Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Cannabinoids in the Treatment of Epilepsy: Current Status and Future Prospects
Authors Morano A, Fanella M, Albini M, Cifelli P, Palma E, Giallonardo AT, Di Bonaventura C ![]()
Received 13 November 2019
Accepted for publication 18 January 2020
Published 7 February 2020 Volume 2020:16 Pages 381—396
DOI https://doi.org/10.2147/NDT.S203782
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Alessandra Morano,1 Martina Fanella,1 Mariarita Albini,1 Pierangelo Cifelli,2,3 Eleonora Palma,2 Anna Teresa Giallonardo,1 Carlo Di Bonaventura1
1Epilepsy Unit, Department of Human Neurosciences, “Sapienza” University of Rome, Rome, Italy; 2Department of Physiology and Pharmacology, Pasteur Institute-Cenci Bolognetti Foundation, University of Rome Sapienza, Rome, Italy; 3IRCCS “Neuromed”, Pozzilli, IS, Italy
Correspondence: Carlo Di Bonaventura
Epilepsy Unit, Department of Human Neurosciences, “Sapienza” University of Rome, P.le A. Moro, 5, Rome 00185, Italy
Email [email protected]
Abstract: Cannabidiol (CBD) is one of the prominent phytocannabinoids found in Cannabis sativa, differentiating from Δ9-tetrahydrocannabinol (THC) for its non-intoxicating profile and its antianxiety/antipsychotic effects. CBD is a multi-target drug whose anti-convulsant properties are supposed to be independent of endocannabinoid receptor CB1 and might be related to several underlying mechanisms, such as antagonism on the orphan GPR55 receptor, regulation of adenosine tone, activation of 5HT1A receptors and modulation of calcium intracellular levels. CBD is a lipophilic compound with low oral bioavailability (6%) due to poor intestinal absorption and high first-pass metabolism. Its exposure parameters are greatly influenced by feeding status (ie, high fat-containing meals). It is mainly metabolized by cytochrome P 450 (CYP) 3A4 and 2C19, which it strongly inhibits. A proprietary formulation of highly purified, plant-derived CBD has been recently licensed as an adjunctive treatment for Dravet syndrome (DS) and Lennox-Gastaut syndrome (LGS), while it is being currently investigated in tuberous sclerosis complex. The regulatory agencies’ approval was granted based on four pivotal double-blind, placebo-controlled, randomized clinical trials (RCTs) on overall 154 DS patients and 396 LGS ones, receiving CBD 10 or 20 mg/kg/day BID as active treatment. The primary endpoint (reduction in monthly seizure frequency) was met by both CBD doses. Most patients reported adverse events (AEs), generally from mild to moderate and transient, which mainly consisted of somnolence, sedation, decreased appetite, diarrhea and elevation in aminotransferase levels, the last being documented only in subjects on concomitant valproate therapy. The interaction between CBD and clobazam, likely due to CYP2C19 inhibition, might contribute to some AEs, especially somnolence, but also to CBD clinical effectiveness. Cannabidivarin (CBDV), the propyl analogue of CBD, showed anti-convulsant properties in pre-clinical studies, but a plant-derived, purified proprietary formulation of CBDV recently failed the Phase II RCT in patients with uncontrolled focal seizures.
Keywords: cannabidiol, cannabidivarin, phytocannabinoids, epileptic encephalopathies, tuberous sclerosis complex, drug-resistance
Introduction
In June 2018, the US Food and Drug Administration (FDA) approved a pharmaceutical preparation of highly purified, plant-derived cannabidiol (CBD) (Epidiolex®, GW Pharmaceuticals, Cambridge, UK) for the treatment of Dravet syndrome (DS) and Lennox-Gastaut syndrome (LGS).1 Despite the later rejection coming from the UK National Institute for Health and Care Excellence (NICE) based on concerns about paucity of long-term studies and economic issues,2 in September 2019 the European Medicine Agency (EMA) granted the approval of CBD (under the trade name of Epidyolex®) as adjunctive treatment for DS and LGS in combination with clobazam (CLB).3 The recent decisions of the regulatory authorities might mark the end of an era of 'much ado' about the use of medical cannabis in the field of epilepsy. Indeed, after decades of legislative restrictions, in the 2010s cannabis potentialities as anti-epileptic medication gained the media attention in the wake of some remarkable cases,4 which paved the way to an international parent-driven quest for CBD-enriched cannabis preparations to treat childhood-onset refractory epilepsies. Such phenomenon further piqued the interest of the scientific community for the therapeutic applications of cannabis derivatives, as clearly demonstrated by the abrupt surge of publications on medical cannabis (which had a 9-fold increase from 2000 to 2017), especially in the fields of psychiatry, oncology and neurology.5 Although CBD, the best-characterized phytocannabinoid (pCB) along with Δ9-tetrahydrocannabinol (Δ9-THC), was soon singled out as a potential anticonvulsant compound based on both preclinical studies6 and the favorable lack of intoxicating effects, initial supporting evidence mostly came from low-quality studies: in fact, in 2012 and 2014 two Cochrane reviews stated that no reliable conclusions could be drawn on cannabinoid effectiveness in epilepsy treatment.7,8 During the following years, three sponsored randomized clinical trials (RCTs) proved CBD to be effective and tolerable in patients suffering from DS and LGS, whereas its use in epileptic subjects with Tuberous Sclerosis Complex (TSC) is currently under investigation.
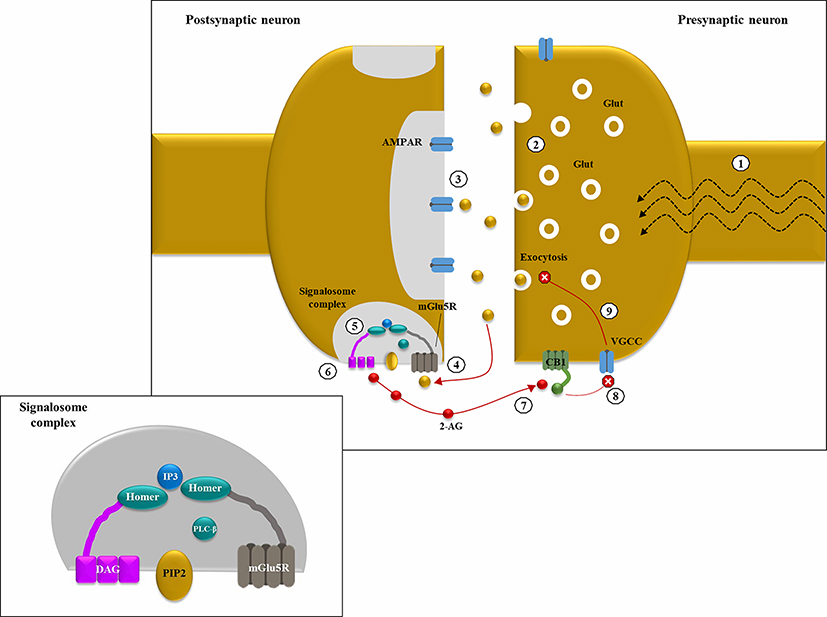 |
Figure 1 Endocannabinoid-mediated negative feedback in epilepsy: (1) in excitatory synapses, depolarization induces Glutamate (Glut) release into the synaptic cleft, thanks to the increase in intracellular Ca2+ levels mediated by the opening of voltage-gated calcium channels (VGCC); (2) in hyperexcitable states, like epileptic seizures, a large amount of neurotransmitter is released from the presynaptic neuron; (3) under basal conditions, Glut binds primarily to intra-synaptic ionotropic receptors (AMPARs); (4) in case of hyperexcitability with Glut “spill over”, group 1 metabotropic Glut receptors (ie, mGlu5Rs) located at pery-synpatic level are activated by ligand binding; (5) mGLU5Rs are anchored together with phospholipase C β (PLCβ) and diacylglycerol lipase α (DGLα) thanks to the scaffolding protein HOMER, forming a sopramolecular complex known as “2-AG signalosome” (illustrated in the smaller panel); (6) mGlu5R activation increases diacyl glycerol (DAG) synthesis by PLCβ and its following conversion into 2-arachydonoyl glycerol (2-AG), catalyzed by DGLα; (7) 2-AG acts as a retrograde messenger and binds pre-synaptic CB1 (coupled with Gi/o); (8) CB1 activation inhibits VGCC thus reducing intracellular Ca2+ levels; (9) ↓ Ca2+ levels determine a decrease in Glut exocytosis. This negative feedback mechanism could be protective against Glut-mediated excitotoxicity in hyperexcitable states. |
In this review, we focused on the pharmacology of CBD and the most solid clinical evidence supporting its use as an anti-seizure medication. We also briefly outlined the current knowledge on cannabidivarin (CBDV) and its therapeutic perspectives in the field of epilepsy. Relevant studies were identified through a literature search of PubMed and the Cochrane databases. RCTs and meta-analysis were mostly considered during the search, and only a few open-label studies were included. Several key-phrases such as “phytocannabinoids”, “endocannabinoids”, “cannabis and epilepsy”, “cannabinoids and epilepsy”, “cannabidiol”, “cannabidiol and epilepsy”, “cannabidivarin”, “cannabidivarin and epilepsy” were used.
Phytocannabinoids and Endocannabinoids: A Brief Overview
The therapeutic potentialities of cannabis plant have been known for millennia: its medical applications were first documented in ancient Chinese books, and over the centuries, cannabis extracts have been used for their anti-emetic, anti-inflammatory, analgesic and anti-convulsant properties.9 Cannabis sativa contains around 540 natural compounds, including over 100 pCBs. pCBs are lipids with a common chemical structure – containing alkylresorcinol and monoterpenes – produced by the flowering tops of the female plant of C. sativa and released in its resin.9 They are synthesized as acids and then decarboxylated to their neutral form when dried, heated or exposed to light. Δ9-THC and CBD are the most abundant pCBs, and share a common precursor, cannabigerol, and similar chemical properties (they are both C21 terpenophenolic compounds with pentyl side-chains).10 Their propyl analogues, derived from cannabigerovarin, are Δ9-tetrahydrocannabivarin and CBDV, respectively.10 Decades after the isolation of Δ9-THC by Mechoulam in 1964,11 specific cannabinoid G protein-coupled receptors, namely, CB1 and CB2, were eventually cloned.12,13 CB1, which appears to be responsible for the psychoactive effects of THC, is mainly found in the central nervous system (CNS), especially in hippocampus, basal ganglia and cerebellum (and, to a lesser extent, in thalamus and lower brainstem), where it is located pre-synaptically in both excitatory and inhibitory synapses. Conversely, CB2 is confined to the peripheral nervous system and the immune system cells, which might explain the anti–inflammatory properties of cannabinoids.9 The cloning of CB1 and CB2 paved the way to the discovery of their endogenous ligands, so-called endocannabinoids, chemically related to pCBs, among which anandamide (AEA, from the Sanskrit word 'ananda', that means bliss)14 and 2-arachydonoyl glycerol (2-AG) appear the most relevant. The endocannabinoid system (ECS) seems to exert a homeostatic function and is thought to be involved in manifold physiological processes (“rest, eat, sleep, forget and protect”);15 therefore, its alteration might be correlated with several neurological diseases as well. The presentation of the complex endocannabinoid signaling is far beyond the aim of this review; nevertheless, some points should be briefly mentioned due to their possible link with epilepsy. Growing evidence suggests that ECS might play a crucial role in modulating neuronal excitability by dynamically regulating neurotransmitter release at the synaptic level. Endocannabinoids are retrograde messengers, synthesized “on demand” in case of increased neuronal activity, probably thanks to the contribution of group 1 metabotropic glutamate receptors (mGluRs). Indeed, in excitatory synapses, mGlu5Rs, that are typically located peri-synaptically, are activated by glutamate (Glut) “spill over” occurring during hyper-excitable states (like epileptic seizures), so producing a feed-forward mechanism.16 After Glut binding, mGlu5Rs (coupled with Gq/11 proteins) activate phospholipase C β (PLCβ), which catalyzes the synthesis of diacylglycerol (DAG), a second messenger and precursor of 2-AG. When high levels of DAG are available, the enzyme diacylglycerol lipase α (DGLα) converts DAG into 2-AG, which migrates in a retrograde way to the presynaptic membrane.16,17 Interestingly, mGlu5Rs, PLCβ and DGLα are all anchored together and structurally organized by HOMER, a scaffold protein, forming a supramolecular complex called “2-AG signalome”.16 When 2-AG binds pre-synaptic CB1 (coupled with Gi/o), its activation triggers various molecular pathways, including the inhibition of adenylate cyclase and of voltage-gated calcium channels (VGCC), which determines the decrease of Ca2+ intracellular levels at the presynaptic terminal, resulting in neurotransmitter (Glut) release reduction.16,17 In one word, the depolarization-induced synthesis of endocannabinoids eventually produces a dampening in neuronal excitability, according to the so-called “synaptic circuit-breaker model”, and might, therefore, be protective against states of hyperexcitability (Figure 1).17 However, CB1 receptors are present not only on excitatory neurons but also on gamma-aminobutyric acid (GABA)-ergic ones, where they are even more abundant and able to produce a depolarization-induced suppression of inhibition. Nevertheless, it has been hypothesized (and partly demonstrated) that a proportion of CB1 receptors on inhibitory interneurons could represent an inactive reservoir, that not all GABAergic cells express CB1, and that CB1 coupling with G proteins could be less effective in GABAergic than in glutamatergic neurons.17 Therefore, CB1-sensitive excitatory synapses are supposed to exceed the inhibitory ones. This would partly account for the possible differential effects of THC, which has been hypothesized to exert an anti-convulsant action at low doses and a pro-convulsant effect at higher concentrations,18 although the latter has been shown only in few animal studies. It would also explain why in mice not expressing CB1 (CB1−/-), that typically develop an epileptic phenotype, kainic-induced status epilepticus (SE) is rescued by the selective reintroduction of CB1 receptors on glutamatergic synapses alone.18
Pharmacodynamic Properties of CBD
The pharmacodynamics of CBD is extremely complex, and only partly elucidated at present. Several putative targets have been identified so far; still, the specific mechanisms of action underlying CBD anti-convulsant effects are not fully clarified, although they are currently supposed to be CB1/CB2-independent. Indeed, CBD has such a low affinity for both cannabinoid receptors that high concentrations (in the micromolar range) are necessary to displace CB1 synthetic ligands.15,19 Nevertheless, CBD can antagonize CB1 action at nanomolar concentrations (lower than those required to significantly interact with the receptor orthosteric site), an unexpected finding suggesting that it may represent an “inverse agonist”.19 Its action as a negative allosteric modulator at CB1 at concentrations <1μM was also demonstrated in a recent in vitro work showing reduced orthosteric ligand (THC and 2-AG) efficacy and negative co-operativity as a result of CBD treatment.20 However, further studies have supported the hypothesis that mechanisms other than CB1 binding are likely to underlie CBD non-competitive antagonism.15,19
In 2007 Ryberg et al identified the orphan G protein-coupled receptor GPR55 as a novel target for CBD and endocannabinoids as well.21 GPR55 is located in excitatory axonal terminals and is thought to facilitate Glut release via intracellular Ca2+ level modulation in an activity-dependent way, probably contributing to short-term potentiation in the hippocampus.9,17 CBD has been proved to act as GPR55 antagonist, thus dampening neuronal excitability by reducing Glut exocytosis.22 Considering that this signaling pathway is not active at baseline conditions, CBD-GPR55 interaction could represent one of the mechanisms underlying CBD anti-convulsant effect, and a potentially safe therapeutic target.22
Another important action of CBD – with respect to epilepsy – is the regulation of the levels of adenosine, a ubiquitous CNS neuromodulator hypothesized to play a role in seizure termination, thanks to CBD-mediated block of the equilibrative nucleotide transporter (ENT), resulting in the inhibition of adenosine re-uptake (and clearance) by astrocytes.23 The elevation of adenosine tone could activate presynaptic A1 receptors (A1Rs), with consequent reduction of Glut release from excitatory terminals. On the other hand, A1Rs have been demonstrated to interact with CB1,24 therefore extracellular adenosine levels might indirectly modulate CB1-dependent glutamatergic inhibition. Moreover, CBD has been also shown to interact with A2A receptors, which could contribute to its anti–inflammatory and neuroprotective action.15,22,23
In spite of CBD low binding affinity (in the micromolar range), relevant targets for its anti-convulsant effect might be 5HT receptors, in particular 5HT1A, that is coupled with Gi/o proteins, and reduces neurotransmitter release.22 Moreover, the modulation of GABAergic transmission as a possible mechanism underlying CBD anti-seizure action has also been suggested by few in vitro studies, performed on human recombinant receptors as well as DS and TSC brain tissues, which demonstrated CBD (at low concentrations) to act as a positive allosteric modulator of GABAA receptors, likely on a different site than benzodiazepines (independent of the presence of γ subunit).25,26
In addition to this, voltage-gated sodium channels (VGSC) have been recently proposed as potential targets for endocannabinoids and pCBs as well: indeed, CBD has been proved to inhibit human Nav1.1.-1.7 currents at therapeutically relevant concentrations;27 moreover, in vitro and in vivo studies have shown CBD to reduce sodium currents in both wild-type and mutant Nav1.6 channels (encoded by SCN8A, whose mutations are associated with a severe epileptic encephalopathy).28 The overall inhibitory effect of CBD on VGSC might contribute to its anti-convulsant properties.
Another class of molecules, namely transient receptor potential (TRP) cation channels, appears to be involved in CBD signaling. More specifically, TRP channels of vanilloid type 1 (TRPV1), activated by heat and capsaicin, are supposed to be phosphorylated in case of neuronal activation.9 CBD is a TRPV1 agonist and is thought to induce TRPV1 activation, dephosphorylation and consequent desensitization, which would decrease calcium levels and neuronal excitability.17,23 TRP channel of ankyrin type 1 (TRPA1), that often co-localizes with TRPV1 and is physiologically activated by menthol and cold, might also be a CBD target, as well as TRP subfamily melastatin type 8 (TRPM8), on which CBD acts as an antagonist.9,22
Finally, CBD is supposed to influence calcium modulation through its action on VGCC, in particular of T- and L-type, and on mitochondrial Na+/Ca2+ exchange.17 Mitochondria might be otherwise involved in CBD signaling, considering that the voltage-dependent anion-selective channel protein 1 (VDCA1), located on the outer mitochondrial membrane, is antagonized by CBD.22
Several other molecules, including ion channels, receptors and enzymes, have been identified as potential targets for CBD, although their specific relevance in determining its pharmacological effects is yet to be clarified. For instance, CBD is an allosteric modulator, either positive or negative, of α3 glycine receptors and μ/δ opioid receptors (which could contribute to its analgesic properties), α1 adrenoreceptors and Dopamine 2 (D2) receptors.22,23 Moreover, CBD could influence endocannabinoid signaling by modulating (in either way) fatty acid amide hydrolase (FAHH), the enzyme responsible for AEA metabolism.22
The anti-inflammatory properties of CBD might be related to manifold mechanisms, including the mobilization of arachidonic acid and the regulation of its metabolite synthesis, namely leukotrienes, thromboxanes and prostaglandins (although conflicting evidence is available at present); the reduction of nitrous oxide (NO) thanks to the inhibition of its inducible synthase (iNOS), and the modulation of cytokine levels.22 Moreover, CBD has well-known anti-oxidant properties: in fact, recent studies suggest that it might induce the synthesis of reactive oxygen species (ROS) in tumoral cells, which could partly justify its anti-carcinogenic potential.15,22 As to that, gene transcription modulation via agonism on peroxisome proliferator-activator receptor γ (PPARγ) might also play a role.
Finally, CBD has been demonstrated to influence neutrophil chemotaxis29 and interact with microglia.30 Microglial lamellipodia express CB2 receptors, and endocannabinoids, in particular 2-AG, which are produced in case of inflammation, have been shown to active microglia cells through CB2, inducing microglia migration towards the inflammation site. CBD might be able to antagonize such mechanism, thus exerting an anti-inflammatory effect.
Pharmacokinetic Properties of Oral CBD
Considering that the recently approved CBD pharmaceutical preparation consists of an oral solution (≥98% pure CBD solubilized in sesame seed oil with additive sucralose and strawberry flavoring, containing CBD 100 mg/mL),31 only this type of formulation will be taken into consideration in the following discussion.
CBD has a complex and rather unfavorable PK profile, which is greatly influenced by fasting/fed status and is prone to develop various (and sometimes clinically relevant) drug–drug interactions. Given its lipophilic nature, it has rather slow oral absorption and a large volume of distribution (20,963 L to 42,849 L in healthy subjects), due to extensive distribution into tissues. Its estimated plasma protein binding is about 94%.23 A pharmacokinetic Phase I, placebo-controlled trial performed on healthy volunteers and comparing single ascending dose (SAD) (1500, 3000, 4500 and 6000 mg) with multiple dose (MD) (750 or 1500 mg/die BID for 7 days), found similar times of peak concentration (tmax 3.5–5 h versus 3 h).32 Steady state was reached after 2 days in the MD arm, and a period effect, with some degree of accumulation, was also documented. In the same study, exposure (area under the concentration–time curve, AUC) to CBD showed a less than proportional increase in the SAD arm of the protocol, which suggested a change in bioavailability possibly due to a solubility-related decrease in the absorption rate.32 Although this finding was not confirmed in the MD group, studies on chronically treated patients, with a daily dose ranging from 5 to 20 mg/kg/d, led to the same conclusion, as indicated in Epidiolex® prescribing information.31,32
Bioavailability of orally administered CBD is estimated at around 6%, probably as a result of poor gastrointestinal absorption and high first-pass metabolism.23 As already anticipated, food can dramatically influence CBD bioavailability: indeed, in the above-mentioned Phase I trial, taking a highly fat-containing meal within 20 mins of drug administration determined a more than 4-fold increase in exposure to CBD, whereas tmax and terminal elimination half-life (t1/2, z) were unchanged.32 Such remarkable influence of feeding status was further demonstrated in 8 epileptic patients by Birnbaum and coworkers, who tested a solid formulation (soft gelatinous capsules) of CBD (200–300 mg) in order to avoid drawbacks related to liquid preparations.33 In this study, the effect of a high-fat meal (840–860 calories, 500–600 of which from fat) consisted in a 14-fold increase of CBD maximal plasma concentration (Cmax), that is a much higher rise than previously detected in healthy volunteers taking CBD oil.33 Moreover, both tmax (2.4 h vs 3.2 h) and t1/2 (24.3 h vs 38.9 h) were shorter during fed status compared with fasting. Various mechanisms might justify the dramatic effect of food on CBD bioavailability, including increased bile salt excretion and prolonged gastric transit time due to fed status, with consequent higher dissolution and absorption of lipophilic compounds.32,33
CBD is extensively metabolized in the liver, mainly by cytochrome P450 (CYP) 3A4 and 2C19, which catalyze its hydroxylation (to the active 7-OH-CBD) and further oxidation, and are strongly inhibited by CBD itself. To a lesser extent, CYP2C9, 1A1, 1A2 and 2D6 are also involved, along with UDP-glucuronosyl-transferases (UGT) 1A9, 1A7, 2B7.22 The prominent metabolite is 7-COOH-CBD, an inactive compound whose serum concentrations greatly exceed (up to 47 times) those of the parent drug. Elimination of CBD (33% of which is unchanged) is almost exclusively via feces, and follows a biphasic pattern, with an initial t 1/2 of 6 h, and a later terminal half-life of 24 h, due to the slow release of the drug from the tissues where it rapidly distributes after absorption.34
Given its extensive liver metabolism, it does not come as a surprise that CBD PK profile is considerably influenced by hepatic function. A recent phase I, open-label, parallel-group study was performed on 22 subjects with hepatic impairment from mild to severe (according to the Child-Pugh score) receiving a single 200-mg dose of CBD, with blood samples being collected pre-dose and over the following 48 h.35 A slight increase in exposure parameters was observed in mildly impaired subjects, whereas the rise was significant in moderately and severely impaired participants. Moreover, t1/2 was prolonged in affected subjects in accordance with clearance reduction. All CBD metabolites showed a comparable increase except for 7-COOH-CBD, the most abundant one, which was the lowest in subjects with severe hepatic dysfunction, pointing to a reduced liver metabolic capacity.35 Based on these findings, a dose adjustment should be always considered in patients with hepatic dysfunction.
Finally, considering the prominent use (and exclusive current indication) of CBD in childhood-onset drug-resistant epilepsy (DRE), its pharmacokinetics has been also evaluated in few pediatric populations. In the multi-center, randomized, placebo-controlled, dose-finding trial by Devinsky et al (GWPCARE1 part A), 34 DS patients aged 4–10 years received CBD at 5, 10 or 20 mg/kg/day BID for 3 weeks (starting from 1.25 mg/kg/day, with a 2.5 or 5 mg/kg increase every other day), followed by 10-day tapering.36 The study documented a remarkable inter-individual variability in the exposure parameters of CBD (% Coefficient of variation (CV) 20–120%) and, even more so, of its metabolites (%CV 57–1570%), although the possible determinants (eg, interactions with other AEDs) were not properly investigated. Exposure to both CBD and its analytes showed a dose-proportional increase. In the open-label INS011-14-029 study, a 99.5% pure synthetic oral formulation of CBD was administered to 61 subjects aged 1 to 17 years suffering from DRE, as both a single dose (5, 10 or 20 mg/kg on day 1, followed by a preset volume of water or clear liquid) and a multiple ascending dose (MAD) (10, 20 or 40 mg/kg BID, from day 4 to day 10).37 During the MAD phase, the median tmax was 2–3 h, independent of the dose, and the steady state was reached between 2 and 6 days. Apart from confirming the high inter-subject variability and the dose-proportional increase in exposure parameters, the authors of this trial found that infants had lower CBD concentrations compared with children and adolescents, regardless of the administered dose.37
Pharmacological Interactions Between CBD and Other AEDs
CBD interaction with Clobazam (CLB) is widely acknowledged and might be particularly relevant in clinical practice since both medications are commonly used in the treatment of epileptic encephalopathies such as DS. CLB is metabolized by CYP3A4 and CYP2C19 into N-desmethylclobazam (norclobazam, nCLB), a compound that is 20–100% as potent as the parent drug, and is further converted by CYP2C19 into inactive metabolites.38 As first demonstrated by Geffrey et al in 13 pediatric DRE patients treated with CLB (0.18–2.24 mg/kg/day), the introduction of CBD (titrated over 4 weeks up to 25 mg/kg/day) was associated with a non-significant mean increase (60±80%) of CLB levels, whereas concomitant nCLB concentrations rose by 500±300%.38 Such remarkable findings might be justified by the strong inhibition exerted by CBD on CYP2C19. In 10 out of 13 subjects, CLB doses were lowered by the investigators, with a reported benefit on side effects (likely related to CLB), although nCLB serum levels did not parallel the parent drug decrease. In order to further assess the interactions between these two medications, a Phase III RCT was later performed on 20 epileptic adult subjects on a stable dose of CLB, receiving either CBD 20 mg/kg/day or placebo (4:1).39 Although complete results have not been published yet, a remarkable increase in nCLB exposure parameters in the CBD group, despite steady CLB concentrations, was confirmed by the investigators, in accordance with previous results. Furthermore, while Geffrey and coworkers ruled out any significant influence of CLB and its metabolites on cannabidiol,38 Morrison and colleagues found that a 21-day treatment with CLB 5 mg BID produced a slight increase in CBD and, more remarkable, in 7-OH-CBD levels, suggesting a possible inhibition of UGTs and other minor CYPs.40 Most interestingly, beside the pharmacokinetic interference, a pharmacodynamic interaction between CBD and CLB has been also hypothesized based on preclinical evidence. Indeed, a recent study on a mouse model of DS demonstrated that a combination of CLB and CBD 100 mg/kg (endowed with intrinsic anti-convulsant properties) was more protective against hyperthermia-induced seizures than either drug alone.41 Conversely, the same effect was not observed when adding CBD at a lower dose (12 mg/kg), although this was proved to increase CLB and nCLB serum concentrations. These findings suggested a pharmacodynamic (as well as pharmacokinetic) interaction between CBD and CLB, which the authors attributed to their action as positive allosteric modulators of GABAA receptors, in accordance with other in vitro experiments.25,26 Unfortunately, the study was unable to demonstrate a synergistic effect between CBD and CLB.
As far as other AEDs are concerned, in the above-mentioned phase I, open-label trial on healthy volunteers by Morrison and coworkers, the authors investigated the possible interactions between CBD (750 mg BID), valproate (VPA) (500 mg BID) and stiripentol (750 mg BID).40 The serum concentrations of neither VPA nor CBD changed when administered in combination. As to stiripentol,42 its exposure parameters rose by 28–55%, likely due to CYP2C19 inhibition by CBD. More importantly, since stiripentol is itself a strong inhibitor of CYP2C19, CYP2D6 and CYP3A4, it is supposed to prevent CBD-mediated increase in nCLB levels, by “neutralizing” its effect on CYP2C19 (whose stiripentol-dependent inhibition would be already maximal at the time of CBD introduction).23 In an open-label trial by Gaston and coworkers, CBD appeared to affect the exposure to other AEDs as well: in particular, a linear increase in the serum levels of topiramate (TPM) and rufinamide (RFD) (in both adult and pediatric patients), eslicarbazepine (ESL) and zonisamide (ZNS) (in adults alone) was documented.43 However, further controlled trials are warranted to confirm the significance of these findings. Finally, in a recent small study on 5 adult DRE patients, the introduction of CBD (up to 50 mg/kg/day) was associated with a 95–280% increase in brivaracetam plasma levels, although the possible mechanisms underlying such interaction are still elusive.44 The effects of CBD on other AEDs are shown in Table 1.
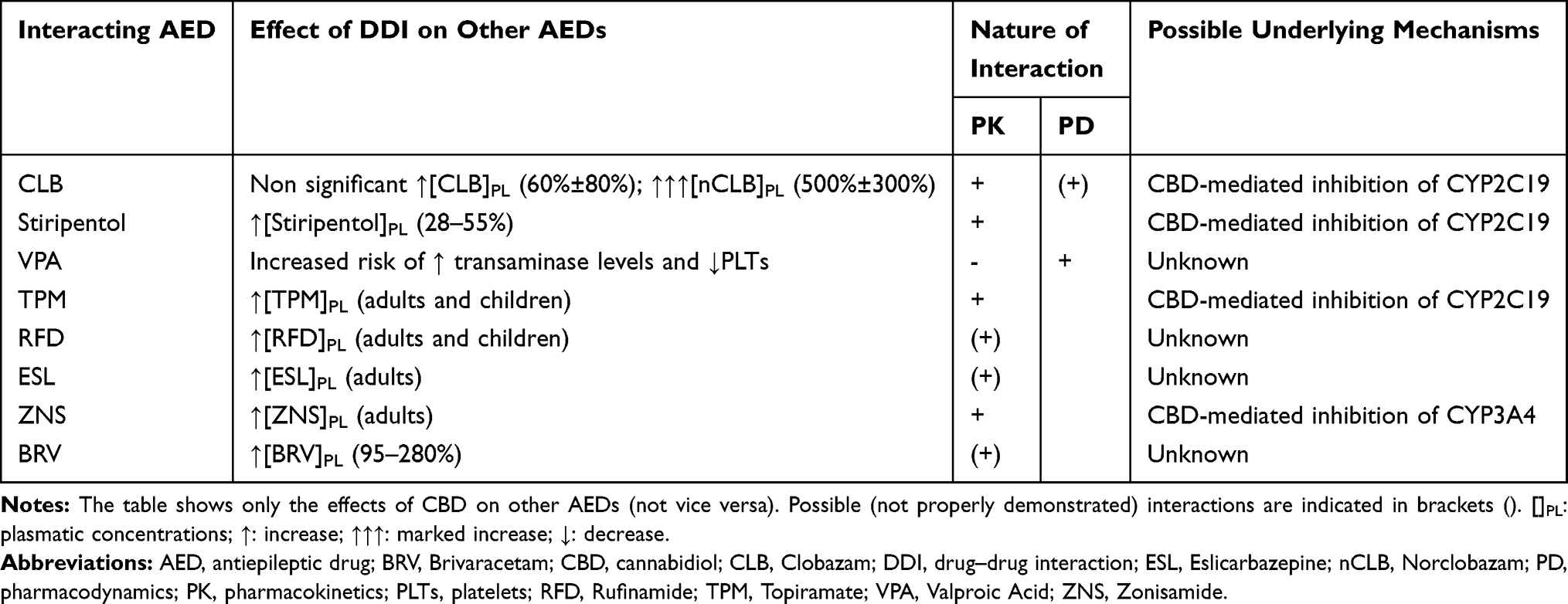 |
Table 1 Interactions Between CBD and Other AEDs |
Effectiveness of CBD in DS and LGS: Findings from RCTs
The effectiveness of CBD as anti-epileptic medication was firstly investigated in 4 randomized, placebo-controlled clinical trials performed from 1978 to 1990 on overall 48 adult patients with DRE (including 12 mentally impaired institutionalized subjects).45–48 Although in half of the studies, a proportion of patients receiving the active treatment (CBD 200–300 mg/day) reported some clinical benefit, the relevance of these findings was affected by several methodological flaws, such as small sample size, short follow-up, lack of clinical details (including baseline seizure frequency and severity), and insufficient information about randomization and blinding. Over the years, numerous papers, mainly case reports, retrospective studies and open-label trials, dealt with the use of cannabis-containing medical products in epileptic patients. However, considering the low quality of these studies and the heterogeneity of the investigational compounds, the following discussion will be mainly focused on pivotal RCTs leading to CBD license for the treatment of epileptic encephalopathies.
The regulatory agencies’ approval of CBD use in patients suffering from DS was granted based on the findings of a Phase II, randomized, double-blind, placebo-controlled trial performed on 120 DS subjects aged 2–18 years (mean age 9.8 years) with ≥4 convulsive seizures per month (GWPCARE1 part B).49 The study design consisted in a 4-week baseline period, followed by 2-week titration, 12-week maintenance and 10-day tapering. Patients were randomized in a 1:1 ratio to either CBD 20 mg/kg/day BID or placebo. The maximal dose of 20 mg/kg/day was recommended based on the safety and PK data from GWPCARE 1 part A study.36 The primary endpoint was the percentage change in convulsive seizure frequency per 28 days during the entire treatment period compared with the 4-week baseline, whereas the responder rate (ie, proportion of subjects achieving ≥50% decrease in convulsive seizures) and the reduction in the frequency of all seizures and nonconvulsive ones were considered as secondary endpoints, along with sleep quality, Caregiver Global Impression of Change (CGIC), etc. CBD group showed a median change in convulsive seizure frequency of −38.9% (from a median of 12.4 seizures/month at baseline to 5.9) compared with −13.3% (from 14.9 to 14.1) in the placebo group, resulting in the adjusted median difference of −22.8% (p=0.01). The most notable reduction in seizure frequency was documented within the first month of the maintenance period. Total seizures per month decreased by 28.6% in patients on CBD compared with 9% in those receiving placebo, with an adjusted median difference of around 19% (p=0.03). However, the responder rate (43% vs 27% in CBD and placebo group, respectively, p=0.08) did not support the superiority of the active drug over placebo, neither did the reduction in nonconvulsive seizures alone (p=0.88). Such findings might suggest that CBD is effective specifically in convulsive seizures; still, the count of nonconvulsive ones could be unreliable, and the study itself could be underpowered to detect differences in the frequency of this seizure type.50 However, the positive global impact of CBD treatment was confirmed by the documented changes in the CGIC scale, showing an improvement in 62% of children receiving active treatment versus 34% of those taking placebo (p=0.02). Another phase III RCT (GWPCARE2) compared the effectiveness and tolerability of CBD 10 and 20 mg/kg/day with placebo in a population of children and young adults diagnosed with DS.51 One-hundred-ninety-eight patients from 38 worldwide sites were randomly assigned in a 2:2:1:1 ratio to receive the active compound (66 and 67 in 10 and 20 mg/kg/day groups, respectively) or placebo (overall 65 subjects) for 14 weeks (2-week titration and 12-week maintenance). Recently released preliminary data showed a significantly higher responder rate in both treatment arms (43.9% and 49.3% in 10 and 20 mg/kg/day groups, respectively) compared with placebo (26.2%). Similarly, remarkable differences favoring CBD over placebo were documented in the frequency change of both convulsive (−48.7% and −45.7% vs −26.9%) and total seizures (−56.4% and −47.3% vs −29.7%).
Apart from DS, CBD is also currently licensed for the treatment of LGS. Two Phase III, randomized, double-blind, placebo-controlled trials demonstrated CBD effectiveness and safety as an adjunctive medication in pediatric and adult LGS subjects.52,53 A total of 396 patients (225 and 171, respectively), aged 2–55 years, with at least two generalized seizure types over the last 6 months and 2 drop seizures over the 4-week baseline period, were enrolled. The first study (GWPCARE3) compared CBD 10 mg/kg/day, CBD 20 mg/kg/day and placebo (with 73, 76 and 76 patients randomly allocated in a 2:2:1:1 ratio), whereas in GWPCARE4 trial the active treatment consisted in CBD 20 mg/kg alone (administered to 86 out of 171 patients). Both studies mainly focused on CBD effectiveness on drop seizures, whose median monthly frequency at baseline was 85 and 73.8, respectively. In GWPCARE3 trial, the percentage change in 28-day frequency of drop seizures was significantly higher in both treatment arms (−37.2% in CBD 10 and −41.9% in CBD 20) compared with placebo (−17.2%), with an estimated median difference of 19.2% for CBD 10 (p=0.002) and 21.6% for CBD 20 (p=0.005). These findings were further confirmed by the GWPCARE4 study, where the reduction in drop seizure frequency was significantly superior in the treated patients than in those receiving placebo (−43.9% vs −21.8%, with an estimated median difference of −17.21%, p=0.0135). Similarly, significant responder rates for drop seizures were also found, regardless of dose (GWPCARE3: 36% in CBD 10, 39% in CBD 20, 14% in placebo, p=0.003 and p<0.001, respectively; GWPCARE4: 43.9% in CBD group vs 21.8% in placebo, p=0.0043). The clinical benefit on drop seizures appeared to be persistent over the entire 12-week maintenance period. Interestingly, CBD proved effective on non-drop seizures as well: in particular, in the dose comparing trial, the estimated median difference with placebo was −28.3% in CBD 10 and −22.4% in CBD 20 (although p-values were not calculated), whereas it amounted to −26.1% (p=0.0044) in favor of patients receiving CBD in GWPCARE4 study. Similar trends were also observed in “total seizures”, supporting CBD effectiveness in treating all seizure types. In a recent meta-analysis gathering data from both RCTs on LGS,54 no dose–response correlation was found due to insufficient data. However, seizure freedom from drop seizures during the maintenance phase appeared more likely in patients on CBD 20 mg/kg/die (Risk Ratio 6.59), although it did not reach statistical significance.
Most patients participating in the above-illustrated RCTs were enrolled in an open-label extension (OLE) study (GWPCARE5), including overall 630 subjects (264 with DS and 366 with LGS) treated with adjunctive CBD (maximal dose 30 mg/kg/day) for 1–3 years, with 12-week follow-up visits.55,56 During the interim analysis period (after data cut in November 2016) CBD was discontinued by 75 DS patients and 67 LGS subjects (corresponding to a withdrawal rate of 28% and 18%, respectively), mainly due to adverse events (AEs) and parent/patient consent withdrawal. In the DS group, the responder rate was about 40% at each visit window, the reduction in convulsive seizure frequency in the first trimester was 37.5% and persisted throughout 48 weeks, whereas the frequency of all seizures decreased by 39–50%.53 Five subjects were free from convulsive seizures during their last 12 weeks. In the LGS population, half of the patients (range 49.2–54.4%) had a ≥50% improvement in drop seizure frequency.56 Moreover, at first follow-up visit, the median percentage change in seizure frequency was −48.2% for drop seizures and −47.7% for all seizure types. The clinical benefit appeared to persist in time, and no tolerance was observed.
A remarkable proportion of participants to pivotal RCTs (and OLE study as well) were taking concomitant CLB (66% in GWPCARE1B, 49% in GWPCARE3, 48% in GWPCARE4, 58% in GWPCARE5), with possible clinically relevant interactions. Indeed, in the first open-label trial on CBD as an add-on medication in a heterogeneous population of childhood-onset DRE, Devinsky and coworkers found the responder rate (for motor seizures) to be higher among subjects taking CLB (51%) compared with the others (27%).57 Besides, the multiple logistic regression analysis showed CLB to be the only independent predictor of motor seizure frequency reduction. Unfortunately, data on CLB and nCLB serum concentrations were not available. By pointing to the possible influence of CLB on seizure outcome, these findings raised concerns about the actual anti-convulsant efficacy of CBD alone.58,59 In fact, a post-hoc analysis performed on LGS patients enrolled in GWPCARE3/4 studies showed CBD to be more effective than placebo regardless of CLB status.60 This conclusion is in accordance with the results of a recent open-label, compassionate-use trial by Gaston and colleagues evaluating the possible clinical impact of CLB and other “interfering” AEDs (namely TPM, ESL, ZNS, RFD, based on a previous study by the same authors), when administered in combination with CBD.43,61 Seizure frequency and severity appeared independent of concomitant treatments, suggesting that relevant interferences were unlikely; nevertheless, these findings should be interpreted with caution considering the intrinsic limitations of the study design.
An overview of the pivotal RCTs leading to CBD license is illustrated in Figure 2.
 |
Figure 2 The figure shows the randomized, double-blind, placebo-controlled trials (followed by ongoing open-label extension studies) performed to evaluate CBD effectiveness and tolerability in DS, LGS and TSC. The percentages shown in the figure indicate the responder rates, ie, the proportion of patients showing >50% seizure reduction (for motor seizures in DS, drop seizures in LGS and convulsive ones in TSC) of CBD 20 mg/kg/day compared with placebo. Abbreviations: DS, Dravet syndrome; LGS, Lennox-Gastaut syndrome; TSC, tuberous sclerosis complex. |
CBD Effectiveness in Other Epileptic Syndromes
Expanded access programs allowed several patients suffering from childhood-onset DRE other than DS and LGS to receive CBD as an adjunctive treatment. Despite the current dearth of solid evidence coming from high-quality studies, this paragraph will provide a brief overview of a few specific epileptic syndromes where CBD might represent a useful therapeutic option. Among them, TSC has been the most extensively investigated so far and deserves a special mention since CBD received Orphan Drug designation for TSC by both FDA and EMA. Eighteen subjects with TSC diagnosis, aged 2–31 years, were enrolled in the expanded assess trial by Hess and colleagues, and followed for ≥6 months.62 The majority of them (14/18) also presented developmental delay. CBD was administered at an initial dose of 5 mg/kg/day, with following 5 mg/week titration up to 25 mg/kg/day: however, further increases to maximal dose of 50 mg/kg/day were allowed for unsatisfactory seizure control, without apparent safety issues. Although only 8/18 subjects achieved a 12-month follow-up, the overall mean seizure frequency showed a decreasing trend over time, of different entity according to seizure types: indeed, frequency reduction was more pronounced for tonic-clonic seizures (−91.4%), infantile spasms (IS) (−87.5%) and atonic seizures (−86.5%) compared with focal ones; moreover, responder rate at 3 months was higher for IS and atonic seizures (75% each). Among 10/18 patients taking concomitant CLB, 58.3% were responding to treatment at 3 months, compared with 33.3% of the others. Besides, cognitive and behavioral improvements were reported in 85.7% and 66.7% of cases, respectively. The authors concluded that CBD might be a tolerable and effective adjunctive treatment for TSC patients, in accordance with preclinical evidence,26 with a possible specific efficacy on IS. Nevertheless, given the open-label design of the study, changes in concomitant AEDs were minimized but not completely avoided for the first 3 months; moreover, seizure aggravation was documented in as well as seven patients at some point during the observation period. In the wake of this study, a phase III RCT (GWPCARE6) was performed on 224 TSC patients, aged 1–65 years (mean age 14 years), to evaluate the safety and efficacy of CBD 25 mg/kg/day (75 patients) and CBD 50 mg/kg/day (73 subjects) versus placebo (76 cases) over a 16-week treatment period (4-w titration and 12-w maintenance).63 Patients concomitantly treated with oral mTOR inhibitors were excluded. The primary endpoint was the change in overall seizure frequency, but several secondary endpoints, including changes in the number of different seizure types (focal seizures with or without awareness impairment, focal to bilateral tonic-clonic seizures, generalized seizures) and in Insulin Growth Factor 1 (IGF1) serum levels were evaluated as well. In May 2019, the manufacturer announced that the study met the primary endpoint, with a seizure frequency reduction significantly higher in both CBD 25 group (48.6%, p=0.0009) and CBD 50 group (47.5%, p=0.0018) when compared with placebo (26.5%).64
Finally, Devinsky and coworkers analyzed data from a subgroup of 55 patients diagnosed with specific epileptic syndromes, namely CDKL5 deficiency disorder (20 subjects), Aicardi syndrome (19 patients), Doose syndrome and Dup15q syndrome (8 cases each), who received CBD (>20 mg/kg) during the expanded access trial.65 The pooled analysis showed a largely significant reduction in convulsive seizure monthly frequency at 12 weeks (from median 59.4 seizures/month at baseline to 22.5 at follow-up), which was persistent over 48 weeks (without further improvement). Accordingly, the responder rate for convulsive seizures was 50% at 12 weeks and 57% at 48. As to retention, five patients withdrew CBD by week 12, 10 by week 48, and 15 by week 144 of extended follow-up, mainly for unsatisfactory seizure control (9) or AEs (4). However, further studies with better design are warranted before any conclusion is drawn on CBD effectiveness as an adjunctive medication for these difficult-to-treat syndromes.
Tolerability Profile of CBD
In the open-label trial by Devinsky and colleagues on the compassionate use of CBD for pediatric and adult DRE patients, 128/162 subjects (79%) reported side effects, the most common being somnolence (25%), decreased appetite (19%), diarrhea (19%), fatigue (13%) and convulsions (11%).57 Moreover, 48 participants developed serious adverse events (SAEs), which the investigators deemed to be causally related to CBD in 20 cases (12%). Interestingly, only 3% of patients withdrew CBD because of AEs, suggesting that either treatment-related benefits outweighed drawbacks, or that parents had too much hope in CBD effectiveness to discontinue it due to tolerability issues. In the safety, PK, dose-finding study on 34 DS subjects, treatment-emergent AEs were reported in each arm, and led to withdrawal in two subjects.49 Based on these results, 20 mg/kg/day was established as the maximal CBD dose for future trials (despite higher doses, up to 50 mg/kg, were used in both open-label studies and in the RCT on TSC subjects, whose safety data are not yet available).
The recent meta-analysis by Lattanzi et al on overall 550 DS and LGS patients participating to three pivotal RCTs showed that 11.1% subjects taking CBD discontinued treatment, compared with 2.6% receiving placebo (p=0.003); moreover, 8.9% of the actively treated population withdrew because of AEs (versus 1.8% in the placebo group).50 Both all-cause withdrawals and those due to AEs appeared significantly more common among subjects taking CBD 20 mg/kg/day compared with CBD 10 mg/kg/day. AEs were reported by a remarkable proportion of patients in both CBD and placebo groups (87.9% and 72.2%, respectively), in line with other AEDs in the same populations, but they were mostly mild to moderate, and generally transient. Besides, SAEs involved 60/323 treated patients (RR 2.61). In accordance with previous findings from expanded access programs, the most common treatment-emergent AEs were somnolence (24.5%), appetite decrease (20.1%), diarrhea (18.2%, which might be related to sesame vehicle),57 and elevation in transaminase levels (16.1%). Moreover, the authors confirmed the correlation between side effects and CBD dose.38 In the LGS subgroup, transaminase level increase was the most common cause of AE-related withdrawal, although none patient met the criteria for drug-induced liver injury.54 Elevation in transaminases was an early finding (documented within the first 90 days in almost all patients), it generally appeared transient, and resolved spontaneously or after down-titration of either CBD or another AED. Interestingly, the pooled analysis confirmed that only patients taking concomitant VPA might develop an increase in transaminase levels. Considering that pharmacokinetic interferences between CBD and VPA (whose serum concentrations were not affected by CBD concomitant administration, nor vice versa) have been clearly ruled out,40 a pharmacodynamic interaction between these two compounds might be advocated to explain the higher risk of hepatotoxicity in patients on combined therapy. As previously stated, somnolence was the most common treatment-emergent AE in nearly all studies, and appeared more likely in subjects receiving concomitant CLB: indeed, over two thirds of LGS patients on CBD reporting somnolence were also taking CLB (66.7% and 69% in GWPCARE3 and 4, respectively),52,63 and an even higher percentage (81.8%) was documented among DS subjects treated with CBD in the pivotal trial (GWPCARE1B).47 Accordingly, Geffrey and colleagues found that all AEs (including somnolence and sedation) improved after CLB down-titration, regardless of nCLB serum levels.37 It should be mentioned that rashes have also been described in epileptic patients receiving CBD (17.6% in the safety trial on DS),36 as well as in healthy volunteers (11%, apparently when the drug was not properly titrated and/or stored).40 Finally, CBD did not appear to affect sleep quality, cognition and behavior, as expected based on its non-intoxicating profile.50,66
Findings from open-label extension studies will add precious information about CBD long-term tolerability: data from interim analysis showed a proportion of SAEs superior to 25% in both DS and LGS population, among which SE was the most common (11% and 7.1% in DS and LGS patients, respectively), although it seldom led to CBD discontinuation.65,66 AEs reported during the GWPCARE5 trial (interim analysis) are shown in Table 2.
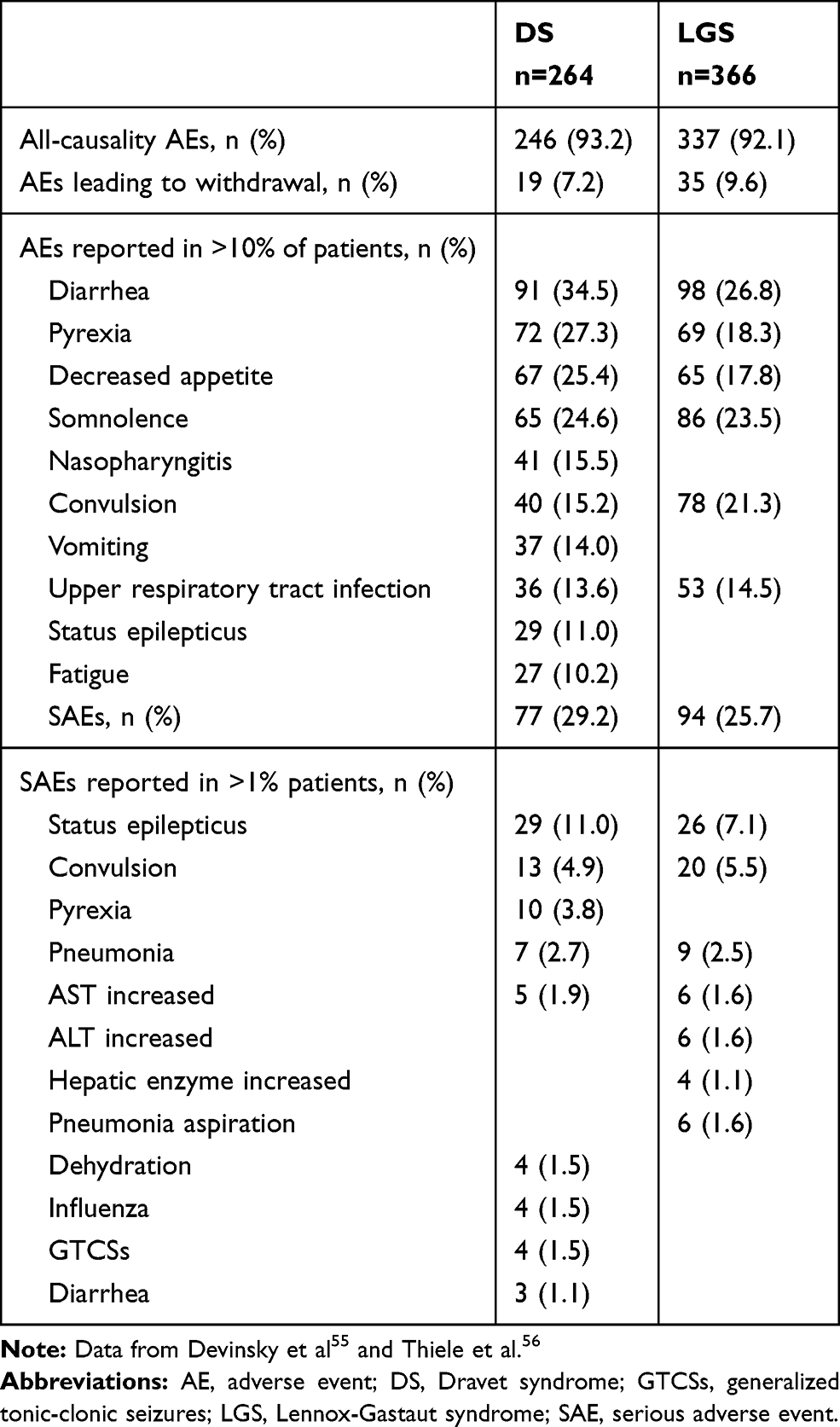 |
Table 2 AEs Reported in the Open-Label Extension Study (Interim Analysis) |
Not Only CBD: A Glance at Cannabidivarin
As previously stated, CBDV is the n-propyl analogue of CBD, and it is being currently investigated as a therapeutic option in both epilepsy and autism spectrum disorder. Its anti-convulsant properties have been documented in several animal models of acute seizures and SE, where it proved widely effective (except for pilocarpine-induced SE).67 Like CBD, CBDV has little affinity for CB1 and CB2, and like CBD it is considered a “multi-target” drug, known to act as an agonist on TRPV1/2 and TRPA1 channels, and as an antagonist on TRPM8 channels.68 It also inhibits DGLα, involved in endocannabinoid synthesis. Interestingly, a recent in vitro study also suggested that GABAA receptors might be a target of CBDV, which was proved to reduce GABA “run-down” at therapeutic doses.69 Nevertheless, the exact mechanisms underlying CBDV anti-convulsant properties are yet to be clarified.
CBDV is a highly liposoluble compound with a large VL (32 L/kg), it is able to rapidly penetrate the blood-brain barrier and has a poor oral bioavailability.70 A single phase I study on healthy volunteers evaluated the PK profile of CBDV oral formulations (25, 75, 200, 400, 800 mg/day over 5 days) and IV preparation (5-mg, single-dose).71 The drug was well tolerated when administered in both routes, and rapidly metabolized in the liver to 7-OH-CBDV and 7-COOH-CBDV, although the exact metabolic pathway is still unknown. In the same study, exposure parameters (Cmax and AUC) also showed a dose-proportional increase (from 200 to 800 mg/day).
A recent phase II trial evaluated PK, safety (part A) and effectiveness (part B) of a proprietary, plant-derived, purified formulation of CBDV (GWP42006) in adult patients with uncontrolled focal seizures. Part A included 34 subjects, randomized in a 4:1 ratio to receive CBDV 400 mg BID or placebo for a 14-day treatment period: participants were divided into three parallel groups, according to concomitant medications (inducer AEDs, inhibitor AEDs and non-interfering AEDs).72 Apparently, no differences in CBDV PK and safety were documented among the three groups (unpublished data).73 On the other hand, 162 subjects (mainly from Eastern Europe) were enrolled in part B and randomly assigned to receive either CBDV 800 mg/day or placebo for 8 weeks (2-week titration and 6-week maintenance), followed by 12-day tapering.74 Although the study results have not been published yet, in February 2018 the manufacturer announced that the trial failed to meet its primary endpoint, ie, the percentage change in focal seizure frequency from baseline to the end of the maintenance period, which amounted to −40% in both actively treated and placebo groups.75 However, the investigators reasonably pointed out that the extent of clinical benefit in subjects taking placebo was exceedingly high when compared with previous trials, which perhaps is in line with the increasing trend in placebo response recently observed in RCTs, that surely represents a matter of concern for clinical research. However, it cannot be ruled out that the use of a purified compound might have influenced the outcome, as already hypothesized concerning CBD-based medications.76 As to tolerability, AEs were reported by a larger proportion of actively treated patients compared with the placebo group (73% versus 48%): most AEs were from mild to moderate in severity, and SAE incidence was generally low (3.7% and 1.2% in CBDV and placebo groups, respectively). In light of these unsatisfactory results, future studies exploring CBDV therapeutic potential in epilepsy should be addressed to specific patient populations.
Future Prospects for Cannabinoid Use in Epilepsy Treatment
In recent times, the anti-epileptic potentialities of cannabis-based medical products have been the focus of intense clinical research as well as the object of overwhelming media attention. After years of “haze”,77 solid evidence supporting CBD effectiveness came from pivotal RCTs on DS and LGS, that showed responder rates ranging from 36% to almost 50%, and a proportion of freedom from convulsive/drop seizures around 5%, which are remarkable outcomes considering the refractoriness observed in epileptic encephalopathies. Although CBD was sufficiently tolerable as to lead most parents/participants to enroll in the OLE trials, the majority of treated subjects reported AEs, mainly CNS symptoms and gastro-intestinal disturbances (likely due to the vehicle); however, they were from mild to moderate and generally transient. Based on these findings, CBD could be rightfully considered a valid tool in the rather poor therapeutic armamentarium of DS and LGS. Still, it is a medication, not a panacea, and, as such, it is not devoid of risks, including the potential for seizure aggravation. Neurologists should keep that in mind when prescribing CBD, taking into consideration its limited therapeutic indications, its possible side effects and, equally important, its remarkable interactions with other AEDs. Moreover, findings from real-world studies on larger populations will help us evaluate the real extent and duration of CBD clinical benefit and its long-term tolerability.
As to future prospects, CBD might find novel clinical applications, as it is currently being evaluated in other conditions, including IS, Rett syndrome and Fragile X syndrome, not to mention several neuropsychiatric diseases such as schizophrenia and autism spectrum disorders. Moreover, in order to maximize its therapeutic potential by increasing bioavailability, formulations other than the commonly used oral preparations (eg, transdermal) are also being tested. Finally, it is still an open question whether cannabis extracts could actually be more effective and tolerable than purified components, as recently suggested by a meta-analysis on 11 studies and overall 670 subjects.76 The reason for the hypothesized superiority of plant extracts over single compounds could lie in the so-called “entourage effect”,78 a phenomenon first described in endocannabinoids and then in pCBs, which refers to the synergistic action of both active and inactive botanical molecules in cannabis plants. Indeed, not only different pCBs can interact with each other (as demonstrated by CBD antagonism with THC,79 and the documented additive anti-convulsant actions of CBD and CBDV),80 but also terpenoids, lipophilic molecules found in cannabis plants and endowed of intrinsic pharmacological actions, could play a role in potentiating pCB effects. Overall, these considerations reflect the great complexity of cannabis pharmacology, which requires extensive researches to explore its real potentialities and to better define its indications in clinical practice.
Abbreviations
AEA, anandamide; AED, antiepileptic drug; AEs, adverse events; 2-AG, 2-arachydonoyl glycerol; A1R, adenosine1 receptor; AUC, area under the curve; BRV, brivaracetam; CBD, cannabidiol; CBDV, cannabidivarin; CLB, clobazam; CNS, central nervous system; CYP, cytochrome P 450; DAG, diacylglycerol; DGLα, diacylglycerol lipase α; DRE, drug-resistant epilepsy; DS, Dravet syndrome; ECS, endocannabinoid system; EMA, European Medicine Agency; ENT, equilibrative nucleotide transporter; ESL, eslicarbazepine; FAHH, fatty acid amide hydrolase; FDA, Food and Drug Administration; GABA, γ-aminobutiryc acid; Glut, glutamate; GTCSs, generalized tonic-clonic seizures; 5HT, 5-hydroxytryptamine; IGF1, Insulin Growth Factor 1; iNOS, inducible nitrous oxide synthase; IS, infantile spasms; LGS, Lennox-Gastaut syndrome; MD, multiple dose; mGluR, metabotropic glutamate receptor; mTOR, mammalian target of rapamycin; nCLB, norclobazam; NICE, National Institute for Health and Care Excellence; NO, nitrous oxide; OLE, open-label extension; pCBs, phytocannabinoids; PK, pharmacokinetic; PLCβ, phospholipase C β; PPARγ, peroxysome proliferator-activator receptor γ; RFD, rufinamide; ROS, reactive oxygen species; RR, risk ratio; SAD, single ascending dose; SAE, serious adverse event; SE, status epilepticus; TEAE, treatment-emergent adverse event; THC, tetrahydrocannabinol; TRPV1, transient receptor potential channel of vanilloid type 1; TRPA1, transient receptor potential channel of ankyrine type 1; TRPM8, transient receptor potential melastatin type 8; TPM, topiramate; TSC, tuberous sclerosis complex; UGT, UDP-glucuronosyl-transferase; VDCA1, voltage-dependent anion selective channel protein 1; VGCC, voltage-gated calcium channels; VGSC, voltage-gated sodium channels; VPA, valproic acid; ZNS, zonisamide.
Disclosure
The authors have no conflicts of interest to report.
References
1. U.S. Food and Drug Administration [website]. FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy.
2. Dijk S, Lok P. NICE rejects cannabidiol for two types of treatment resistant epilepsy in children. BMJ. 2019;366:l5280. doi:10.1136/bmj.l5280
3. GW Pharmaceuticals. GW pharmaceuticals receives European Commission approval for EPIDYOLEX® (cannabidiol) for the treatment of seizures in patients with two rare, severe forms of childhood-onset epilepsy.
4. Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783–786. doi:10.1111/epi.2014.55.issue-6
5. Treister-Goltzman Y, Freud T, Press Y, Peleg R. Trends in publications on medical cannabis from the year 2000. Popul Health Manag. 2019;22(4):362–368. doi:10.1089/pop.2018.0113
6. Klein BD, Jacobson CA, Metcalf CS, et al. Evaluation of cannabidiol in animal seizure models by the Epilepsy Therapy Screening Program (ETSP). Neurochem Res. 2017;42(7):1939–1948. doi:10.1007/s11064-017-2287-8
7. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2012;13(6):CD009270.
8. Gloss D, Vickrey B. Cannabinoids for epilepsy. Cochrane Database Syst Rev. 2014;5(3):CD009270.
9. Amin MR, Ali DW. Pharmacology of medical cannabis. Adv Exp Med Biol. 2019;1162:151–165.
10. Hill AJ, Williams CM, Whalley BJ, Stephens GJ. Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol Ther. 2012;133(1):79–97. doi:10.1016/j.pharmthera.2011.09.002
11. Mechoulam R, Gaoni Y. A total synthesis of dl-Δ1-tetrahydrocannabinol, the active constituent of hashish”. J Am Chem Soc. 1964;87(14):3273–3275. doi:10.1021/ja01092a065
12. Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–564. doi:10.1038/346561a0
13. Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–65. doi:10.1038/365061a0
14. Cheer JF, Hurd YL. A new dawn in cannabinoid neurobiology: the road from molecules to therapeutic discoveries. Neuropharmacol. 2017;124:1–2. doi:10.1016/j.neuropharm.2017.07.004
15. McPartland JM, Duncan M, Di Marzo V, Pertwee RG. Are cannabidiol and Δ(9) -tetrahydrocannabivarin negative modulators of the endocannabinoid system? A systematic review. Br J Pharmacol. 2015;172(3):737–753. doi:10.1111/bph.12944
16. Olmo IG, Ferreira-Vieira TH, Ribeiro FM. Dissecting the signaling pathways involved in the crosstalk between metabotropic glutamate 5 and cannabinoid type 1 receptors. Mol Pharmacol. 2016;90(5):609–619. doi:10.1124/mol.116.104372
17. Katona I. Cannabis and endocannabinoid signaling in epilepsy. Handb Exp Pharmacol. 2015;231:285–316.
18. Ruehle S, Remmers F, Romo-Parra H, et al. Cannabinoid CB1 receptor in dorsal telencephalic glutamatergic neurons: distinctive sufficiency for hippocampus-dependent and amygdala-dependent synaptic and behavioral functions. J Neurosci. 2013;33(25):10264–10277. doi:10.1523/JNEUROSCI.4171-12.2013
19. Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215. doi:10.1038/sj.bjp.0707442
20. Laprairie RB, Bagher AM, Kelly ME, Denovan-Wright EM. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharmacol. 2015;172(20):4790–4805. doi:10.1111/bph.13250
21. Ryberg E, Larsson N, Sjögren S, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152(7):1092–1101. doi:10.1038/sj.bjp.0707460
22. Ibeas Bih C, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12(4):699–730. doi:10.1007/s13311-015-0377-3
23. Franco V, Perucca E. Pharmacological and therapeutic properties of cannabidiol for epilepsy. Drugs. 2019;79(13):1435–1454. doi:10.1007/s40265-019-01171-4
24. Lupica CR, Hu Y, Devinsky O, Hoffman AF. Cannabinoids as hippocampal network administrators. Neuropharmacol. 2017;124:25–37. doi:10.1016/j.neuropharm.2017.04.003
25. Bakas T, van Nieuwenhuijzen PS, Devenish SO, McGregor IS, Arnold JC, Chebib M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol Res. 2017;119:358–370. doi:10.1016/j.phrs.2017.02.022
26. Ruffolo G, Cifelli P, Roseti C, et al. A novel GABAergic dysfunction in human dravet syndrome. Epilepsia. 2018;59(11):2106–2117. doi:10.1111/epi.2018.59.issue-11
27. Ghovanloo MR, Shuart NG, Mezeyova J, Dean RA, Ruben PC, Goodchild SJ. Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J Biol Chem. 2018;293(43):16546–16558. doi:10.1074/jbc.RA118.004929
28. Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain. 2016;139(Pt 8):2164–2181. doi:10.1093/brain/aww129
29. McHugh D, Tanner C, Mechoulam R, Pertwee RG, Ross RA. Inhibition of human neutrophil chemotaxis by endogenous cannabinoids and phytocannabinoids: evidence for a site distinct from CB1 and CB2. Mol Pharmacol. 2008;73(2):441–450. doi:10.1124/mol.107.041863
30. Walter L, Franklin A, Witting A, et al. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23(4):1398–1405. doi:10.1523/JNEUROSCI.23-04-01398.2003
31. Epidiolex® (Cannabidiol) Oral Solution [Prescribing Information]. Carlsbad, CA:Greenwich Biosciences, Inc. 2018.
32. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double-blind, placebo-controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053–1067. doi:10.1007/s40263-018-0578-5
33. Birnbaum AK, Karanam A, Marino SE, et al. Food effect on pharmacokinetics of cannabidiol oral capsules in adult patients with refractory epilepsy. Epilepsia. 2019;60(8):1586–1592. doi:10.1111/epi.v60.8
34. Perucca E. Cannabinoids in the treatment of epilepsy: hard evidence at last? J Epilepsy Res. 2017;7(2):61–76. doi:10.14581/jer.17012
35. Taylor L, Crockett J, Tayo B, Morrison G. A phase 1, open-label, parallel-group, single-dose trial of the pharmacokinetics and safety of Cannabidiol (CBD) in subjects with mild to severe hepatic impairment. J Clin Pharmacol. 2019;59(8):1110–1119. doi:10.1002/jcph.v59.8
36. Devinsky O, Patel AD, Thiele EA, GWPCARE1 Part A Study Group. Randomized, dose-ranging safety trial of cannabidiol in dravet syndrome. Neurology. 2018;90(14):e1204–e1211. doi:10.1212/WNL.0000000000005254
37. Wheless JW, Dlugos D, Miller I, INS011-14-029 Study Investigators. Pharmacokinetics and tolerability of multiple doses of pharmaceutical-grade synthetic cannabidiol in pediatric patients with treatment-resistant epilepsy. CNS Drugs. 2019;33(6):593–604. doi:10.1007/s40263-019-00624-4
38. Geffrey AL, Pollack SF, Bruno PL, Thiele EA. Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia. 2015;56(8):1246–1251. doi:10.1111/epi.13060
39. GW Research Ltd. A randomized controlled trial to investigate possible drug-drug interactions between clobazam and cannabidiol. ClinicaTrials.gov identifier: NCT02565108. Available from: https://clinicaltrials.gov/ct2/show/NCT02565108?term=NCT02565108&draw=2&rank=1.
40. Morrison G, Crockett J, Blakey G, Sommerville K, Phase A. 1, open-label, pharmacokinetic trial to investigate possible drug-drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:1009–1031. doi:10.1002/cpdd.665
41. Anderson LL, Absalom NL, Abelev SV, et al. Coadministered cannabidiol and clobazam: preclinical evidence for both pharmacodynamic and pharmacokinetic interactions. Epilepsia. 2019;60:2224–2234. doi:10.1111/epi.16355
42. Diacomit® (Stiripentol) Capsule [Prescribing Information]. Beauvais, France:Biocodex, 2018.
43. Gaston TE, Bebin EM, Cutter GR, Liu Y, Szaflarski JP, UAB CBD Program. Interactions between cannabidiol and commonly used antiepileptic drugs. Epilepsia. 2017;58(9):1586–1592. doi:10.1111/epi.2017.58.issue-9
44. Klotz KA, Hirsch M, Heers M, Schulze-Bonhage A, Jacobs J. Effects of cannabidiol on brivaracetam plasma levels. Epilepsia. 2019;60(7):e74–e77. doi:10.1111/epi.16071
45. Mechoulam R, Carlini EA. Toward drugs derived from cannabis. Naturwissenschaften. 1978;65:174–179. doi:10.1007/BF00450585
46. Cunha JM, Carlini EA, Pereira AE, et al. Chronic administration of cannabidiol to healthy volunteers and epileptic patients. Pharmacology. 1980;21:175–185. doi:10.1159/000137430
47. Ames FR, Cridland S. Anticonvulsant effect of cannabidiol. S Afr Med J. 1985;69:14.
48. Trembly B, Sherman M Double-blind clinical study of cannabidiol as a secondary anticonvulsant. Proceedings of: marijuana ’90 international conference on cannabis and cannabinoids; 1990 July 8–11; Kolympari, Crete.
49. Devinsky O, Cross JH, Laux L, Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug-resistant seizures in the dravet syndrome. N Engl J Med. 2017;376(21):2011–2020. doi:10.1056/NEJMoa1611618
50. Lattanzi S, Brigo F, Trinka E, et al. Efficacy and safety of cannabidiol in epilepsy: a systematic review and meta-analysis. Drugs. 2018;78(17):1791–1804. doi:10.1007/s40265-018-0992-5
51. GW Research Ltd. A study to investigate the efficacy and safety of cannabidiol (GWP42003-P) in children and young adults with dravet syndrome. ClinicalTrials.gov identifier: NCT02224703. Available from: https://clinicaltrials.gov/ct2/show/NCT02224703?term=GWPCARE+2&cond=Dravet&draw=2&rank=1.
52. Devinsky O, Patel AD, Cross JH, GWPCARE3 Study Group. Effect of cannabidiol on drop seizures in the lennox-gastaut syndrome. N Engl J Med. 2018;378(20):1888–1897. doi:10.1056/NEJMoa1714631
53. GWPCARE4 Study Group. Cannabidiol in patients with seizures associated with lennox-gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled Phase 3 trial. Lancet. 2018;391(10125):1085–1096. doi:10.1016/S0140-6736(18)30136-3
54. Lattanzi S, Trinka E, Russo E, et al. Cannabidiol as adjunctive treatment of seizures associated with lennox-gastaut syndrome and dravet syndrome. Drugs Today (Barc). 2019;55(3):177–196. doi:10.1358/dot.2019.55.3.2909248
55. Devinsky O, Nabbout R, Miller I, et al. Long-term cannabidiol treatment in patients with dravet syndrome: an open-Label extension trial. Epilepsia. 2019;60(2):294–302. doi:10.1111/epi.2019.60.issue-2
56. Thiele E, Marsh E, Mazurkiewicz-Beldzinska M, et al. Cannabidiol in patients with lennox-gastaut syndrome: interim analysis of an open-label extension study. Epilepsia. 2019;60(3):419–428. doi:10.1111/epi.2019.60.issue-3
57. Devinsky O, Marsh E, Friedman D, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016;15(3):270–278. doi:10.1016/S1474-4422(15)00379-8
58. Mandelbaum DE. Cannabidiol in patients with treatment-resistant epilepsy. Lancet Neurol. 2016;15(6):544–545. doi:10.1016/S1474-4422(16)00122-8
59. Tang R, Fang F. Trial of cannabidiol for drug-resistant seizures in the dravet syndrome. N Engl J Med. 2017;377(7):699.
60. Thiele EA, Devinsky O, Checketts D, Knappertz V Cannabidiol (CBD) treatment responders analysis in patients with lennox– gastaut syndrome (LGS) on and off clobazam (CLB) [abstract no.1.436].
61. Gaston TE, Bebin EM, Cutter GR, et al. Drug-drug interactions with cannabidiol (CBD) appear to have no effect on treatment response in an open-label expanded access program. Epilepsy Behav. 2019;98(Pt A):201–206. doi:10.1016/j.yebeh.2019.07.008
62. Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(10):1617–1624. doi:10.1111/epi.2016.57.issue-10
63. GW Research Ltd. A randomized controlled trial of cannabidiol (GWP42003-P, CBD) for seizures in tuberous sclerosis complex (GWPCARE6). ClinicalTrial.gov Identifier: NCT02544763. Available from: https://clinicaltrials.gov/ct2/show/NCT02544763?term=GWPCARE6&draw=2&rank=1.
64. GW Pharmaceuticals. GW pharmaceuticals reports positive phase 3 pivotal trial results for EPIDIOLEX® (cannabidiol) Oral solution in patients with seizures associated with tuberous sclerosis complex.
65. Devinsky O, Verducci C, Thiele EA, et al. Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and aicardi, Dup15q, and doose syndromes. Epilepsy Behav. 2018;86:131–137. doi:10.1016/j.yebeh.2018.05.013
66. Martin RC, Gaston TE, Thompson M, et al. Cognitive functioning following long-term cannabidiol use in adults with treatment-resistant epilepsy. Epilepsy Behav. 2019;97:105–110. doi:10.1016/j.yebeh.2019.04.044
67. De Caro C, Leo A, Citraro R, et al. The potential role of cannabinoids in epilepsy treatment. Expert Rev Neurother. 2017;17:1069–1079. doi:10.1080/14737175.2017.1373019
68. Devinsky O, Cilio MR, Cross H, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014;55(6):791–802. doi:10.1111/epi.2014.55.issue-6
69. Morano A, Cifelli P, Nencini P, et al. Cannabis in epilepsy: from clinical practice to basic research focusing on the possible role of cannabidivarin. Epilepsia Open. 2016;1:145–151. doi:10.1002/epi4.12015
70. Greco M, Varriale G, Coppola G, Operto F, Verrotti A, Iezzi ML. Investigational small molecules in phase II clinical trials for the treatment of epilepsy. Expert Opin Investig Drugs. 2018;27(12):971–979. doi:10.1080/13543784.2018.1543398
71. Bialer M, Johannessen SI, Levy RH, et al. Progress report on new antiepileptic drugs: a summary of the thirteenth Eilat conference on new antiepileptic drugs and devices (EILAT XIII). Epilepsia. 2017;58:181–221. doi:10.1111/epi.13634
72. GW Research Ltd. A study of GWP42006 in people with focal seizures - Part A. ClinicalTrial.gov Identifier: NCT02369471. Available from: https://clinicaltrials.gov/ct2/show/NCT02369471?term=cannabidivarin&draw=2&rank=5.
73. Bialer M, Johannessen SI, Koepp MJ, et al. Progress report on new antiepileptic drugs: A summary of the Fourteenth Eilat conference on new antiepileptic drugs and devices (EILAT XIV). I. Drugs in preclinical and early clinical development. Epilepsia. 2018;59(10):1811–1841. doi:10.1111/epi.14557
74. GW Research Ltd. A study of GWP42006 in people with focal seizures - part B. ClinicalTrial.gov Identifier: NCT02365610. Available from: https://clinicaltrials.gov/ct2/show/NCT02365610?term=cannabidivarin&draw=2&rank=6.
75. GW Pharmaceuticals. GW Pharmaceuticals Announces Preliminary Results of Phase 2a Study for its Pipeline Compound GWP42006.
76. Pamplona FA, da Silva LR, Coan AC. Potential clinical benefits of CBD-rich cannabis extracts over purified CBD in treatment-resistant epilepsy: observational data meta-analysis. Front Neurol. 2018;9:759. doi:10.3389/fneur.2018.00759
77. Detyniecki K, Hirsch LJ. Cannabidiol for epilepsy: trying to see through the haze. Lancet Neurol. 2016;15(3):235–237. doi:10.1016/S1474-4422(16)00002-8
78. Russo EB. The case for the entourage effect and conventional breeding of clinical cannabis: no “strain,” no gain. Front Plant Sci. 2019;9:1969. doi:10.3389/fpls.2018.01969
79. Russo EB. Taming THC: potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br J Pharmacol. 2011;163(7):1344–1364. doi:10.1111/j.1476-5381.2011.01238.x
80. Hill TD, Cascio MG, Romano B, et al. Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br J Pharmacol. 2013;170(3):679–692. doi:10.1111/bph.12321
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.