Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
Cardiorenal syndrome type 2: from diagnosis to optimal management
Authors De Vecchis R, Baldi C
Received 11 July 2014
Accepted for publication 1 September 2014
Published 12 November 2014 Volume 2014:10 Pages 949—961
DOI https://doi.org/10.2147/TCRM.S63255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Renato De Vecchis,1 Cesare Baldi2
1Cardiology Unit, Presidio Sanitario Intermedio “Elena d’Aosta”, Napoli, Italy; 2Heart Department, Interventional Cardiology, AOU “San Giovanni di Dio e Ruggi D’Aragona”, Salerno, Italy
Abstract: The deterioration of renal function, which is linked to chronic heart failure by a chronological and causal relationship (ie, the so-called cardiorenal syndrome [CRS] type 2), has recently become a matter of growing debate. This debate has concerned the efficacy, safety, and cost effectiveness of the therapies that have been implemented thus far for this syndrome (for example, the intravenous [IV] loop diuretics, such as repeated IV boluses or slow IV infusions, as well as mechanical fluid removal, particularly by means of isolated ultrafiltration [IUF]). Further controversies have also emerged concerning the optimal dosage and timing of some evidence-based drugs, such as angiotensin-converting-enzyme inhibitors. The present review summarizes the currently used diagnostic tools for detecting renal damage in CRS type 2. Subsequently, the meaning of worsening renal function is outlined, as well as the sometimes inconsistent therapeutic schemes that have been implemented in order to prevent or counteract worsening renal function. The need to elaborate upon more detailed and comprehensive scientific recommendations for targeted prevention and/or therapy of CRS type 2 is also underlined. The measures usually adopted (such as the more accurate modulation of loop diuretic dose, combined with the exploitation of other diuretics that are able to achieve a sequential blockade of the nephron, as well as the use of IV administration for loop diuretics) are briefly presented. The concept of diuretic resistance is illustrated, along with the paramount operational principles of IUF in diuretic-resistant patients. Some controversies regarding the comparison of IUF with stepped diuretic therapy in patients with CRS type 2 are also addressed.
Keywords: cardiorenal syndrome type 2, worsening renal function, diuretic resistance, intravenous diuretics, isolated ultrafiltration
Classification of cardiorenal syndromes
In recent years, biomedical research has focused on a group of clinical syndromes known as cardiorenal syndromes (CRSs) in which both dysfunction of the heart and kidneys are present and connected by a causal link, with a variable degree of intensity of functional harm that can range from mild dysfunction to severe impairment of cardiac pump function, as well as of renal function.1,2 Indeed, according to its original meaning, the term “cardiorenal syndrome” would indicate a condition in which cardiac dysfunction or decompensation induces damage and/or dysfunction of the kidneys.3 However, considerable emphasis has recently been placed on the fact that the cardiac involvement – rather than being primary – can be secondary to a condition of renal failure (for example, the variable degree of cardiac injury that consistently occurs in patients suffering from advanced chronic renal failure undergoing renal replacement therapy by hemodialysis).4 Therefore, it has been necessary to provide more detailed categorization by distinguishing those conditions in which renal dysfunction clearly appears as a consequence of heart dysfunction or failure (CRS types I and II) from the conditions characterized by the chronological antecedence of renal dysfunction (CRS types III and IV). The currently accepted scheme was developed by Ronco et al1,2 which allows for the division of CRS into five types, as briefly summarized in Table 1. This classification into five categories should be integrated with the respective definitions of the terms for “heart failure”, “renal failure”, and “worsening renal function”, all of which come into play in the setting of CRS type 2.
 | Table 1 Five-part classification system for CRSs proposed by Ronco et al |
Heart failure (HF), often used to denote chronic heart failure (CHF), could be defined as a pathologic condition in which the heart is unable to exert its pump function in an effective manner (ie, it does not provide a blood flow sufficient to meet the needs of the various organs and apparatuses of the body. With regards to renal dysfunction, it may be appropriate to keep the concept of acute kidney injury distinct from that of worsening renal function (WRF) in this review. Acute kidney injury (AKI), previously known as acute renal failure, is a rapidly progressive loss of renal function,5 which is generally characterized by oliguria (decreased urine production, quantified as <400 mL/day in adults or <0.5 mL/kg/hour in children), increased serum creatinine (Cr) ie, Cr>1.3 mg/dL, and fluid and electrolyte imbalance.
Instead, the term “worsening renal function” applies to an alteration in the biochemical pattern consisting only of an increase in Cr of >0.3 mg/dL compared to baseline (for example, compared with a previous determination made before the beginning of a pharmacologic therapy or a cycle of ultrafiltration).6 In contrast, according to the criteria adopted by some other authors, WRF would be defined by an increase in Cr of ≥25% with respect to basal measurements.7,8
The present paper will mainly focus on CRS type 2 – ie, the condition characterized by preexisting chronic cardiac disease, which propitiates or provokes progressive renal damage and/or dysfunction. It is noteworthy that the mere coexistence of cardiovascular disease and chronic kidney disease (CKD) is not sufficient to make a diagnosis of true CRS type 2. In the specific setting of stable CHF, the following two prerequisites to make a diagnosis of CRS type 2 have been proposed:9 first, that CHF and CKD coexist in the patient; and second, that CHF causally underlies the occurrence or progression of CKD. The latter should be supported by both temporal association (ie, the documented or presumed onset of congestive HF temporally precedes the occurrence or progression of CKD) and by pathophysiological plausibility – that is, the manifestation and degree of kidney disease is plausibly explained by the underlying heart condition. However, the available studies are frequently unable to determine which of the two disease processes is primary versus secondary, presenting challenges when attempting to classify patients into the CRS subtype definitions. In these situations, it has been suggested that the term CRS “type 2/4” be used.10
Cardiorenal syndrome type 2: some considerations about the currently available biochemical diagnostic tools
Traditionally, the glomerular filtration rate (GFR) remains the gold standard for assessing renal function. However, measuring true, real-time GFR remains difficult in the setting of acute decompensated heart failure (ADHF) or CHF because formulas estimating GFR have been validated when Cr is in a steady state. Moreover, Cr represents an imperfect surrogate that entails important limitations. First, Cr reflects only GFR and not tubular injury directly, whereas tubular injury may help to better predict and characterize AKI and the progression of chronic kidney damage.11 In addition, Cr presents a relatively belated rise on occasion of an episode of AKI. In fact, serum concentration of this marker begins to rise many hours after AKI stabilization, when very little can be done to avoid or counteract the renal worsening in a timely manner.2
Furthermore, Cr levels are also influenced by a series of variables such as age, sex, ethnicity, and muscle mass. Consequently, in patients hospitalized with HF, especially women and the elderly who often have decreased muscle mass, seemingly low Cr levels may cause an under-recognition of renal insufficiency.12
In addition, under-appreciation of the exponential relationship between Cr and GFR, where small elevations in Cr in the near-normal range can indicate large reductions in GFR, may further cause the under-recognition of new-onset renal impairment. A kinetic study has also shown significant variability in and a low overall rate of increase of Cr after AKI, with substantial increases often not being observed until 48–72 hours after the initial damage; also, a new steady state is sometimes not reached for up to 7 days, rendering Cr a relatively belated marker of AKI.13 For the early diagnosis of CRS type 2, novel biomarkers of acute or subacute renal deterioration have been proposed.
Neutrophil gelatinase-associated lipocalin
One of the most interesting new biomarkers of AKI appears to be neutrophil gelatinase-associated lipocalin (NGAL). This 25 kDa protein, comprised of 178 amino acids, belongs to the lipocalin family and was first identified in 1993.14 It is involved in immune modulation, inflammation, and neoplastic transformation. In humans, the NGAL protein is produced and secreted by neutrophils as a result of the activation of so-called toll-like receptors; its role is to bind iron by removing it from bacteria, thereby inhibiting their growth. Moreover, it is physiologically expressed at reduced levels in several human tissues, including the kidney, lung, stomach, and colon. The expression of NGAL is markedly increased in epithelial cells that have suffered damage – for example, in the renal tubular epithelium following any type of stress or injury. The increase in NGAL serum levels occurs earlier compared to that of the classic markers of kidney damage (primarily creatinine).15 Moreover, irrespective of the other indexes, it correlates with the extent of renal tubular damage; for this reason, NGAL is used today, albeit still experimentally, as an early marker of AKI.
Cystatin C
Among the markers of renal function applicable to the study of CRSs, cystatin C is one of the most widely exploited as a diagnostic tool for use as a replacement or supplement for Cr. This proteic substance with a chain structure consisting of 120 amino acids is present in almost all tissues and body fluids, and is a marker of proximal renal tubule injury. Serum levels of cystatin C are a more precise test of kidney function (as represented by the GFR) than Cr levels.16 However, the cystatin C values alone were not found to be more accurate than the formula-adjusted estimates of GFR calculated on the basis of Cr.17 Furthermore, contrary to previous reports, cystatin C was recently proven to be influenced by body composition.18
Other biomarkers of AKI
Kidney injury molecule-1 is a protein that is detectable in the urine after ischemic or nephrotoxic insult to proximal tubular cells, and it appears to be highly specific for ischemic AKI, where it may play a clinical role.19
Other biomarkers evaluated for detecting AKI and/or for predicting the progression of CKD include cytokines/interleukins (IL)-6, IL-8, and IL-18, and N-acetyl-(D)glucosaminidase.20,21 While these various biomarkers may contribute to our pathophysiological understanding of AKI and cardiorenal disease, as yet, their relevance to routine clinical practice remains unknown.
Clinical and instrumental approach for the diagnosis of CRS type 2
A diagnosis of CRS type 2 should be based on a clinical picture of either CHF with preserved or reduced left ventricular ejection fraction on echocardiogram, joined with biochemical signs of renal dysfunction, the onset or progression of which is reasonably secondary to congestive HF. The findings indicative of renal dysfunction in the context of a CRS type 2 should include increased Cr or, in subjects with poor muscle mass, a Cr value in the near-normal range, provided that it is associated with low values (<60 mL/minute/1.73 m2) of eGFR, calculated using the Modified Diet in Renal Diseases study (MDRD) or Cockcroft–Gault equations. Further laboratory findings that are useful for a better diagnostic definition of renal damage are represented by the coexistence of albuminuria or anemia, or both. Moreover, in recent times, Maisel et al22 have highlighted the opportunity to increase the accuracy of the prognostic assessment of both CRS type 1 and 2 through the integrated use of simultaneous biochemical assays of B-type natriuretic peptide and NGAL. In fact, the latter approach would have greater accuracy when compared to traditional criteria evaluating renal function (Cr and eGFR) for identifying a possible increased risk of HF-related hospitalization or death from all causes.22
Worsening renal function: general considerations
Various definitions have been provided for the term “worsening renal function”; consequently, in recent times, there has been some effort made to establish order with the semantic confusion that has been created. In reality, the most frequently used definition refers specifically to a “worsening renal function proven by an increase in serum creatinine >0.3 mg/dL(ie, 26.4 μmol/L) compared to baseline values”.23,24 The term is usually abbreviated to the acronym WRF, which has taken the place of the now-obsolete abbreviation “ARD” (“aggravated renal dysfunction”), which was used until a few years ago to designate the same pathological phenomenon.7 It is also worth pointing out that WRF refers not to a generic renal deterioration but rather, in the ordinary meaning of the term, it indicates mostly reversible acute renal functional damage, whether spontaneous (ie, related to cardiac illness per se) or iatrogenic (ie, propitiated or generated by the therapy). In the second scenario, this means that renal harm ensues from therapies used to counteract the pulmonary and systemic venous congestion in patients presenting with an exacerbation of CHF,25 the so-called ADHF. However, outside a possible acute exacerbation of symptoms, the term WRF can also be applied to cases of CHF in which a sharp and rapid decline of the Cr (>0.3 mg/dL within 24 hours) is detected in the context of a clinically stable picture.
The first drawback of the definition given here is the fact that it does not identify an upper limit for the elevation of Cr. However, really, the worsening of Cr pertaining to this term should be relatively exiguous because a very marked rise has to be more appropriately designated as AKI (other terms include acute renal failure, acute renal insufficiency, and so on). Therefore, a new, more detailed definition of this term would be useful in order to make it more rigorous and able to distinguish the WRF picture from that of the properly termed AKI. Moreover, another issue to be considered is that WRF is a deterioration of renal function that would be naturally reversible26 and primarily driven by changes in intrarenal hemodynamics rather than by phenomena such as inflammation, ischemia, or necrosis of the renal tubules and/or renal glomeruli (so-called “vasomotor nephropathy”).23 So, the markers of inflammation, cytolysis, or apoptosis may not consistently be involved in WRF, which is in contrast to that which happens in the case of the acute tubular necrosis or contrast-induced nephropathy. As previously mentioned,17 the more suitable markers of WRF are those that explore glomerular filtration (ie, Cr) and, above all, the estimated GFR (eGFR) calculated from Cr by applying the simplified four-variable MDRD formula. Instead, the use of cystatin C does not appear to offer substantial advantages compared to the Cr-derived eGFR.18 A point to be noted is that some authors have attempted to replace the operational definition of WRF that has barely been outlined by instead adopting the alternative definition of a reduction in eGFR of ≥20% from baseline.27 However, the two definitions are not interchangeable, because there is a poor concordance between the two methods with regard to the detection of renal dysfunction. In other words, the loss of glomerular filtrate that occurs when an increase in Cr of 0.3 mg/dL takes place in the normal or near-normal range of Cr levels can even amount to an eGFR reduction of 30%–35%, while it is much less pronounced if the same increase (0.3 mg/dL – ie, 26.4 μmol/L) is realized in a patient with impaired basal renal function due to preexisting chronic parenchymal nephropathy. Therefore, the same Cr increase (0.3 mg/dL) may be indicative of a negligible decrease or a serious fall of the eGFR, depending on whether it is realized within a state of CKD (whose progression is driven by the cardiac decompensation) or in the context of normal renal function.
Worsening renal function and prognosis: the debate continues
There are still some differences of opinion among scholars regarding the prognostic significance that should be attributed to WRF. This phenomenon affects no less than 20% of patients with CHF who undergo a single course of intravenous (IV) infusions with loop diuretics; however, it has been demonstrated that in at least half of the cases, the iatrogenic increase in Cr is reversible within approximately 1 week after the end of the course of infusional therapy.7 The ever-changing evolution of theories has not spared this topic, since some have concluded by rejecting an unfavorable prognostic significance for this finding28 which, in contrast, had been emphasized in the past.7,29 Indeed, according to some, this elevation in Cr may simply be the epiphenomenon of a condition of greater clinical severity and hemodynamic instability of cardiac or combined (renal and cardiac) disease.26,28 In fact, the therapy itself, with IV diuretics and angiotensin-converting-enzyme (ACE) inhibitors at full doses, is one of the main precipitating causes of WRF. However, in the opinion of some authors,28 persistent hemodynamic congestion, with or without any overt clinical signs, should consistently be treated by increasing the dose of diuretics, since intensive decongestive treatments in CHF patients would have the potential to achieve better cardiovascular outcomes and increased survival.28,30
Diuretics for acute and chronic heart failure: preliminary concepts
Although loop diuretics are the usual treatment for counteracting clinical or hemodynamic congestion in patients with CHF, as well as any type of CRS, in reality, their efficacy and safety has never been tested by means of randomized controlled trials. Even the optimal dose of diuretics is still controversial.
In the context of CRS type 1, when exacerbation of dyspnea at rest and orthopnea and/or worsening of peripheral edema are present, with poor response to oral diuretics, the IV administration of loop diuretics (furosemide or another drug of the same class) is usually practiced for a brief period (usually 3–12 days) at doses ranging from 40–2,000 mg of furosemide per day.31 However, a specific therapeutic behavior regarding this clinical problem (ie, dosing, timing, and way of administration of furosemide and similar compounds) has not yet been accurately established on the basis of dedicated scientific guidelines. Hypokalemia, ototoxicity, hypotension, myocardial fibrosis, activation of neurohormones, and the paradoxical further impairment of CRS are all reported as potential adverse effects of IV diuretic therapy at high doses.32 According to some,33 the previously mentioned side effects should discourage the use of high doses of IV loop diuretics in patients with HF in whom signs and symptoms are adequately controlled. If IV loop diuretics are necessary due to the exacerbation of dyspnea or widespread edema, they should be used at the minimum efficacious dose (Table 2). In patients with acute decompensation, vast literature would seem to argue an increased efficacy and safety of the continuous IV infusion of furosemide compared to its intermittent administration by means of repeated bolus doses.32,34,35 However, judging by a relatively recent randomized trial,36 the continuous infusion of furosemide would not be superior to repeated IV boluses with regard to the primary endpoints of this study (patients’ global assessment of symptoms and the changes in renal function).
 | Table 2 Diuretic dosing for acute HF according to the ASCEND-HF model |
Therapies for preventing and/orcounteracting cardiac and renal deterioration in CRS type 2: a rather controversial issue
With regard to the therapeutic problems of CRS type 2, the main issues are represented either by the need to prevent new-onset renal dysfunction, emerging in a setting of CHF, or by the need to adequately counteract renal dysfunction once it has developed, by providing suitable measures in order to promote the attenuation or regression of cardiac and renal damage whenever possible. In CRS type 1, WRF occurs in the context of an acute exacerbation of HF (ADHF). Thus, this ensues from the overlapping and interaction of acutely reduced cardiac output and increased central venous pressure (resulting in renal venous congestion); the result that emerges is a reduction in renal blood flow with a decrease of eGFR, and the consequent increased concentration of metabolic wastes in the blood (for example, creatinine). Instead, in CRS type 2, the pathogenetic mechanism by which a cardiac dysfunction elicits a new-onset renal dysfunction or induces the detrimental progression of a preexisting CKD is not so obvious; in fact, it has not yet been completely elucidated. Indeed, in this case, the deterioration of renal filtration occurs in patients who are not affected by the clinical signs and symptoms of hemodynamic destabilization (in fact, patients who develop a CRS type 2 are free from ADHF by definition). Thus, the mechanisms underlying WRF probably differ based on acute versus chronic HF. Chronic HF is likely to be characterized by a long-term situation of renal venous congestion and reduced intrarenal perfusion and filtration gradients. In addition, microvascular and macrovascular renal disorders (so-called chronic ischemic nephropathy) may be present and contribute to harm renal function. In this context, a very important pathogenetic role should be ascribed to the pharmacotherapies used in the management of CHF that may worsen renal function when applied in a nonrigorous and appropriate manner. In fact, diuresis-associated hypovolemia, the early introduction of renin–angiotensin–aldosterone system (RAAS) blockade and drug-induced hypotension, have all been suggested for a long time as being very important contributing factors37 to the genesis or aggravation of CRS type 2. In addition, the so-called “resistance to diuretics” may play a crucial role in propitiating overzealous therapeutic approaches with excessive increases in diuretic dosing, so as to unfortunately induce various harmful phenomena such as exaggerated stimulation of the tubuloglomerular feedback mechanism and activation of the RAAS, with consequent reactive vasoconstriction of the renal afferent arterioles and fall of GFR, which then results in increased Cr.
In addition, the concept of resistance to diuretics is controversial, and it is still far from having a clear and universally accepted definition. In general, compared with normal individuals, patients with CHF need higher doses of loop diuretics to achieve similar sodium excretion, and the magnitude of the “maximal” response is attenuated.38 In patients with CHF, this relatively weak response to loop diuretics is further attenuated in the case of very prolonged oral diuretic therapy (ie, dating from many months or years earlier, so the so-called “braking phenomenon” frequently ensues).
This term applies to the long-term use of diuretics and refers to a decline in the magnitude of natriuresis after the administration of sequential doses. Diuretic resistance can be reasonably suspected when the urine output is relatively poor (for example, <1,000 mL per day), in spite of the maximal tolerated oral dose of a loop diuretic (for example, 250 mg of furosemide per day), and in the presence of signs and symptoms of seemingly refractory hydrosaline retention. This should prompt the physician to overcome the apparent condition of refractoriness to oral diuretics by altering the diuretic regimen in the following ways: 1) by combining thiazide diuretics with loop diuretics (to block increased distal sodium reabsorption); 2) by preferably adopting the IV method of administration for loop diuretics (to be given at the same doses or at higher doses compared to those given orally); 3) using continuous diuretic infusions to avoid the phenomenon of postdiuretic salt retention;39 and 4) aldosterone receptor antagonists should be taken into consideration as an adjunctive treatment to resolve congestion and reduce the diuretic dose.37
Another aspect that is worth considering is the widespread use of combination therapy with IV loop diuretics plus RAAS inhibitors (ie, ACE inhibitors and/or angiotensin II receptor blockers [ARBs]) in CHF patients. As mentioned earlier, the use of high doses of IV loop diuretics in patients with CHF, in whom signs and symptoms are adequately controlled, should be strongly discouraged because of the possible IV loop diuretic-related side effects (hypokalemia, hypotension, marked neurohormonal activation, and possible renal impairment). Furthermore, the risk of IV loop diuretic-related renal impairment (WRF) may be further aggravated when an ACE inhibitor or ARB at full dose is maintained in the therapeutic schedule, in combination with an IV diuretic regimen. This has been interpreted as a consequence of impaired constrictive tone of the glomerular efferent arteriole (due to the angiotensin II blockade) joined with an exaggerated fall in the effective intravascular volume, due in turn to the IV diuretic. Thus, these pharmacological actions are able to elicit tubular dysfunction and tubuloglomerular feedback, resulting in a sustained decrease in intraglomerular pressure.40 Thus, even though combined therapy with loop diuretics and renin–angiotensin blockers at high doses is able to relieve congestion in the setting of CHF, it may also decrease renal flow and lower the glomerular filtration fraction, thereby eliciting a deleterious marked impairment of GFR. It should be noted that renal insufficiency in CRS type 2 can be caused not only by renal venous congestion, but also by decreased renal perfusion due to the decrease in cardiac output and/or hypotension (decreased preload), and/or activation of the neurohormonal cascade, leading to a “vasomotor nephropathy” with pronounced and sustained renal reactive vasoconstriction. Thus, in the setting of CRS type 2, iatrogenic influences may account for renal damage as much as the congestive nephropathy itself.25,41,42 However, higher doses of certain drugs (loop diuretics and RAAS inhibitors, such as ACE inhibitors and ARBs) may simply detect patients with severe hemodynamic impairment and, thus, a propensity to renal dysfunction rather than being responsible per se for worsening renal function.40,43,44 It should be noted that ACE inhibitors do not harm the kidney, but instead they change the intrarenal hemodynamics and reduce the filtration fraction. This is a protective mechanism in the case of hypertension, diabetic nephropathy, chronic inflammatory disease of glomeruli, or any nephropathy with albuminuria.2,45 Nevertheless, the reduction in glomerular filtration fraction (ACE inhibitor-related) loses any renal protective significance when occurring in the context of reduced renal flow due to a pathological decrease in the perfusion gradient across the kidney (ie, congestive nephropathy typical of congestive CHF). Similarly, any drug-induced fall in filtration fraction is surely harmful when occurring in CHF patients characterized by markedly diminished effective intravascular volume after undergoing overtreatment for hemodynamic congestion, such as those becoming relatively hypovolemic following overzealous diuretic treatment at high doses.7,32,46
Other therapies available for combined cardiac and renal impairment in chronic heart failure
Several drugs of ancillary importance, or that exhibit unproven or controversial efficacy have been introduced in the therapeutic armamentarium for HF complicated by renal impairment. They serve as a testament to the considerable effort performed in recent decades to achieve innovative pharmacological tools with complementary or alternative mechanisms of action with respect to loop diuretics and evidence-based drugs for HF, such as RAAS blockers (namely, ACE inhibitors and ARAs) and beta-blockers. With respect to the number of novel drugs that have been proposed for the treatment of HF with renal compromise, some have entered into routine use (as is the case for nesiritide, which is approved for ADHF treatment in the United States, but not in Europe, while others have received official consent for clinical indications other than the heart failure (the case of tolvaptan, which has been approved in Europe for the treatment of syndrome of inappropriate antidiuretic hormone secretion (SIADH), but not specifically for therapy of hyponatriemic heart failure, with or without renal impairment47). Finally, some of these drugs are of primarily historical importance because they have triggered research on pulmonary hypertension (either primary or secondary to diseases that exclusively engage the right heart), but they have not received any validation for the treatment of left ventricular failure, or for the treatment of CRS type 2 (as is the case for bosentan and the other endothelin receptor antagonists [ERAs]).48
Nesiritide
Nesiritide is a synthetic polypeptide of 32 amino acids that reproduces the structure of B-type natriuretic peptide, normally secreted by the ventricular myocardium. It has the typical functions of the natural peptide; that is, it induces improved relaxation of the smooth muscle cells of the venous and arterial vessels in response to acute increases in ventricular volume, and it antagonizes the vasoconstriction, sodium retention, and antidiuretic effects of the activated RAAS. In particular, nesiritide-induced relaxation of vascular smooth muscle cells arises from its stimulation of cyclic guanosine monophosphate. Nesiritide has been introduced for a long time in the treatment of ADHF in the US, while its use has not been validated by the regulatory authority in Europe (the European Medicines Agency [EMEA]). Its clinical indications are limited to the treatment of acute episodes of cardiac failure because it is only available in IV formulation. Indeed, the renal safety profile of this drug does not contraindicate its use in patients with CRS type 2 who are affected by episodes of symptom exacerbation.49,50 Nesiritide reproduces some of the hemodynamic effects of nitroglycerin (in particular, a rapid and effective reduction in ventricular preload due to the hematic pooling effect at the level of the great venous vessels; this is a very helpful hemodynamic adjustment in the setting of acute or subacute pulmonary congestion).33 However, the purchase price of nesiritide is approximately 40 times higher than that of the standard vasodilator agent for this indication, namely nitroglycerin. Given this relatively high financial burden, even in the US, specific restrictive protocols have been implemented by some institutions in order to recommend the administration of nitroglycerin and IV diuretics (using ≥2 times the usual daily diuretic dose) before using nesiritide.51
Antidiuretic hormone antagonists: tolvaptan
The identification of hypervolemic hyponatremia as a potential target for HF therapy has led to the clinical development of vasopressin receptor antagonists, such as tolvaptan, conivaptan, and lixivaptan. In particular, the potential role played by tolvaptan in the treatment of HF was extensively investigated by means of the EVEREST Clinical Status Trials.52,53 In these studies, a thorough exploration of the effects of tolvaptan on the cardiovascular outcomes in patients with decompensated HF was conducted with regard to multiple endpoints, such as weight loss, clinical status (dyspnea and other symptoms), renal function, long-term mortality, rehospitalization for HF, and so on. The addition of tolvaptan to standard therapy in patients hospitalized for ADHF resulted in greater weight loss and significant improvement in dyspnea without adversely affecting renal function.52 However, tolvaptan therapy initiated during hospitalization for ADHF had no effect on the long-term mortality or rehospitalization for HF.53 Tolvaptan was approved for use in the US in May 2009 for patients with hypervolemic and euvolemic hyponatremia, including those with HF, cirrhosis, and SIADH. In contrast, in Europe, tolvaptan was approved only for the treatment of adult patients with hyponatremia secondary to SIADH.47
Endothelin receptor antagonists: bosentan
ERAs have been proposed in the late 1990s in the treatment of HF. However, the initial enthusiasm of the supposed therapeutic potential of ERAs in HF has considerably declined because of unfavorable outcomes of the ENABLE study,54 conducted with the use of bosentan (a nonselective ERA) in a large cohort of patients with severe HF. In fact, treatment with bosentan appeared to entail an early risk of worsening HF requiring hospitalization, as a consequence of fluid retention. It was suggested that further studies using even lower doses of bosentan or more aggressive concomitant diuretic therapy might prevent this unfavorable outcome.48,54 At present, bosentan or other ERAs are exclusively exploited for treatment of pulmonary hypertension, provided that it is not related to any type of left ventricular failure.55
Serelaxin
Serelaxin, a recombinant form of human relaxin-2, triggers adaptive cardiovascular effects during pregnancy that could be helpful in the HF population, mainly through nitric oxide-mediated vasodilation. Serelaxin has shown promise in the treatment of HF, even complicated by renal impairment (CRS type1).56 However, serelaxin has recently been rejected by both the EMEA and the US Food and Drug Administration for use in patients with HF.57 The reason for these rejections would primarily consist of concerns about the benefits of the drug, considering that there are not many studies powered for “hard” outcomes.
Ultrafiltration for cardiorenal syndrome type 2
The main rationale for use of isolated ultrafiltration (IUF) in the setting of CRS type 2 would be for the rapid correction of fluid overload when standard management (for example, high-dose IV diuretics with or without inotrope agents) has been unsuccessful.58,59 Indeed, the current American Heart Association/American College of Cardiology and the European Society of Cardiology treatment guidelines establish that IUF is a reasonable option in patients with congestion when the altered fluid status in decompensated patients has been shown not to respond satisfactorily to medical therapy (class IIa, level of evidence B).60,61
Adverse reactions and resistance to diuretics have caused researchers to explore the possibility of using IUF instead. As in all modalities of renal replacement therapy, it is also the case for IUF that an extracorporeal circuit is provided through which blood is pump-driven from a venous access into the filter, and then returned to the patient. This procedure almost always requires blood anticoagulation.40,43,44 During IUF, water crosses a semipermeable membrane in the filter by means of a convective process, which is driven by the hydrostatic pressure difference across the filter membrane. Solutes that are smaller than the membrane pores, such as electrolytes and urea contained in that amount of plasma water, are removed concurrently and at the same concentration as the plasma water (Figure 1). Thus, by allowing for only isotonic removal, IUF leaves unchanged the plasma concentration of low-molecular-weight solutes, such as sodium and other small solutes.
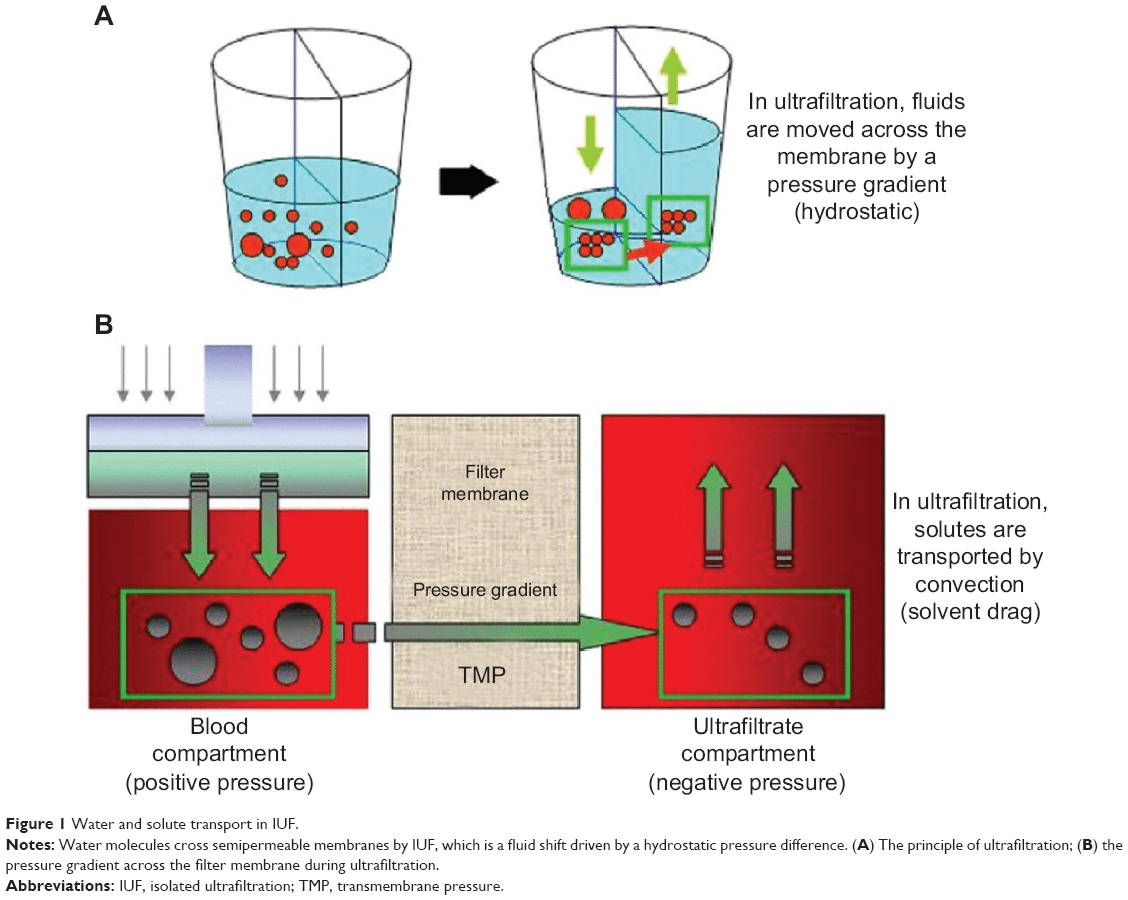 | Figure 1 Water and solute transport in IUF. |
Notably, this method of convective removal of plasma water does not include the partial or total replacement by a clean solution with known electrolyte concentrations, as in the case for hemofiltration (usually 2–3 L/hour in continuous forms of hemofiltration, or up to 6–8 L/hour in high-volume hemofiltration/hemodiafiltration). Obviously, hemofiltration has a depurative efficiency that is far superior to IUF.44,45 In the latter case, the electrolyte concentration across the filter does not change. Thus, IUF should not have any effect on blood electrolyte levels or on binding urea nitrogen (Table 3).43,44 The volume of fluid removed by IUF can be accurately predicted since it can be determined beforehand by adjusting the instrument parameters. Ultrafiltration does not cause any neurohormonal activation mediated by the macula densa, unlike diuretics, because the subtraction of the fluids is achieved through a circuit that does not involve the urinary tract or the renal chemoreceptor apparatus of the macula densa (Table 3).
 | Table 3 Rationale and therapeutic targets of IUF in heart failure |
Comparing IV diuretics and IUF for fluid removal in CRS type 2
In the literature, there are only a small number of randomized controlled trials38,62–65 that have been designed to compare IUF with IV diuretics for treating exacerbations of CHF and/ordiuretic-resistant cardiac decompensation. Moreover, they include a relatively small number of patients. Considerable methodological differences, such as different deadlines of the adopted endpoints in interstudy comparisons, do not allow for the pooling and subsequent meta-analysis of the data from some studies.66 However, a qualitative analysis is possible, even in the absence of criteria for methodological homogeneity that would be needed to conduct a meta-analysis (quantitative analysis). A brief description is provided for the two largest studies (UNLOAD38 and CARRESS-HF65), which compared IV diuretics with ultrafiltration in CHF.
In the UNLOAD trial conducted by Costanzo et al38 the early application of IUF was compared to conventional therapy with diuretics in the setting of ADHF (200 patients in total) with regard to weight loss and dyspnea assessment 48 hours after randomization (primary outcome endpoints); moreover, a comparison was made between the two arms regarding net fluid loss at 48 hours, functional capacity, HF-related rehospitalizations, and unscheduled visits in a period of 90 days (secondary outcome endpoints). At 48 hours, weight (5.0±3.1 kg versus 3.1±3.5 kg; P=0.001) and net fluid loss (4.6 L versus 3.3 L; P=0.001) were greater in the IUF arm when compared to the conventional therapy arm, respectively, whereas dyspnea scores were similar. At 90 days, the IUF group reported fewer unscheduled visits to the emergency department (14 of a total of 65 visits [21%] versus 29 of 66 visits [44%]; P=0.009), fewer patients rehospitalized for HF (16 of 89 [18%] versus 28 of 87 [32%]; P=0.037), and shorter hospital stays for rehospitalization (1.4±4.2 days per patient versus 3.8±8.5 days per patient; P=0.022).38,67,68 No statistically significant difference in Cr increase at 48 hours was identified by comparing the IUF patients with those undergoing IV diuretic therapy.
In the CARRESS-HF study by Bart et al65 a total of 188 patients with ADHF complicated by worsened renal function were randomized to a stepped therapy with diuretics (94 patients) or IUF (94 patients). The primary endpoint was the bivariate change from baseline in the Cr level and body weight, as assessed 96 hours after randomization. Patients were followed for 60 days. At 96 hours, the mean change in the creatinine level was −0.04±0.53 mg/dL (−3.5±46.9 μmol/L) in the diuretics group versus +0.23±0.70 mg/dL (20.3±61.9 μmol/L) in the IUF group (P=0.003). At the same time point, a higher percentage of IUF patients had one or more serious adverse event compared to the diuretics arm (72% versus 57%, respectively; P=0.03). In contrast, no significant difference was detected regarding weight loss 96 hours after enrolment when comparing diuretic-treated patients with those assigned to the IUF (5.5±5.1 kg in the former versus 5.7±3.9 kg in the latter; P=0.58).
Based on this study, ADHF patients with CRS assigned to the IUF group had a more pronounced increase in Cr level at 96 hours, as well as a higher rate of serious adverse events when compared to those in the diuretics arm. Identifying the main grounds able to explain the differences between the UNLOAD38 and CARRESS-HF65 trials would be interesting. In particular, the collected baseline values were sufficient to allow us to suspect that a more deteriorated renal condition was present at baseline in the CARRESS-HF patients when compared to those recruited for the UNLOAD study. In fact, in the former, the basal Cr values were 2.09 mg/dL (1.71–2.65 mg/dL) and 1.90 mg/dL (1.57–2.37 mg/dL; median plus interquartile range [IQR]) for the IV loop diuretics and IUF arms, respectively (Table 4). In contrast, a much lower Cr level of 1.5±0.5 mg/dL (mean ± standard deviation [SD]) for both the IV loop diuretics and IUF arms was observed at baseline in UNLOAD patients.38
 | Table 4 Baseline characteristics of the renal parameters found in the UNLOAD trial and CARRESS-HF trial |
In the CARRESS-HF study65 the blood urea nitrogen (BUN) was 50.5 mg/dL (39–64 mg/dL) and 48.7 mg/dL (39.5–66 mg/dL; median plus IQR) in the IV loop diuretics and IUF arms, respectively. Instead, a blood urea nitrogen (BUN) of only 33±20 mg/dL and 32±16 mg/dL (mean ± SD) in the IV loop diuretics and IUF arms, respectively, was found in UNLOAD patients at baseline. Based on these data, a CRS was likely present at baseline in the majority of CARRESS-HF patients, while similar renal dysfunction was mostly absent in the UNLOAD patients at entry. Although a condition of chronic renal insufficiency at baseline does not contraindicate the ultrafiltration per se, it could likely enhance the risk of unfavorable renal consequences resulting from the relatively rapid fluid subtraction achieved by IUF. Indeed, in the presence of frank renal insufficiency, it is usually recommended that IUF be avoided, and that other renal replacement techniques that have higher depurative efficiency, such as continuous hemofiltration or high-volume hemofiltration/hemodiafiltration, be used instead. In the CARRESS-HF trial,65 the increase in Cr observed at 96 hours in the IUF arm was +0.23±0.7 mg/dL, while the variation in Cr found in the diuretics arm was −0.04±0.53 mg/dL. Although sufficient to generate statistical significance (P=0.003), these results were not enough to systematically attain a properly termed “worsening renal function” (defined by an increase higher than 0.3 mg/dL from baseline). In fact, the seemingly disappointing IUF results from the CARRESS-HF trial should be reevaluated, taking into consideration that enrolled patients, all of whom had serious basal renal dysfunction, should have undergone a different kind of renal replacement therapy. In contrast, IUF should be regarded as a very valuable therapeutic tool40 in cases of ADHF characterized by nonadvanced renal dysfunction, especially when diuretic failure or diuretic resistance has been detected.
Conclusion
The CHF-related deterioration of renal function (ie, CRS type 2) found in at least 25% of cases of CHF has become a matter of growing debate. In fact, there are some relevant issues concerning efficacy, safety, and cost effectiveness that still remain unresolved with regard to therapy with IV loop diuretics, as well as with mechanic fluid removal (in particular, with the use of IUF). Further controversies have also emerged concerning the optimal dosage and timing of some evidence-based drugs, such as ACE inhibitors. In the future, an increased understanding of the hemodynamic derangements and underlying mechanistic pathways of CRS type 2 will hopefully help guide the development of novel therapeutic strategies and allow for the more careful characterization of patient subpopulations in which targeted therapies will have the greatest benefit.
Disclosure
The authors have no conflicts of interest to declare.
References
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52(19):1527–1539. | ||
Ronco C, McCullough P, Anker SD, et al; Acute Dialysis Quality Initiative (ADQI) consensus group. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31(6):703–711. | ||
Ronco C, House AA, Haapio M. Cardiorenal syndrome: refining the definition of a complex symbiosis gone wrong. Intensive Care Med. 2008;34(5):957–962. | ||
Schrier RW. Cardiorenal versus renocardiac syndrome: is there a difference? Nat Clin Pract Nephrol. 2007;3(12):637. | ||
Moore EM, Bellomo R, Nichol AD. The meaning of acute kidney injury and its relevance to intensive care and anaesthesia. Anaesth Intensive Care. 2012;40(6):929–948. | ||
Krumholz HM, Chen YT, Vaccarino V, et al. Correlates and impact on outcomes of worsening renal function in patients > or =65 years of age with heart failure. Am J Cardiol. 2000;85(9):1110–1113. | ||
Cioffi G, Tarantini L, Pulignano G, et al. Prevalence, predictors and prognostic value of acute impairment in renal function during intensive unloading therapy in a community population hospitalized for decompensated heart failure. J Cardiovasc Med (Hagerstown). 2007;8(6):419–427. | ||
De Vecchis R, Ciccarelli A, Ariano C, et al. [Renoprotective effect of small volumes of hypertonic saline solution in chronic heart failure patients with marked fluid retention: results of a case-control study]. Herz. 2011;36(1):12–17. German. | ||
Cruz DN, Schmidt-Ott KM, Vescovo G, et al. Pathophysiology of cardiorenal syndrome type 2 in stable chronic heart failure: workgroup statements from the eleventh consensus conference of the Acute Dialysis Quality Initiative (ADQI). Contrib Nephrol. 2013;182:117–136. | ||
Bagshaw SM, Cruz DN, Aspromonte N, et al; Acute Dialysis Quality Initiative Consensus Group. Epidemiology of cardio-renal syndromes: workgroup statements from the 7th ADQI Consensus Conference. Nephrol Dial Transplant. 2010;25(5):1406–1416. | ||
Aghel A, Shrestha K, Mullens W, Borowski A, Tang WH. Serum neutrophil gelatinase-associated lipocalin (NGAL) in predicting worsening renal function in acute decompensated heart failure. J Card Fail. 2010;16(1):49–54. | ||
Amsalem Y, Garty M, Schwartz R, et al. Prevalence and significance of unrecognized renal insufficiency in patients with heart failure.Eur Heart J. 2008;29(8):1029–1036. | ||
Waikar SS, Bonventre JV. Creatinine kinetics and the definition of acute kidney injury. J Am Soc Nephrol. 2009;20(3):672–679. | ||
Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993;268(14):10425–10432. | ||
Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet. 2005;365(9466):1231–1238. | ||
Roos JF, Doust J, Tett SE, Kirkpatrick CM. Diagnostic accuracy of cystatin C compared to serum creatinine for the estimation of renal dysfunction in adults and children–a meta-analysis. Clin Biochem. 2007;40(5–6):383–391. | ||
Stevens LA, Coresh J, Schmid CH, et al. Estimating GFR using serum cystatin C alone and in combination with serum creatinine:a pooled analysis of 3,418 individuals with CKD. Am J Kidney Dis. 2008;51(3):395–406. | ||
Macdonald J, Marcora S, Jibani M, et al. GFR estimation using cystatin C is not independent of body composition. Am J Kidney Dis. 2006;48(5):712–719. | ||
Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV. Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol. 2006;290(2):F517–F529. | ||
Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. 2005;16(10):3046–3052. | ||
Liangos O, Perianayagam MC, Vaidya VS, et al. Urinary N-acetyl-beta-(D)-glucosaminidase activity and kidney injury molecule-1 level are associated with adverse outcomes in acute renal failure. J Am Soc Nephrol. 2007;18(3):904–912. | ||
Maisel AS, Mueller C, Fitzgerald R, et al. Prognostic utility of plasma neutrophil gelatinase-associated lipocalin in patients with acute heart failure: the NGAL EvaLuation Along with B-type NaTriuretic Peptide in acutely decompensated heart failure (GALLANT) trial. Eur J Heart Fail. 2011;13(8):846–851. | ||
Gottlieb SS, Abraham W, Butler J, et al. The prognostic importance of different definitions of worsening renal function in congestive heart failure. J Card Fail. 2002;8(3):136–141. | ||
Metra M, Nodari S, Parrinello G, et al. Worsening renal function in patients hospitalised for acute heart failure: clinical implications and prognostic significance. Eur J Heart Fail. 2008;10(2):188–195. | ||
MacFadyen RJ, Ng Kam Chuen MJ, Davis RC. Loop diuretic therapy in left ventricular systolic dysfunction: has familiarity bred contempt for a critical but potentially nephrotoxic cardio renal therapy? Eur J Heart Fail. 2010;12(7):649–652. | ||
Testani JM, McCauley BD, Kimmel SE, Shannon RP. Characteristics of patients with improvement or worsening in renal function during treatment of acute decompensated heart failure. Am J Cardiol. 2010;106(12):1763–1769. | ||
Testani JM, McCauley BD, Chen J, Shumski M, Shannon RP. Worsening renal function defined as an absolute increase in serum creatinine is a biased metric for the study of cardio-renal interactions. Cardiology. 2010;116(3):206–212. | ||
Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP. Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation. 2010;122(3):265–272. | ||
Smith GL, Lichtman JH, Bracken MB, et al. Renal impairment and outcomes in heart failure: systematic review and meta-analysis. J Am Coll Cardiol. 2006;47(10):1987–1996. | ||
Lucas C, Johnson W, Hamilton MA, et al. Freedom from congestion predicts good survival despite previous class IV symptoms of heart failure. Am Heart J. 2000;140(6):840–847. | ||
De Vecchis R, Esposito C, Ariano C, Cantatrione S. Hypertonic saline plus i.v. furosemide improve renal safety profile and clinical outcomes in acute decompensated heart failure: A meta-analysis of the literature. Herz. Epub 2014 Mar 30. | ||
Brandimarte F, Mureddu GF, Boccanelli A, et al. Diuretic therapy in heart failure: current controversies and new approaches for fluid removal. J Cardiovasc Med (Hagerstown). 2010;11(8):563–570. | ||
Ezekowitz JA, Hernandez AF, Starling RC, et al. Standardizing care for acute decompensated heart failure in a large megatrial: the approach for the Acute Studies of Clinical Effectiveness of Nesiritide in Subjects with Decompensated Heart Failure (ASCEND-HF). Am Heart J. 2009;157(2):219–228. | ||
Howard PA, Dunn MI. Effectiveness of continuous infusions of loop diuretics for severe heart failure. J Cardiovasc Med (Hagerstown). 2006;7(1):5–10. | ||
Leto L, Aspromonte N, Feola M. [Continuous infusion versus bolus injection of loop diuretics in acute heart failure: a literature review]. G Ital Cardiol (Rome). 2012;13(4):263–272. Italian. | ||
Felker GM, Lee KL, Bull DA, et al; NHLBI Heart Failure Clinical Research Network. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. 2011;364(9):797–805. | ||
Liang KV, Williams AW, Greene EL, Redfield MM. Acute decompensated heart failure and the cardiorenal syndrome. Crit Care Med. 2008;36(1 Suppl):S75–S88. | ||
Costanzo MR, Guglin ME, Saltzberg MT, et al; UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol. 2007;49(6):675–683. | ||
Shah SU, Anjum S, Littler WA. Use of diuretics in cardiovascular diseases: (1) heart failure. Postgrad Med J. 2004;80(942):201–205. | ||
Kazory A, Ross EA. Contemporary trends in the pharmacological and extracorporeal management of heart failure: a nephrologic perspective. Circulation. 2008;117(7):975–983. | ||
Butler J, Forman DE, Abraham WT, et al. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J. 2004;147(2):331–338. | ||
Knight EL, Glynn RJ, McIntyre KM, Mogun H, Avorn J. Predictors of decreased renal function in patients with heart failure during angiotensin-converting enzyme inhibitor therapy: results from the studies of left ventricular dysfunction (SOLVD). Am Heart J. 1999;138(5 Pt 1):849–855. | ||
Kazory A. Ultrafiltration does not affect certain predictors of outcome in heart failure. Int J Cardiol. 2010;143(1):1–3. | ||
Fiaccadori E, Regolisti G, Maggiore U, et al. Ultrafiltration in heart failure. Am Heart J. 2011;161(3):439–449. | ||
Ravid M, Lang R, Rachmani R, Lishner M. Long-term renoprotective effect of angiotensin-converting enzyme inhibition in non-insulin-dependent diabetes mellitus. A 7-year follow-up study. Arch Intern Med. 1996;156(3):286–289. | ||
De Vecchis R, Ciccarelli A, Pucciarelli A. Unloading therapy by intravenous diuretic in chronic heart failure: a double-edged weapon? J Cardiovasc Med (Hagerstown). 2010;11(8):571–574. | ||
Reilly T, Schork MR. Vasopressin antagonists: Pharmacotherapy for the treatment of heart failure. Ann Pharmacother. 2010;44(4):680–687. | ||
Packer M, McMurray J, Massie BM, et al. Clinical effects of endothelin receptor antagonism with bosentan in patients with severe chronic heart failure: results of a pilot study. J Card Fail. 2005;11(1):12–20. | ||
Witteles RM, Kao D, Christopherson D, et al. Impact of nesiritide on renal function in patients with acute decompensated heart failure and pre-existing renal dysfunction a randomized, double-blind, placebo-controlled clinical trial. J Am Coll Cardiol. 2007;50(19):1835–1840. | ||
van Deursen VM, Hernandez AF, Stebbins A, et al. Nesiritide, renal function, and associated outcomes during hospitalization for acute decompensated heart failure: results from the Acute Study of Clinical Effectiveness of Nesiritide and Decompensated Heart Failure (ASCEND-HF). Circulation. 2014;130(12):958–965. | ||
Noviasky JA, Kelberman M, Whalen KM, Guharoy R, Darko W. Science or fiction: use of nesiritide as a first-line agent? Pharmacotherapy. 2003;23(8):1081–1083. | ||
Gheorghiade M, Konstam MA, Burnett JC, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA. 2007;297(12):1332–1343. | ||
Konstam MA, Gheorghiade M, Burnett JC Jr, et al; Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study with Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA. 2007;297(12):1319–1331. | ||
Kalra PR, Moon JC, Coats AJ. Do results of the ENABLE (Endothelin Antagonist Bosentan for Lowering Cardiac Events in Heart Failure) study spell the end for non-selective endothelin antagonism in heart failure? Int J Cardiol. 2002;85(2–3):195–197. | ||
Galiè N, Rubin LJ, Hoeper M, et al. Treatment of patients with mildly symptomatic pulmonary arterial hypertension with bosentan (EARLY study): a double-blind, randomised controlled trial. Lancet. 2008;371(9630):2093–2100. | ||
Metra M, Cotter G, Davison BA, et al; RELAX-AHF Investigators. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61(2):196–206. | ||
Neverova N, Teerlink JR. Serelaxin: a potential new drug for the treatment of acute heart failure. Expert Opin Investig Drugs. 2014;23(7):1017–1026. | ||
Costanzo MR. Ultrafiltration in the management of heart failure. Curr Opin Crit Care. 2008;14(5):524–530. | ||
Wertman BM, Gura V, Schwarz ER. Ultrafiltration for the management of acute decompensated heart failure. J Card Fail. 2008;14(9):754–759. | ||
Hunt SA, Abraham WT, Chin MH, et al; American College of Cardiology Foundation; American Heart Association. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53(15):e1–e90. | ||
McMurray JJ, Adamopoulos S, Anker SD, et al; Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology; ESC Committee for Practice Guidelines. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14(8):803–869. | ||
Bart BA, Boyle A, Bank AJ, et al. Ultrafiltration versus usual care for hospitalized patients with heart failure: the Relief for Acutely Fluid-Overloaded Patients With Decompensated Congestive Heart Failure (RAPID-CHF) trial. J Am Coll Cardiol. 2005;46(11):2043–2046. | ||
Rogers HL, Marshall J, Bock J, et al. A randomized, controlled trial of the renal effects of ultrafiltration as compared to furosemide in patients with acute decompensated heart failure. J Card Fail. 2008;14(1):1–5. | ||
Giglioli C, Landi D, Cecchi E, et al. Effects of ULTRAfiltration vs DIureticS on clinical, biohumoral and haemodynamic variables in patients with deCOmpensated heart failure: the ULTRADISCO study. Eur J Heart Fail. 2011;13(3):337–346. | ||
Bart BA, Goldsmith SR, Lee KL, et al; Heart Failure Clinical Research Network. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med. 2012;367(24):2296–2304. | ||
De Vecchis R, Esposito C, Ariano C. Efficacy and safety assessment of isolated ultrafiltration compared to intravenous diuretics for acutely decompensated heart failure: a systematic review with meta-analysis. Minerva Cardioangiol. 2014;62(2):131–146. | ||
Costanzo MR, Saltzberg MT, Jessup M, Teerlink JR, Sobotka PA; Ultrafiltration Versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure (UNLOAD) Investigators. Ultrafiltration is associated with fewer rehospitalizations than continuous diuretic infusion in patients with decompensated heart failure: results from UNLOAD. J Card Fail. 2010;16(4):277–284. | ||
Bradley SM, Levy WC, Veenstra DL. Cost-consequences of ultrafiltration for acute heart failure: a decision model analysis. Circ Cardiovasc Qual Outcomes. 2009;2(6):566–573. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.