness and improve endothelial function in rheumatoid arthritis. J Am Coll Cardiol. 2007;50(9):852–858. | ||
Sin DD, Lacy P, York E, Man SF. Effects of fluticasone on systemic markers of inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2004;170(7):760–765. | ||
Miller J, Edwards LD, Agustí A, et al. Comorbidity, systemic inflammation and outcomes in the ECLIPSE cohort. Respir Med. 2013;107(9):1376–1384. | ||
Wu YW, Kao HL, Huang CL, et al. The effects of 3-month atorvastatin therapy on arterial inflammation, calcification, abdominal adipose tissue and circulating biomarkers. Eur J Nucl Med Mol Imaging. 2012;39(3):399–407. | ||
Criner GJ, Connett JE, Aaron SD, et al. Simvastatin for the prevention of exacerbations in moderate-to-severe COPD. N Engl J Med. 2014;370(23):2201–2210. | ||
Armitage J, Bowman L, Wallendszus K, et al. Intensive lowering of LDL cholesterol with 80 mg versus 20 mg simvastatin daily in 12,064 survivors of myocardial infarction: a double-blind randomised trial. Lancet. 2010;376(9753):1658–1669. | ||
Laurent S, Cockcroft J, Van Bortel L, et al. Expert consensus document on arterial stiffness: methodological issues and clinical applications. Eur Heart J. 2006;27(21):2588–2605. | ||
Crapo RO, Casaburi R, Coates AL, et al. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med. 2002;166(1):111–117. | ||
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18(6):499–502. | ||
Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55(13):1318–1327. | ||
Van Bortel LM, Laurent S, Boutouyrie P, et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J Hypertens. 2012;30(3):445–448. | ||
Boutouyrie P, Hofman A, Verwoert GC, et al. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J. 2010;31(19):2338–2350. | ||
Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2013;34(28):2159–2219. | ||
John M, Hussain S, Prayle A, et al. Target renal damage: the microvascular associations of increased aortic stiffness in patients with COPD. Respir Res. 2013;14:31. | ||
Dransfield MT, Cockcroft JR, Townsend RR, et al. Effect of fluticasone propionate/salmeterol on arterial stiffness in patients with COPD. Respir Med. 2011;105(9):1322–1330. | ||
Sin DD, Man SFP. Why are patients with chronic obstructive pulmonary disease at increased risk of cardiovascular diseases? The potential role of systemic inflammation in chronic obstructive pulmonary disease. Circulation. 2003;107(11):1514–1519. | ||
Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347(20):1557–1565. | ||
Pinto-Plata VM, Mullerova H, Toso JF, et al. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax. 2006;61(1):23–28. | ||
Lowrey GE, Henderson N, Blakey JD, Corne JM, Johnson SR. MMP-9 protein level does not reflect overall MMP activity in the airways of patients with COPD. Respir Med. 2008;102(6):845–851. | ||
Basta G. Receptor for advanced glycation endproducts and atherosclerosis: from basic mechanisms to clinical implications. Atherosclerosis. 2008;196(1):9–21. | ||
O’Driscoll G, Green D, Taylor RR. Simvastatin, an HMG-coenzyme A reductase inhibitor, improves endothelial function within 1 month. Circulation. 1997;95(5):1126–1131. | ||
Mason RP, Walter MF, Jacob RF. Effects of HMG-CoA reductase inhibitors on endothelial function: role of microdomains and oxidative stress. Circulation. 2004;109(21 Suppl 1):II34–II41. | ||
Lee TM, Lin MS, Chang NC. Usefulness of C-reactive protein and interleukin-6 as predictors of outcomes in patients with chronic obstructive pulmonary disease receiving pravastatin. Am J Cardiol. 2008;101(4):530–535. | ||
Yasmin, McEniery CM, Wallace S, et al. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler Thromb Vasc Biol. 2005;25(2):372. | ||
Coulson JM, Rudd JHF, Duckers JM, et al. Excessive aortic inflammation in chronic obstructive pulmonary disease: an 18F-FDG PET pilot study. J Nucl Med. 2010;51(9):1357–1360. | ||
Alexeeff SE, Litonjua AA, Sparrow D, Vokonas PS, Schwartz J. Statin use reduces decline in lung function: VA Normative Aging Study. Am J Respir Crit Care Med. 2007;176(8):742–747. |
Supplementary materials
Inclusion criteria (protocol available on request)
- Male or female patients aged 45–80 years
- Confirmed chronic obstructive pulmonary disease: forced expired volume in one second (FEV1) 30%–80% predicted, FEV1 to forced vital capacity (FVC) ratio (FEV1/FVC) <0.7, salbutamol reversibility <12% and 200 mL, supportive smoking history
- If female and of childbearing potential, have a negative serum pregnancy test at screening and use a medically acceptable form of contraception
- Able to attend for regular clinic appointments
- In opinion of investigator, the patient will be able to comply with the requirements of the protocol
- Provide written informed consent
Exclusion criteria
- Known hypersensitivity to or side effects relating to previous statin treatment, or current therapy which includes a statin, ezetimibe or fibrate
- Clinically significant liver function abnormality; alcohol excess (defined as >21 units per week for males or 14 units for females)
- Hypercholesterolemia ≥6.5 mmol/L
- Females who are pregnant, breast feeding, or at risk of pregnancy and not using a medically acceptable form of contraception
- Any condition judged by investigator that would cause the study to be detrimental to patient
- Conditions:
- rheumatoid disease/other collagen vascular disease requiring therapy
- diabetes mellitus
- untreated hypothyroidism
- inflammatory bowel disease
- other respiratory disease
- malignancy
- documented history of ischemic heart disease; cor pulmonale or known congestive heart failure
- known alpha-1 antitrypsin deficiency
- patients planning to undergo elective surgery during the study period.
- Exacerbation in the last 4 weeks
- Significant hypoxia (PaO2 <7.3 kPa)
- Known lactose intolerance
- Therapies:
- oral prednisolone for more than one week in the last 6 months
- disease-modifying drugs (eg, gold/sulfasalazine)
- weight loss drugs
- concomitant use of warfarin, cyclosporine
- concomitant administration of potent cytochrome 3A4 inhibitors, eg, itraconazole, ketoconazole, human immunodeficiency virus protease inhibitors, erythromycin, clarithromycin, telithromycin and nefazodone.
- use of any investigational drug within 4 weeks of the baseline visit.
Sputum
Sputum results are not available in all subjects as the induction procedure could not be performed in everyone. Baseline sputum results are only available for 27 subjects (active, n=11; placebo, n=16). Of the 43 subjects without results, 23 could not perform the induction procedure because they had a post-bronchodilator FEV1 of <50%; nine subjects were unable to produce a sputum sample; five produced a sample but the slide was uncountable and therefore results could not be obtained; and six declined the sputum induction procedure. Sputum was induced by ultrasonic nebulizer (NE-U17, Omron, Milton Keynes, UK) using increasing concentrations of saline solution −3%, 4%, and 5%.
Sputum plugs were isolated from saliva macroscopically. The resulting plugs were mixed with Dulbecco’s phosphate-buffered saline (1:1) and protease inhibitor (150 μg per gram of isolated sputum) by vortexing (FB15012 TopMix, Fisher Scientific, Waltham, MA, USA) for 15 seconds before pulse sonicating for a further 15 seconds. The suspension was then filtered and a quota was removed to assess both cell number and viability by mixing with Trypan blue (1:1) and analyzing using a hemocytometer. The remaining cell suspension was centrifuged (600 g for 10 minutes at 4°C; Eppendorf 5702R).
Exhaled nitric oxide
Airway inflammation can be assessed using the fraction of exhaled nitric oxide (FeNO), which is a repeatable measure in subjects with stable chronic obstructive pulmonary disease.1 Exhaled nitric oxide levels could not be performed in every subject at both visits as technical issues were frequently encountered. Exhaled nitric oxide was measured using the five-flow method (Aerocrine, Solna, Sweden). Two measurements were recorded at each flow rate, and the mean used for analysis. Alveolar gradient and bronchial intercept were not measured directly, but parameters estimated from a model such as the slope-intercept model.2 The alveolar gradient and bronchial intercept refer to the regression line through the nitric oxide output for each of the flow rates (−10, 30, 50, 100, and 200 mL/sec). The alveolar gradient corresponds to the alveolar concentration and the bronchial intercept corresponds to the bronchial nitric oxide flux.3 The machine was calibrated every 14 days in accordance with manufacturer’s guidance.
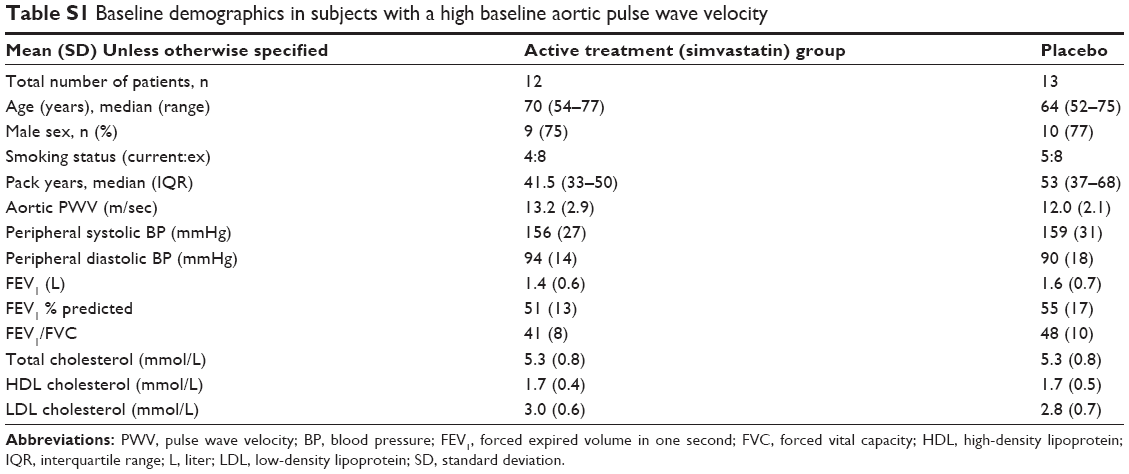 | Table S1 Baseline demographics in subjects with a high baseline aortic pulse wave velocity |
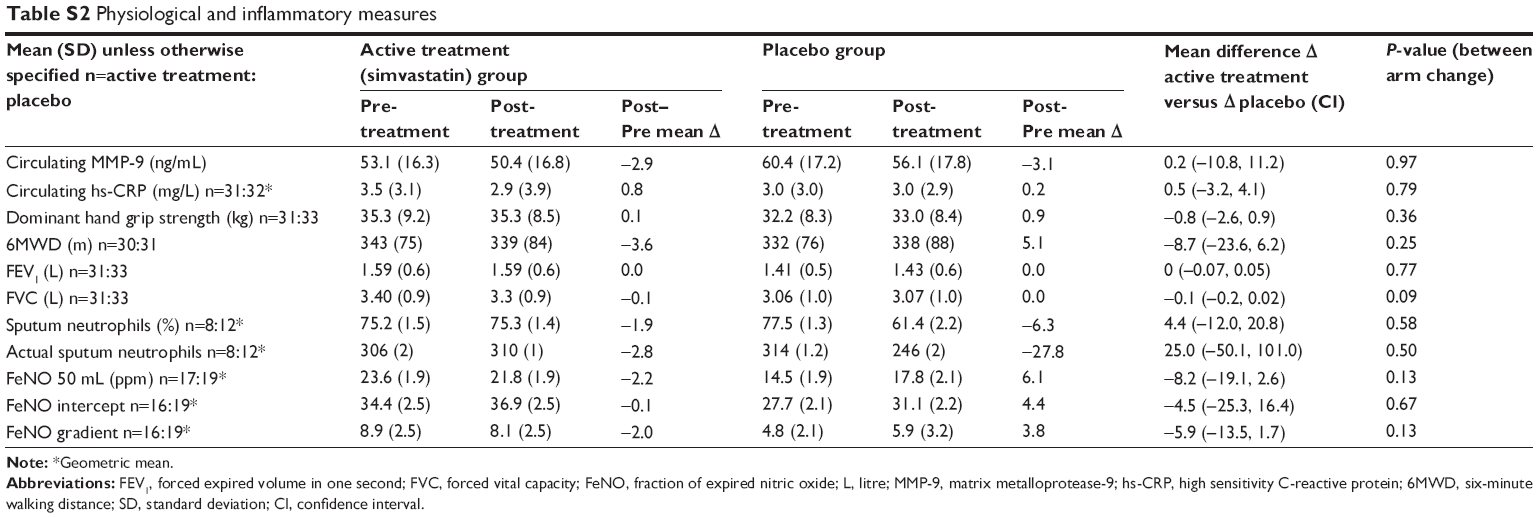 | Table S2 Physiological and inflammatory measures |
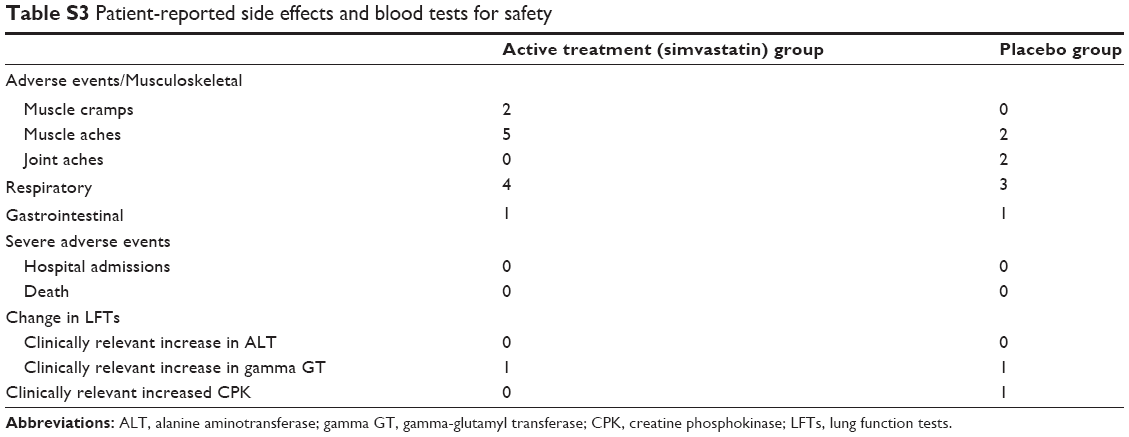 | Table S3 Patient-reported side effects and blood tests for safety |
References
Rouhos A, Kainu A, Piirila P, et al. Repeatability of exhaled nitric oxide measurements in patients with COPD. Clin Physiol Funct Imaging. 2011;31(1):26–31. | ||
Tsoukias NM, George SC. A two-compartment model of pulmonary nitric oxide exchange dynamics. J Appl Physiol. 1998;85(2):653–666. | ||
Malinovschi A, Janson C, Holm L, Nordvall L, Alving K. Basal and induced NO formation in the pharyngo-oral tract influences estimates of alveolar NO levels. J Appl Physiol. 2009;106(2):513–519. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.