Back to Journals » Clinical Ophthalmology » Volume 20
Candidate Genes for Non-Syndromic Pediatric Cataracts
Authors Rossen JL, Drackley A, Goetsch Weisman A, Ing A, Bohnsack BL
Received 31 October 2025
Accepted for publication 26 February 2026
Published 17 March 2026 Volume 2026:20 555904
DOI https://doi.org/10.2147/OPTH.S555904
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Bharat Gurnani
Jennifer L Rossen,1,2 Andy Drackley,3,4 Allison Goetsch Weisman,4,5 Alexander Ing,3,4 Brenda L Bohnsack1,2
1Division of Ophthalmology, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA; 2Department of Ophthalmology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA; 3Center for Genomics Department of Pathology and Laboratory Medicine, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, USA; 4Department of Pediatrics, Northwestern University Feinberg School of Medicine, Chicago, IL, 60611, USA; 5Division of Genetics, Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, IL, 60611, USA
Correspondence: Jennifer L Rossen, Division of Ophthalmology, Ann & Robert H. Lurie Children’s Hospital of Chicago, 225 E. Chicago Ave, Box 70, Chicago, IL, 60611, USA, Tel +1312 227 6180, Fax +1312 227 9411, Email [email protected]
Introduction: Pediatric cataracts are a significant cause of vision loss in children and may present in isolation or in association with other ocular or systemic diseases. Despite advances in molecular diagnostics, the underlying etiology of cataracts in most patients remains unknown, even in the setting of a positive family history. Genetic testing for pediatric cataracts is neither standardized nor widely utilized. Lack of standardization is multifold, including limited published clinical and experimental reports and the absence of a comprehensive list of candidate genes with grading of the strength of gene-disease relationships.
Areas Covered: The purpose of this review is to provide a comprehensive list of the 81 candidate genes potentially associated with non-syndromic pediatric cataracts and the accompanying case-based and experimental literature support in order to start the process of developing a standardized approach to genetic testing. Inheritance patterns, other associated ocular findings, and proposed mechanisms of pathogenesis will be described for the candidate genes. Genes that are associated with two distinct phenotypes, one syndromic and one characterized by non-syndromic cataracts, will also be presented. The types of cataracts and age of onset are often highly variable at both the gene and variant level, so they will not be the focus of this review, but are of interest for future studies.
Future Work: Future work is needed to formalize a standardized list of established and candidate genes for non-syndromic pediatric cataracts and to systematically grade our confidence in the gene-disease relationships through the ClinGen framework. An improvement in genetic testing for pediatric cataracts will improve clinical care of these patients and their families regarding prognostication, personalized medical management, and clarification of recurrence risk for reproductive decision making. Further, a better understanding of the pathogenesis of pediatric cataracts can lead to targets for novel treatment development.
Keywords: congenital cataracts, pediatric cataracts, ocular genetics, isolated cataracts, inherited eye diseases
Introduction
Pediatric cataracts have been reported to cause 5–20% of low vision or blindness in children, with an estimated 314,000 new cases worldwide every year.1 There are many potential etiologies of cataracts in children such as congenital infections, trauma, medication/treatment-related, inflammation, and genetic.2 It has been reported that 22–50% of pediatric cataracts are inherited, however due to the low rate of genetic testing for pediatric cataracts, the true rate is likely underreported.3,4 Additionally, genetic testing practices for pediatric cataracts, including gene panel content, are not standardized across laboratories. Consequently, the specific genes analyzed vary substantially between laboratories. Without a standardized list of candidate genes, there is significant variability in which genes are analyzed for individuals undergoing genetic testing for pediatric cataracts. Notably, among seven major commercial laboratories, pediatric cataract gene panels range in size from 66 to 171 genes each and collectively encompass 227 unique genes with only modest overlap across panels (Figure 1). Moreover, 55% (125/227) of these genes are included on only one panel, and only one laboratory’s panel includes analysis of mitochondrial DNA genes. The emergence of broad genomic sequencing such as whole exome sequencing (WES) and whole genome sequencing (WGS) exacerbates this issue; data filtration of these assays is highly dependent on an established gene-disease relationship. The absence of a known relationship of a gene to manifestation of cataracts may preclude thorough analysis, leaving potentially causative variants as unrecognized and unreported. Adding to the difficulty in establishing or clarifying these gene-disease associations, the type of cataract and other ocular and systemic findings often differ for patients with pathogenic variants in the same gene, including intrafamilial phenotypic variability.
 |
Figure 1 Percentage of Genes Tested on Each U.S.-Based Commercial Test. Abbreviation: PG, Prevention Genetics. |
A standardized list of genes with formalized grading of the strength of the gene-disease relationships is needed to improve genetic testing for patients with pediatric cataracts. The Clinical Genome Resource (ClinGen) is an NIH-funded organization developed in 2013 to standardize genetic testing analysis through expert panels, such as gene curation expert panels (GCEPs). The GCEPs follow a rigorous protocol to grade the strength of gene-disease relationships, known as gene curation. The majority of candidate genes for pediatric cataracts have not yet been addressed in current GCEPs. In order to prepare for gene curation of pediatric cataracts, a thorough examination of the literature is required. The purpose of this review is to provide a summary of the case-based and experimental literature support for the 81 identified candidate genes associated with non-syndromic pediatric cataracts as we prepare for future gene-disease validation work. The genes associated with syndromic pediatric cataracts will be the focus of another review.
Methods
The genes tested in these seven commercial panels, the National Health Service (NHS) panel in England, and an online reference database5 identified over 500 established and candidate genes for pediatric cataracts. A literature review was completed by utilizing the The Human Gene Mutation Database (HGMD) to identify reports of gene variants and querying PubMed for each gene. Reports on case-based human data and in vitro and in vivo non-human studies that described a possible gene-disease relationship with non-syndromic pediatric cataracts were included. Around 300 of the genes had at least one literature report of an association with pediatric cataracts. The 81 candidate genes that were reported to have a potential association with non-syndromic pediatric cataracts were included in this paper. Genes were included if they were reported to have a possible association with isolated pediatric cataracts and non-syndromic pediatric cataracts with other anterior segment and retinal diseases. Genes that are associated with non-syndromic cataracts in some reports and in association with other systemic diseases in other reports were also included. The remainder of the genes associated with syndromic pediatric cataracts will be reported in a subsequent review paper.
Crystallin Genes
Previous reports predict that about half of isolated cataracts are due to variants in one of the 13 crystallin genes (Table 1).6–8 Crystallins, of which there are three subtypes (α, β, and γ), occupy about 90% of the protein content of the human lens. While pediatric cataracts associated with pathogenic variants in the crystallin genes are predominantly associated with an autosomal dominant inheritance pattern, some forms are autosomal recessive. Additionally, pathogenic variants in crystallin genes are generally associated with isolated cataracts, though some have also been linked to other systemic manifestations. Most notably CRYAB is associated with cardiac and musculoskeletal abnormalities in addition to cataracts.
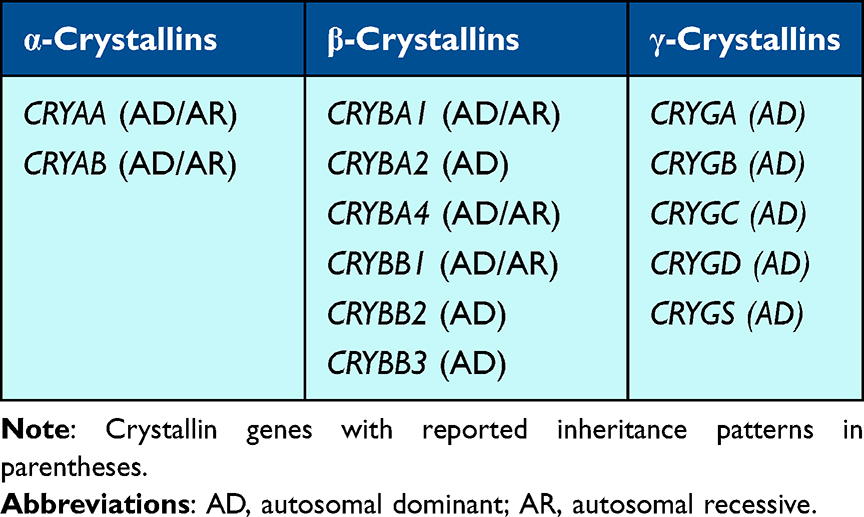 |
Table 1 Crystallin Genes |
α-Crystallins
There are two α-Crystallin subunits, αA and αB, encoded by CRYAA and CRYAB, respectively.6,9,10 The α-Crystallins are heat shock proteins that act as molecular chaperones to maintain lens clarity by preventing other proteins from aggregating.9,11 Heterozygous pathogenic variants in CRYAA have been associated with variable types of bilateral cataracts presenting from birth to adulthood.6,12–35 Many of the pediatric cataracts are also associated with other ocular abnormalities/diseases including microcornea, microphthalmia, and secondary glaucoma.13–17 Notably, missense variants replacing an arginine appear to be enriched in individuals with cataracts; substitutions of arginine by an amino acid of a different charge may alter heat shock protein structure, decreasing solubility as well as impacting protein expression.6,13,14,17–25 Patients with heterozygous protein-elongating (eg, stop-loss) variants appear to have more severe ocular abnormalities, including congenital aphakia and retinal and optic nerve anomalies.26 Biallelic pathogenic variants in CRYAA have also been reported in association with pediatric cataracts, with some heterozygous “carriers” also having subtle lens changes.27,28 Functional in vitro and in vivo mouse and zebrafish studies support this gene-disease relationship.15,22,29–35
CRYAB is the crystallin gene with the strongest and best characterized link to non-cataract phenotypes, particularly cardiac and skeletal muscle abnormalities.6 Heterozygous CRYAB pathogenic variants, mostly missense, have been linked to pediatric cataracts occurring either in isolation14,36–43 or in conjunction with cardiac and/or muscular defects.44–46 Like CRYAA, biallelic pathogenic variants in CRYAB have also been reported in individuals with isolated pediatric cataracts.47,48 However, many reports of individuals with CRYAB variants focus predominantly on cardiac disease and do not describe pediatric cataracts as part of the phenotype.49–71 Functional studies with in vitro assays and in vivo zebrafish and mouse models have validated the pathogenesis of CRYAB variants and elucidated the role of αB-Crystallin as a heat shock protein in the lens.49,50,72–77
β-Crystallins
Six β-Crystallin genes – 3 acidic (CRYBA1, CRYBA2, and CRYBA4) and 3 basic (CRYBB1, CRYBB2, and CRYBB3) – encode proteins that are not heat shock proteins, but rather function to maintain the refractive index of the lens.50,51 Of the β-Crystallin genes, CRYBA1, CRYBB1, and CRYBB2 are the most well studied and supported with respect to their involvement in cataracts. Heterozygous pathogenic variants in CRYBA1, encoding both the βA1-Crystallin and βA3-Crystallin proteins, have been reported in many individuals and families with isolated bilateral pediatric cataracts of various types, with considerable intra-familial variability.30,31,78–89 However, one group identified a potential association between CRYBA1 variation and cardiomyopathy or learning disabilities,52 warranting further studies on phenotypic associations. Whereas missense variants make up the majority of variants identified in the isolated cataract-associated CRYAA and CRYAB variants, the CRYBA1 mutational spectrum comprises mainly nonsense, frameshift, and splicing variants predicted to result in protein truncation or loss of protein expression, with missense variants reported much more rarely.24,53–65 Similar to CRYAA and CRYAB, biallelic pathogenic variants in CRYBA1 have been reported in association with cataracts, with one group reporting a homozygous frameshift pathogenic variant in association with isolated cataracts in a family.66 Functional studies, including mouse models and in vitro assays, have strengthened the association of CRYBA1 with pediatric cataracts.65,67–71 Heterozygous missense variants in CRYBA2 have been associated with pediatric cataracts of variable types in individuals, with incomplete penetrance demonstrated in at least one family.54,90–93 Compared to CRYBA2, there have been more reports of heterozygous missense variants in CRYBA4 associated with pediatric cataracts of various types (often nuclear/lamellar when described) and frequently associated with microcornea/microphthalmia.54,64,78–81,94–98 Homozygous CRYBA4 missense variants have also been reported in two families with pediatric cataracts.82,83
There are numerous reports of CRYBB1 heterozygous missense, nonsense, frameshift, and start-loss variants associated with pediatric cataracts of various types; these cataracts often require surgery at a young age, and sometimes present with microcornea/microphthalmia and glaucoma, and, rarely, posterior scleral staphylomas and colobomas.6,27,32,88,99–113 There have also been five reports of patients with CRYBB1 biallelic variants associated with cataracts.84–87,97 In vitro and in vivo mouse studies have also supported the association of CRYBB1 variants with cataracts.88,89,114,115 Similarly, CRYBB2 also has significant clinical evidence supporting the role of heterozygous missense and nonsense variants in development of pediatric cataracts; additionally, some individuals presented with microcornea/microphthalmia, and one individual each with coloboma and microphthalmia.14,15,27,31,81,108,116–127 Of interest, several variants have been determined to be the result of pseudogene conversions with the pseudogene CRYBB2P1.128–130 Overall, there is less published support for an association between CRYBB3 and cataracts; the majority of studies report heterozygous missense variants in individuals with various types of cataracts14,79,90–92,99,131,132 and the same homozygous variant in several families with isolated cataracts.14,133
γ-Crystallins
The five γ-Crystallins are encoded by genes mainly clustered on the long arm of chromosome 2 (CRYGC, CRYGD, CRYGA, and CRYGB are located at 2q33.3 and CRYGS is located at 3q27.3) and, like the β-Crystallins, maintain the refractive index of the lens.11 Of the γ-Crystallin-encoding genes, CRYGC and CRYGD are the best described and most well supported in their association with pediatric cataracts, whereas CRYGA, CRYGB, and CRYGS have very limited data available. There are a few reports of heterozygous missense CRYGA variants associated with pediatric cataracts18,134–136 with in vitro and in vivo mouse models reported.116,137,138 While two studies report a possible association of heterozygous CRYGB variants with pediatric cataracts, their causal relationship remains inconclusive;139,140 however, in vitro and in vivo mouse studies have provided more support by showing abnormal protein development and cataract formation due to the Crygbnop variant.116,138 CRYGC has substantial evidence linking heterozygous variants to pediatric cataracts, often with microcornea or microphthalmia.6,28,88,99,108,114,135,141–155 Similarly, CRYGD heterozygous variants have been reported frequently in the literature in association with pediatric cataracts, also sometimes with microcornea or microphthalmia.6,12,19,27,46,100,114,144,148,151,156–174 Heterozygous CRYGS variants have also been reported with progressive congenital cataracts in multiple families18,39,41,54,56,93,100,175–178 and the role of the gene and variants in cataract formation have been supported by in vitro and in vivo mouse studies.100–103,178
Additional Non-Syndromic Cataract Genes
In addition to the crystallin genes, there are 22 genes associated with isolated cataracts in the literature, many of which encode proteins vital to lens development and maintaining clarity (Table 2). The amount of literature support is variable, ranging from one case report to several identified pathogenic variants.
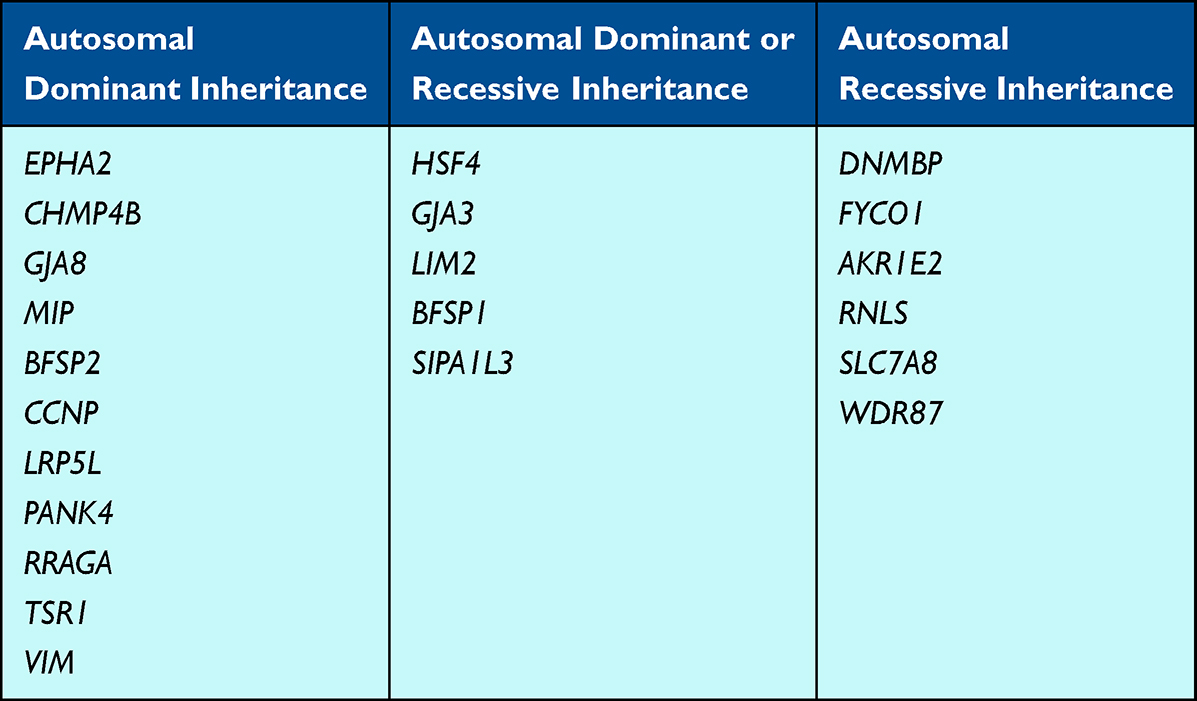 |
Table 2 Non-Crystallin Genes Associated with Non-Syndromic Pediatric Cataracts |
EPHA2 encodes a membrane bound tyrosine kinase receptor expressed in lens epithelial cells.104 Heterozygous variants in EPHA2 have been identified with pediatric cataracts in many patients and are suspected to account for 5% of inherited cataracts in South-Eastern Australia.105,106 Multiple in vivo and in vitro studies have suggested that the absence of EphA2 disrupts cell–cell junctions, thereby disrupting communication and ultimately lens homeostasis.105,107
CHMP4B encodes a key protein with multivesicular bodies that regulates membrane remodeling within the lens epithelial cells.108 Shiels et al (2007) first reported on the association of CHMP4B heterozygous variants with pediatric cataracts with further in vitro studies showing the importance of this gene’s expression in maintaining lens clarity.109 Other groups have also reported heterozygous variants in CHMP4B in association with pediatric cataracts and our institution has identified two unrelated families with isolated cataracts due to heterozygous variants in CHMP4B.110,111 Additionally, a conditional Chmp4b-knockdown mouse deficient in lens CHMP4B results in variable lens changes,108 providing functional data supporting the role of deficient CHMP4B protein in cataract development.
HSF4 encodes a heat-shock transcription factor required for lens cell growth and differentiation.112 At least 16 heterozygous or biallelic HSF4 variants have been reported in association with pediatric cataracts across geographical and ethnic populations; the heterozygous variants reside in the α-helical DNA-binding-domain, whereas the biallelic variants are outside of it.112 Hsf4 knockout mice develop cataracts due to abnormal lens development113 and zebrafish models also support this gene’s importance for lens development.179
Variants in two connexin genes, GJA3 (Cx46) and GJA8 (Cx50) have been linked to pediatric cataracts due to their disruption of hemichannel formation and function, leading to disordered crystallin proteins and cataract development.180,181 Hassan et al (2021) reviewed the literature and identified 48 variants present in the heterozygous state in individuals with isolated pediatric cataracts, and two instances caused by a homozygous GJA3 variant.180 To date, more than 30 heterozygous variants in GJA8 have been reported in association with pediatric cataracts.182
Genes encoding membrane proteins within the lens have also been associated with cataracts. LIM2 encodes the protein MP20, which colocalizes with Cx46 and is expressed in lens fiber cells as the second most prevalent membrane protein in the lens.117 Both heterozygous and biallelic variants in LIM2 have been reported in association with pediatric cataracts.117–120 MIP encodes MIP, an intrinsic membrane protein that comprises 45% of the membrane protein in lens fiber cells.121 At least 22 heterozygous MIP variants have been reported in association with pediatric cataracts and functional in vitro studies support their role in disease pathogenesis.121
The lens-specific beaded filament structural proteins (BFSPs) within the cytoskeleton are essential for lens clarity and homeostasis; both BFSP1 (filesin) and BFSP2 (phakinin) have been associated with pediatric cataracts.122 There has been one report of a homozygous BFSP1 variant and one heterozygous BFSP1 variant in individuals with pediatric cataracts with supporting in vitro studies.122–124 A few groups have also identified heterozygous BFSP2 variants associated with pediatric cataracts.125–127 Knockout Bfsp1 and Bfsp2 mouse models support their importance in preserving lens clarity, but not always cataract formation.141,183,184
Pediatric cataracts, sometimes also with microphthalmia and anterior segment dysgenesis, have been reported to be associated with both heterozygous and biallelic variants in SIPA1L3, which encodes a GTPase activating protein.185,186 Pathogenicity has been suggested to be due to the role of the encoded protein in Rap1 signaling of the regulation of lens epithelial cell polarity and morphogenesis, as shown in mouse studies.185,186
Ansar et al (2018) identified homozygous DNMBP frameshift variants in three consanguineous families with bilateral pediatric cataracts; functional studies in Drosophila examined the role of the DNMBP-encoded dynamic binding protein in tight junctions and regulation of E-cadherin in lens vesicle separation and lens epithelial cell survival.187
Iqbal et al (2020) reported that homozygous variants in FYCO1, a gene involved with transport of microtubule vesicles, were found in 15% of a cohort of patients with inherited cataracts in Pakistan.188 The group also reviewed the literature and identified many additional individuals with biallelic FYCO1 variants (mostly frameshift, nonsense, or affecting splicing) identified with pediatric cataracts worldwide.188
Some candidate genes have only one or two cases reported in the literature of heterozygous variants identified in individuals with pediatric cataracts, including CCNP, LRP5L, PANK4, RRAGA, TSR1, and VIM, and therefore less strong support for their gene-disease relationship. Khaliq et al (2002) reported on a CCNP heterozygous variant that segregated with cataracts in members of a four-generation family.189 Sun et al (2020) identified a heterozygous variant in LRP5L associated with a membranous congenital cataract in a four-generation family.190 Functional studies of the variant supported pathogenicity and proposed that inhibition of laminin γ1 and c-MAF resulted in cataract formation.190 Sun et al (2019) identified an intronic heterozygous PANK4 variant in a four-generation family with pediatric cataracts.191 Furthermore, the authors developed a Pank4-mutant mouse that also developed cataracts and demonstrated that this variant affects crystallin expression, suggesting a possible mechanism for disease.191 Chen et al (2016) identified three heterozygous variants in RRAGA associated with pediatric cataracts and performed functional in vitro studies that suggested that their pathogenicity may be due to effects on mTORC1 signaling.192 Yu et al (2020) reported a TSR1 heterozygous variant in a family with pediatric cataracts and performed studies in mice and fetal lens tissue to characterize TSR1 expression.193 An association of VIM, which encodes vimentin, an intermediate filament expressed in the lens, with pediatric cataracts has been suggested based on identification of heterozygous VIM variants in two patients.14,142
There are also candidate genes with limited case reports in the literature of biallelic variants associated with pediatric cataracts, including AKR1E2, RNLS, SLC7A8, and WDR87, and like the genes described in the paragraph above, have less strong support for gene-disease association. Aldahamesh et al (2012) reported a homozygous canonical splice site variant in AKR1E2 that segregated with pediatric cataracts in one family.143 AKR1E2 has been shown to have lens-enriched gene expression in mice,144 and the authors suggest that the variant results in 1,5-anhydro-D-fructose accumulation, which increases osmotic pressure in the lens and leads to cataract development.143 The same group reported a homozygous truncating variant in RNLS associated with pediatric cataracts in a family, yet the mechanism of pathogenesis is unclear.143 Knopfel et al (2019) identified a homozygous variant in SLC7A8, which encodes LAT2 that is highly expressed in the lens, in a family with pediatric cataracts and conducted in vivo mouse studies to support pathogenesis.145 Khan et al (2017) reported a homozygous WDR87 variant in a family with pediatric cataracts with associated mouse studies.66,146 Importantly, the heterozygous parents in the aforementioned families were not reported to have any ocular manifestations, supporting a true autosomal recessive inheritance pattern for these genes, at least with respect to the specific identified variants.
Genes Associated with Non-Syndromic Pediatric Cataracts and Other Anterior Segment Diseases
Pediatric cataracts may also be one component of a more severe anterior segment dysgenesis (ASD) phenotype. While microcornea/microphthalmia is considered a standard feature associated with many pediatric cataracts, other anterior segment disorders such as posterior embryotoxon, iris hypoplasia, congenital corneal opacities, and glaucoma may also be seen in association with 10 genes (Table 3).
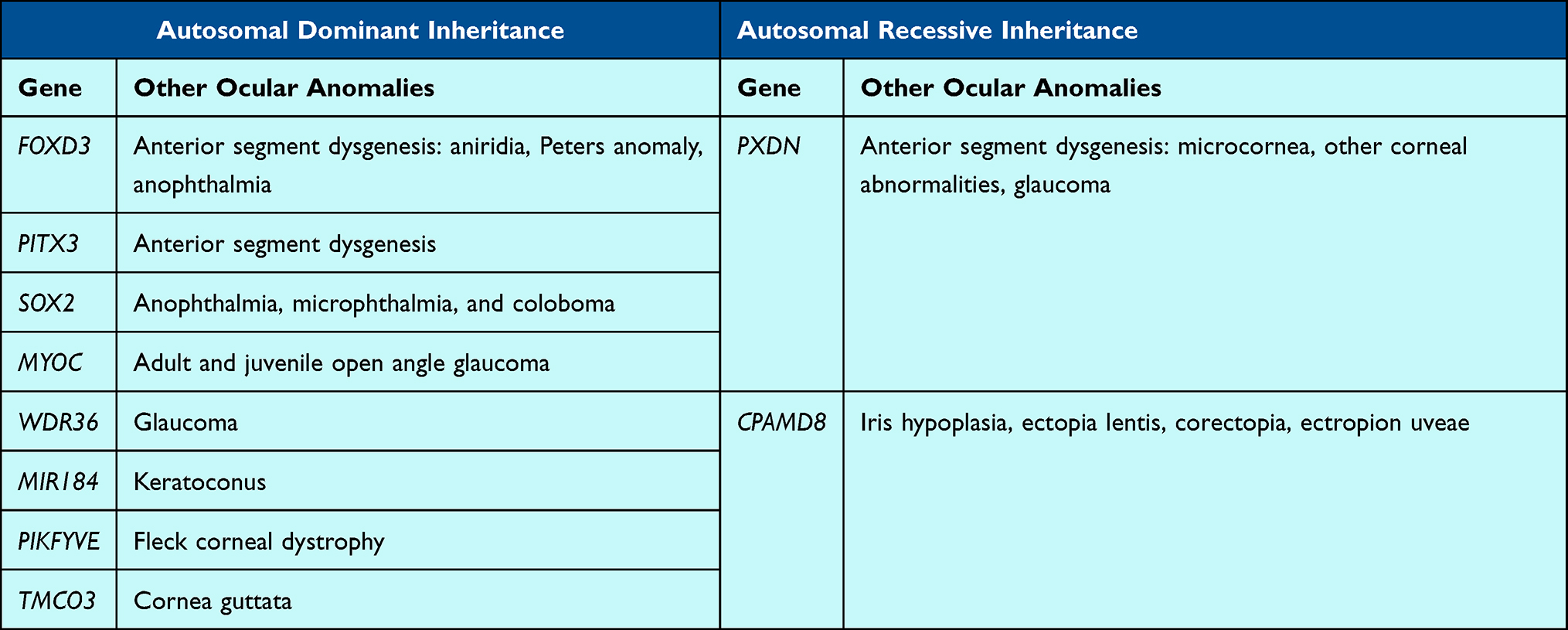 |
Table 3 Genes Associated with Non-Syndromic Pediatric Cataracts and Other Anterior Segment Diseases |
There are several genes associated with either anterior segment or global ocular anomalies and pediatric cataracts, including FOXD3, PITX3, PXDN, and SOX2. Heterozygous variants in FOXD3, which encodes a forkhead transcription factor that is an early marker for neural crest cell specification, have been shown to be associated with various forms of anterior segment dysgenesis, including aniridia, Peters anomaly, anophthalmia, and at least one case of isolated congenital cataracts.147 PITX3 heterozygous variants have been reported in several reports of patients with pediatric cataracts with or without anterior segment dysgenesis, and functional in vitro studies have shown expression within the early lens vesicle and regulation of lens epithelial cell differentiation.148 Biallelic variants in PXDN, which is in the same signaling pathway as PITX3 and FOXE3, have also been reported in individuals with various ASD phenotypes, including congenital cataract-microcornea, corneal abnormalities, and glaucoma.149 Although the mechanism is not fully known, it has been suggested that the lack of PXDN protein in the cornea and lens allows for the buildup of reactive oxygen intermediates that leads to clouding of the cornea and lens.150 SOX2 pathogenic heterozygous variants can result in multiple ocular anomalies including anophthalmia, microphthalmia, and coloboma, with the latter two commonly found in association with pediatric cataracts.151
Several genes traditionally associated with glaucoma have also been reported with pediatric cataracts in a few instances, including CPAMD8, MYOC, and WDR36. Cheong et al (2016) reported on three unrelated families with biallelic variants in CPAMD8 associated with iris hypoplasia, ectopia lentis, corectopia, ectropion uveae and pediatric cataracts; in vitro and in vivo (zebrafish and mice) functional studies support the role of CPAMD8 in anterior segment development.152 Siggs et al (2020) identified 11 patients with either childhood or juvenile glaucoma and biallelic variants in CPAMD8, the majority of whom also had iris abnormalities and cataracts, further supporting its role in anterior segment development.153 MYOC is a gene with a well-known association with adult and juvenile onset open-angle glaucoma.154 In the Li et al (2016) evaluation of patients with non-familial sporadic pediatric cataracts, they identified one pediatric patient with bilateral cataracts and a heterozygous MYOC variant, though it is unclear whether this patient also had glaucoma.91 The same group identified heterozygous WDR36 variants in two patients with pediatric cataracts, another gene previously implicated in glaucoma only.91,155
Additionally, variants in genes associated with corneal diseases have also been rarely identified in patients with pediatric cataracts. These genes include MIR184, PIKFYVE, and TMCO3. Hughes et al (2011) evaluated a large family with autosomal dominant keratoconus and early onset anterior polar cataract, identifying a heterozygous MIR184 variant; functional studies conducted support the pathogenicity of this variant.156 PIKFYVE, previously implicated in fleck corneal dystrophy, is also associated with pediatric cataracts; Mei et al (2022) identified a heterozygous variant segregating with pediatric cataracts in a four-generation family and demonstrated lens defects in a zebrafish model.157 Chen et al (2016) identified a heterozygous variant in TMCO3 segregating with cornea guttata and anterior polar cataract, sometimes presenting at birth, in 17 members of a family.158
Genes Associated with Non-Syndromic Pediatric Cataracts and Retinal Dystrophies
Pediatric patients with inherited retinal dystrophies may also have cataracts as an ocular manifestation of their disease as has been previously reported for 14 genes, although the literature support is sparse and mainly include one or two cases (Table 4). For example, a few genes associated with early onset Leber Congenital Amaurosis (LCA) also contain variants associated with pediatric cataracts. Biallelic AIPL1 variants are associated with LCA and the development of cataracts in teen years or early adulthood in addition to the retinal findings.159 Biallelic GUCY2D variants are also well known to be associated with LCA and have been shown to cause pediatric cataracts as well.160 Ahmad et al (2011) also reported on a family with LCA due to a homozygous variant in LCA5 that led to the typical retinal findings in addition to cataract development during the teen years.161
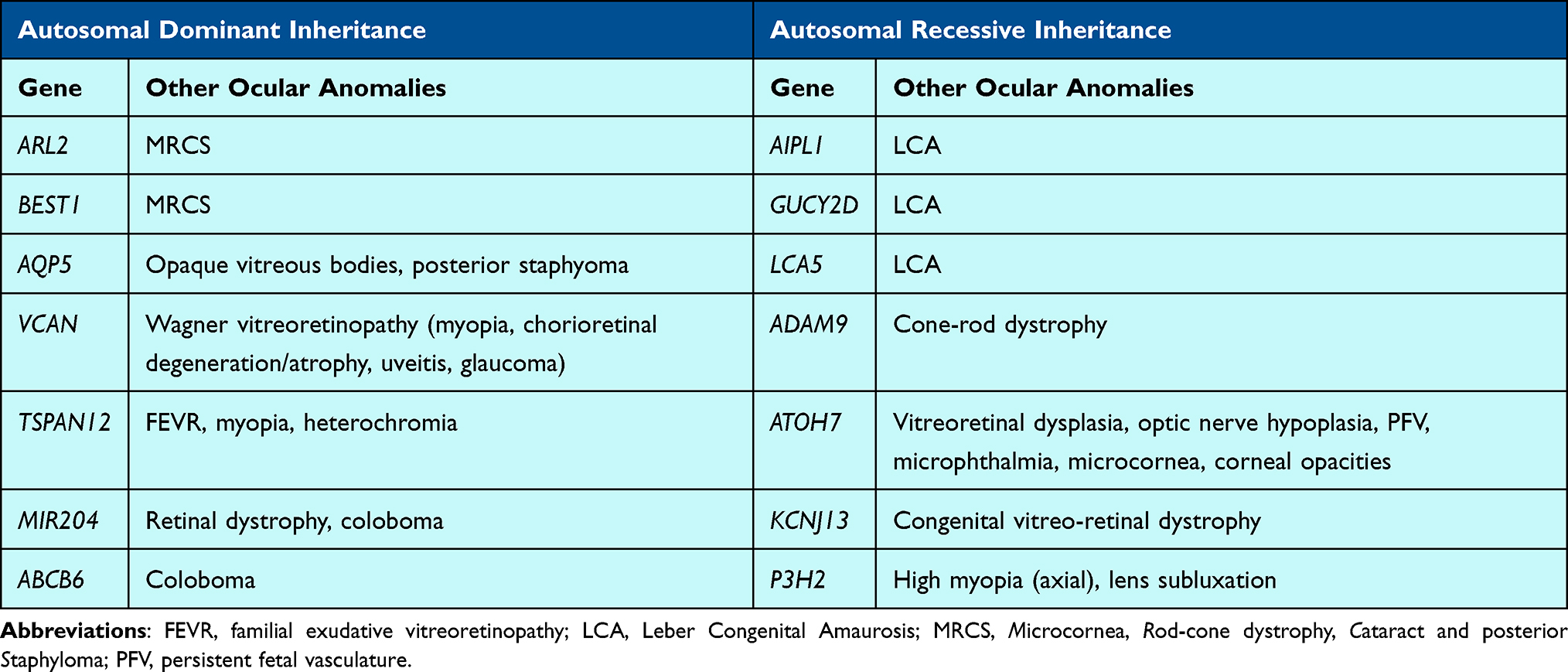 |
Table 4 Genes Associated with Non-Syndromic Pediatric Cataracts and Retinal Dystrophies |
Microcornea, Rod-cone dystrophy, Cataract and posterior Staphyloma (MRCS), an autosomal dominant ocular disease characterized by reduced visual acuity, congenital cataract, microcornea, posterior staphyloma, and reduced scotopic and photopic responses on electroretinography, is thought to be due to variation in one of at least two genes. Cai et al (2019) reported a heterozygous variant in ARL2 that segregated within a family with MRCS. ARL2 encodes a GTP-binding protein belonging to the RAS superfamily; in vitro functional studies demonstrated that abnormal ARL2 resulted in mitochondrial dysfunction and a mouse model largely recapitulated the ocular findings seen in humans.162 While variation in BEST1 is most commonly known to be associated with Best vitelliform macular dystrophy, heterozygous variants in BEST1 have also been linked to MRCS.163
There are several other genes associated with retinal and vitreoretinal disorders as well as pediatric cataracts. Biallelic pathogenic variants in ADAM9 can cause cone-rod dystrophy in addition to pediatric cataracts; in vitro studies have shown ADAM9 to be downregulated in anterior polar cataracts.164–166 Khan et al (2012) reported 2 different homozygous variants in ATOH7 in families with a variety of findings, including vitreoretinal dysplasia, optic nerve hypoplasia, persistent fetal vasculature, microphthalmia, congenital cataracts, microcornea, and corneal opacities.167 Although mice models do not directly mirror human clinical data, they show some ocular anomalies and PAX6, gene known to be key in ocular dysgenesis, is an upstream regular of ATOH7.167 Khan et al (2015) also reported two unrelated patients with a congenital vitreo-retinal dystrophy and pediatric or early-onset cataracts who were identified to have the same homozygous variant in KCNJ13.168 Additionally, Tang et al (2021) reported on a heterozygous missense variant in AQP5 that segregated with congenital cataracts in a 4-generation family also presenting with opaque vitreous bodies and sometimes posterior staphylomas.169 They further supported this association through development of an Aqp5 knockout mouse that recapitulated the early-onset cataract phenotype seen in humans.169 The group showed that AQP5 helps maintain lens clarity and protects against cataract formation, with the suggestion that AQP5 may regulate vimentin expression via miR-124-3p.1.169 Wagner vitreoretinopathy, due to heterozygous variants in VCAN, may also present with cataract, myopia, chorioretinal degeneration/atrophy, uveitis or glaucoma.170 Elhusseiny et al (2022) described a child with familial exudative vitreoretinopathy (FEVR), myopia, pediatric cataract, and heterochromia who was found to have a heterozygous in TSPAN12, a gene most closely linked to FEVR.171,172
Patients with inherited colobomas may also have cataracts as described in a couple families. Conte et al (2015) reported a heterozygous MIR204 variant that segregated in a large 5-generation family with autosomal dominant retinal dystrophy and coloboma; some individuals in this family also developed pediatric cataracts.173 ABCB6, associated with autosomal dominant coloboma, was also implicated in a family with pediatric cataracts.132,174
Homozygous truncating variants in P3H2 (previously LEPREL1, which encodes prolyl 3-hydroxylase 2, an enzyme involved in hydroxylation of collagens, were identified in multiple families with various ocular features including lens subluxation, lens opacities and axial high myopia.194,195
Genes Associated with Non-Syndromic Cataracts and/or Systemic Diseases
In addition to CRYAB, as discussed above, 22 genes have been reported in association with isolated cataracts with or without other ocular findings in some patients, syndromic cataracts in others, and systemic manifestations without cataracts in yet others (Table 5).
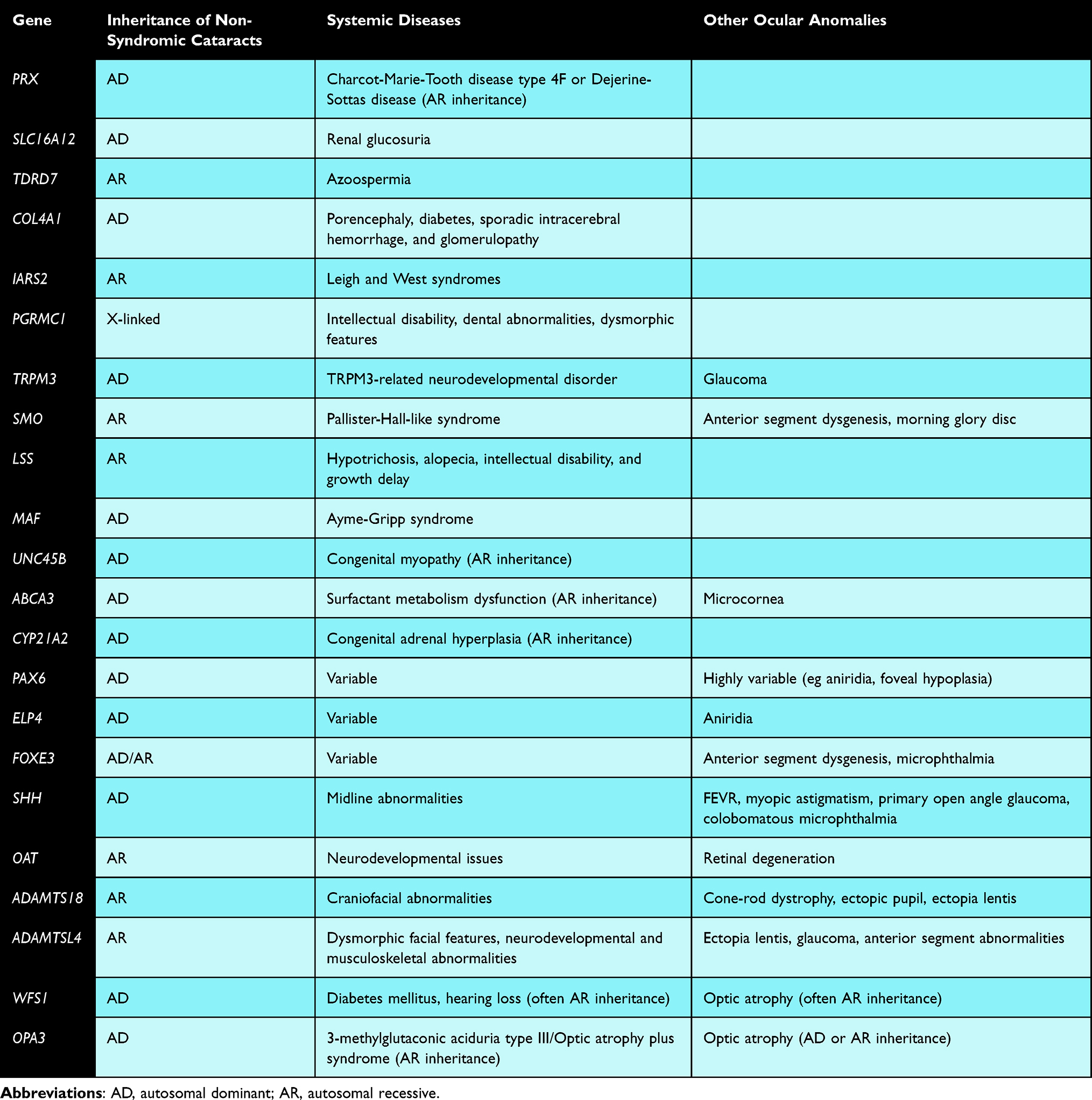 |
Table 5 Genes Associated with Non-Syndromic Cataracts and/or Systemic Diseases |
Several genes have been reported to be associated with either isolated or syndromic cataracts. For example, Yuan et al (2016) identified a heterozygous PRX variant in a 4-generation family with pediatric cataracts and incomplete penetrance196 but did not have systemic findings of Charcot-Marie-Tooth disease type 4F or Dejerine-Sottas disease, which have been associated with biallelic PRX variants.196 Prx knockout mice have abnormal lenses, suggesting an importance of this gene in lens development.197 SLC16A12 heterozygous variants have been associated with pediatric cataracts, sometimes in isolation and others with renal glucosuria with pathogenicity supported by rat studies.91,198 Biallelic TDRD7 variants have been associated with pediatric cataracts in isolation or in combination with azoospermia, with supportive functional in vitro and in vivo studies completed.83,132,199–201 Patients with COL4A1 heterozygous variants often have syndromic cataracts in addition to porencephaly, diabetes, sporadic intracerebral hemorrhage, and glomerulopathy, yet non-syndromic pediatric cataracts have been identified in a few families.93,202 Biallelic variants in IARS2 have been associated with severe conditions like Leigh and West syndromes, though more rarely with isolated cataracts.64,203 Hemizygous variants in PGRMC1 can result in non-syndromic pediatric cataracts or with syndromic features such as intellectual disability, dental abnormalities and dysmorphic features; notably, pgrmc1 knockdown zebrafish develop cataracts.204 The role of TRPM3 in human disease is not well understood but thought to be related to a neurodevelopmental disorder; however, Bennett et al (2014) reported on a family with pediatric cataracts and glaucoma and a heterozygous variant in TRPM3.205 Trpm3 is co-expressed with Mir204 in mice ocular tissues, and Pax6 has been shown to regulate both.205 In addition to its known association with Pallister-Hall-like syndrome, SMO and its encoded smoothened protein are known to be an important regulator of eye development, and biallelic variants were identified in an individual with pediatric cataracts, anterior segment dysgenesis and morning glory disc, comparable to findings in mouse models.206
In some instances, the location of the variant determines whether systemic manifestations accompany the cataracts. Biallelic variants in LSS have been associated with cataracts in isolation or with systemic manifestations including hypotrichosis, alopecia, intellectual disability, and growth delay; missense variants and those on the N-terminus tend to lead to hair loss, whereas those in the C-terminus usually are associated with cataracts.207 For variants in MAF, at least 17 heterozygous variants in the C-terminus region of the encoded transcription factor have been identified in individuals with isolated cataracts compared to those in the N-terminus, which lead to cataracts as a manifestation of Ayme-Gripp syndrome.208
For some genes, biallelic variants are associated with systemic diseases but monoallelic/heterozygous variants have been linked to isolated cataracts. A heterozygous UNC45B variant has been linked to autosomal dominant cataracts in one patient, while a homozygous UNC45B variant was suggested as the cause of congenital myopathy in another, with in vivo models supporting both phenotypes.209,210 Though biallelic ABCA3 variants have been associated with surfactant metabolism dysfunction, multiple different heterozygous ABCA3 variants were reported by Chen et al (2014) in individuals with pediatric cataract and microcornea.211 Similarly, biallelic pathogenic variants in CYP21A2 cause congenital adrenal hyperplasia, while heterozygous variants have been linked to isolated cataracts.212
Some genes associated with anterior segment dysgenesis are also associated with cataracts and variably with systemic diseases. Heterozygous PAX6 variants are associated with a wide spectrum of disease, ranging from pediatric cataracts in several cases to aniridia, foveal hypoplasia, amongst other ocular findings with or without systemic manifestations (even in absence of subsequent deletion of WT1).213,214 Heterozygous variants in ELP4, which is downstream of PAX6 and involved in PAX6 regulation, can lead to aniridia and pediatric cataracts.215,216 Variation in FOXE3 is linked to ocular phenotypes including an array of anterior segment dysgenesis, microphthalmia, and pediatric cataracts, with both autosomal dominant and recessive inheritance, sometimes with ocular only phenotypes and other times with variable systemic manifestations.217
Genes associated with vitreoretinal abnormalities may also be associated with cataracts, sometimes with systemic findings. Young et al (2022) reported on a large family with multiple variable ocular manifestations, including pediatric cataracts in most, as well as FEVR, myopic astigmatism, and primary open-angle glaucoma; a heterozygous intronic variant in SHH was found to segregate with these phenotypes.218 SHH plays an important role in eye development through well-characterized pathways involving PTCH1 and PAX6, amongst others.218 SHH variants have also been reported in association with non-syndromic colobobomatous microphthalmia219 in addition to other more well-described midline abnormalities.218 In vivo studies have similarly confirmed the importance of SHH in ocular development along with allowing for better understanding of its mechanism of pathogenesis.218,220 Gyrate atrophy is caused by biallelic variants in OAT, leading to retinal degeneration and childhood cataracts, with or without neurodevelopmental issues.221 Biallelic ADAMTS18 variants have been associated with various ocular abnormalities, including cone-rod dystrophy, ectopic pupils, pediatric cataracts, and ectopia lentis in multiple families with or without craniofacial abnormalities.222,223 Patients with biallelic ADAMTSL4 variants have ocular findings such as ectopia lentis, early-onset cataracts, glaucoma and other anterior segment abnormalities; though the majority of individuals with ADAMTSL4-related disease do not have systemic involvement, dysmorphic facial features as well as neurodevelopmental and musculoskeletal abnormalities have been reported in some.224
Additionally, two genes associated with optic atrophy may also be associated with cataracts and variable systemic manifestations. Wolfram syndrome, associated with pathogenic variants in WFS1 and characterized clinically by diabetes mellitus, hearing loss, and optic atrophy, can be inherited in either autosomal dominant or recessive manners.78,93,225 Pediatric cataracts are more common in the autosomal dominant form and a heterozygous WFS1 variant was identified in a family with cataracts only.78,93,225 Biallelic variants in OPA3 are associated with 3-methylglutaconic aciduria type III, also referred to as “optic atrophy plus syndrome”, while heterozygous pathogenic variants lead to a less severe form of disease with optic atrophy and cataract, and rarely with neurologic features.226
Discussion
At least81 genes have been reported to be associated with pediatric cataracts without systemic manifestations, though the amount of literature support varies widely. Some genes have only one variant identified, while others have dozens with accompanying functional studies. Additionally, not all have known pathogenic mechanisms for how variation leads to disease. Accordingly, clinicians and laboratories should be cautious in assigning pathogenicity to variants associated with genes described within this manuscript with only minimal literature support. In order to have greater confidence in providing genetic diagnoses for patients with pediatric cataracts, ClinGen gene curations are needed to formally grade the strength of each gene-disease relationship. This work is particularly critical given the number of cataract-associated genes potentially presenting with multiple inheritance patterns and with syndromic and non-syndromic phenotypes. Variant interpretation is also inherently tied to a thorough understanding of the gene, as numerous criteria that are directly influenced by the gene curation process (eg mechanism of disease, minor allele frequency thresholds, mutational hotspots).
However, the quality of gene-disease curation is only as good as the literature available. The limited support for many of the genes included in this paper may not be due to lack of pathogenicity, but due to the sparsity of clinical testing and reporting on potential genetic associations with pediatric cataracts. In order to better understand the strength and scope of gene-disease relationships as well as to the development of genotype-phenotype correlations, more reports on genetic testing results of patients with pediatric cataracts with detailed ocular phenotypes are needed. With improved knowledge, we can begin developing more individualized screening and treatment guidelines for patients and families based on their specific molecular etiology. In addition to increasing our clinical and medical genetic knowledgebase, a better understanding of the pathogenesis of pediatric cataracts through functional studies to help elucidate gene-disease relationships, as well as the impact of individual variants, can lead to advances in treatment.
Funding
The authors have received funding from the Ann & Robert H. Lurie Children’s Pilot Project Funding Genomics Network (GeNe) and from the Knights Templar Eye Foundatin Pediatric Ophthalmology Career-Starter Research grant for their research on cataract genetics.
Disclosure
Dr Jennifer Rossen reports personal fees from Mirum Pharmaceuticals, outside the submitted work. Dr Brenda L Bohnsack reports personal fees from Luminopia, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Sheeladevi S, Lawrenson JG, Fielder AR, et al. Global prevalence of childhood cataract: a systematic review. Eye. 2016;30(9):1160–19. doi:10.1038/eye.2016.156
2. Reis LM, Semina EV. Genetic landscape of isolated pediatric cataracts: extreme heterogeneity and variable inheritance patterns within genes. Hum Genet. 2019;138(8–9):847–863. doi:10.1007/s00439-018-1932-x
3. Li J, Chen X, Yan Y, et al. Molecular genetics of congenital cataracts. Exp Eye Res. 2020;191:107872. doi:10.1016/j.exer.2019.107872
4. Francis PJ, Moore AT. Genetics of childhood cataract. Curr Opin Ophthalmol. 2004;15(1):10–15. doi:10.1097/00055735-200402000-00003
5. Shiels A, Bennett TM, Hejtmancik JF. Cat-Map: putting cataract on the map. Mol Vis. 2010;16:2007–2015.
6. Berry V, Ionides A, Pontikos N, et al. The genetic landscape of crystallins in congenital cataract. Orphanet J Rare Dis. 2020;15(1):333. doi:10.1186/s13023-020-01613-3
7. Shiels A, Hejtmancik JF. Molecular genetics of cataract. Prog Mol Biol Transl Sci. 2015;134:203–218.
8. Shiels A, Hejtmancik JF. Mutations and mechanisms in congenital and age-related cataracts. Exp Eye Res. 2017;156:95–102. doi:10.1016/j.exer.2016.06.011
9. Wistow GJ, Piatigorsky J. Lens crystallins: the evolution and expression of proteins for a highly specialized tissue. Annu Rev Biochem. 1988;57:479–504. doi:10.1146/annurev.bi.57.070188.002403
10. Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76(2):145–153. doi:10.1016/S0014-4835(02)00278-6
11. Wistow G. The human crystallin gene families. Human Genom. 2012;6(1):26. doi:10.1186/1479-7364-6-26
12. Rossen JL, Bohnsack BL, Zhang KX, et al. Evaluation of genetic testing in a cohort of diverse pediatric patients in the United States with congenital cataracts. Genes. 2023;14(3):608. doi:10.3390/genes14030608
13. Hansen L, Yao W, Eiberg H, et al. Genetic heterogeneity in microcornea-cataract: five novel mutations in CRYAA, CRYGD, and GJA8. Invest Ophthalmol Vis Sci. 2007;48(9):3937–3944. doi:10.1167/iovs.07-0013
14. Ma AS, Grigg JR, Ho G, et al. Sporadic and familial congenital cataracts: mutational spectrum and new diagnoses using next-generation sequencing. Hum Mutat. 2016;37(4):371–384. doi:10.1002/humu.22948
15. Song Z, Si N, Xiao W. A novel mutation in the CRYAA gene associated with congenital cataract and microphthalmia in a Chinese family. BMC Med Genet. 2018;19(1):190. doi:10.1186/s12881-018-0695-5
16. Khidiyatova I, Khidiyatova I, Zinchenko R, et al. Study of the molecular nature of congenital cataracts in patients from the volga-ural region. Curr Issues Mol Biol. 2023;45(6):5145–5163. doi:10.3390/cimb45060327
17. Litt M, Kramer P, LaMorticella DM, et al. Autosomal dominant congenital cataract associated with a missense mutation in the human alpha crystallin gene CRYAA. Hum Mol Genet. 1998;7(3):471–474. doi:10.1093/hmg/7.3.471
18. Javadiyan S, Craig JE, Souzeau E, et al. High-throughput genetic screening of 51 pediatric cataract genes identifies causative mutations in inherited pediatric cataract in South Eastern Australia. G3. 2017;7(10):3257–3268. doi:10.1534/g3.117.300109
19. Mackay DS, Andley UP, Shiels A. Cell death triggered by a novel mutation in the alphaA-crystallin gene underlies autosomal dominant cataract linked to chromosome 21q. Eur J Hum Genet. 2003;11(10):784–793. doi:10.1038/sj.ejhg.5201046
20. Graw J, Klopp N, Illig T, et al. Congenital cataract and macular hypoplasia in humans associated with a de novo mutation in CRYAA and compound heterozygous mutations in P. Graefes Arch Clin Exp Ophthalmol. 2006;244(8):912–919. doi:10.1007/s00417-005-0234-x
21. Su D, Guo Y, Li Q, et al. A novel mutation in CRYAA is associated with autosomal dominant suture cataracts in a Chinese family. Mol Vis. 2012;18:3057–3063.
22. Laurie KJ, Dave A, Straga T, et al. Identification of a novel oligomerization disrupting mutation in CRYΑA associated with congenital cataract in a South Australian family. Hum Mutat. 2013;34(3):435–438. doi:10.1002/humu.22260
23. Yang Z, Su D, Li Q, et al. A R54L mutation of CRYAA associated with autosomal dominant nuclear cataracts in a Chinese family. Curr Eye Res. 2013;38(12):1221–1228. doi:10.3109/02713683.2013.811260
24. Bell S, Malka S, Lloyd IC, et al. Clinical spectrum and genetic diagnosis of 54 consecutive patients aged 0-25 with bilateral cataracts. Genes. 2021;12(2):131. doi:10.3390/genes12020131
25. Kessel L, Bach-Holm D, Al-Bakri M, et al. Genetic disease is a common cause of bilateral childhood cataract in Denmark. Ophthalmic Genet. 2021;42(6):650–658. doi:10.1080/13816810.2021.1941128
26. Marakhonov AV, Voskresenskaya AA, Ballesta MJ, et al. Expanding the phenotype of CRYAA nucleotide variants to a complex presentation of anterior segment dysgenesis. Orphanet J Rare Dis. 2020;15(1):207. doi:10.1186/s13023-020-01484-8
27. Pras E, Frydman M, Levy-Nissenbaum E, et al. A nonsense mutation (W9X) in CRYAA causes autosomal recessive cataract in an inbred Jewish Persian family. Invest Ophthalmol Vis Sci. 2000;41(11):3511–3515.
28. Khan AO, Aldahmesh MA, Meyer B. Recessive congenital total cataract with microcornea and heterozygote carrier signs caused by a novel missense CRYAA mutation (R54C). Am J Ophthalmol. 2007;144(6):949–952. doi:10.1016/j.ajo.2007.08.005
29. Bhagyalaxmi SG, Srinivas P, Barton KA, et al. A novel mutation (F71L) in alphaA-crystallin with defective chaperone-like function associated with age-related cataract. Biochim Biophys Acta. 2009;1792(10):974–981. doi:10.1016/j.bbadis.2009.06.011
30. Liang C, LIANG H, YANG YU, et al. Mutation analysis of two families with inherited congenital cataracts. Mol Med Rep. 2015;12(3):3469–3475. doi:10.3892/mmr.2015.3819
31. Hsu CD, Kymes S, Petrash JM. A transgenic mouse model for human autosomal dominant cataract. Invest Ophthalmol Vis Sci. 2006;47(5):2036–2044. doi:10.1167/iovs.05-0524
32. Xi JH, Bai F, Gross J, et al. Mechanism of small heat shock protein function in vivo: a knock-in mouse model demonstrates that the R49C mutation in alpha A-crystallin enhances protein insolubility and cell death. J Biol Chem. 2008;283(9):5801–5814. doi:10.1074/jbc.M708704200
33. Wu SY, Zou P, Mishra S, et al. Transgenic zebrafish models reveal distinct molecular mechanisms for cataract-linked αA-crystallin mutants. PLoS One. 2018;13(11):e0207540. doi:10.1371/journal.pone.0207540
34. Posner M, Murray KL, Andrew B, et al. Impact of α-crystallin protein loss on zebrafish lens development. Exp Eye Res. 2023;227:109358. doi:10.1016/j.exer.2022.109358
35. Zou P, Wu S-Y, Koteiche HA, et al. A conserved role of αA-crystallin in the development of the zebrafish embryonic lens. Exp Eye Res. 2015;138:104–113. doi:10.1016/j.exer.2015.07.001
36. Berry V, Francis P, Reddy MA, et al. Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet. 2001;69(5):1141–1145. doi:10.1086/324158
37. Liu M, Ke T, Wang Z, et al. Identification of a CRYAB mutation associated with autosomal dominant posterior polar cataract in a Chinese family. Invest Ophthalmol Vis Sci. 2006;47(8):3461–3466. doi:10.1167/iovs.05-1438
38. Liu Y, Zhang X, Luo L, et al. A novel alphaB-crystallin mutation associated with autosomal dominant congenital lamellar cataract. Invest Ophthalmol Vis Sci. 2006;47(3):1069–1075. doi:10.1167/iovs.05-1004
39. Devi RR, Yao W, Vijayalakshmi P, et al. Crystallin gene mutations in Indian families with inherited pediatric cataract. Mol Vis. 2008;14:1157–1170.
40. Chen Q, Ma J, Yan M, et al. A novel mutation in CRYAB associated with autosomal dominant congenital nuclear cataract in a Chinese family. Mol Vis. 2009;15:1359–1365.
41. Sun W, Xiao X, Li S, et al. Mutation analysis of 12 genes in Chinese families with congenital cataracts. Mol Vis. 2011;17:2197–2206.
42. Xia XY, Wu Q-Y, An L-M, et al. A novel P20R mutation in the alpha-B crystallin gene causes autosomal dominant congenital posterior polar cataracts in a Chinese family. BMC Ophthalmol. 2014;14:108. doi:10.1186/1471-2415-14-108
43. Yu Y, Xu J, Qiao Y, et al. A new heterozygous mutation in the stop codon of CRYAB (p.X176Y) is liable for congenital posterior pole cataract in a Chinese family. Ophthalmic Genet. 2021;42(2):139–143. doi:10.1080/13816810.2020.1855665
44. Sacconi S, Féasson L, Antoine JC, et al. A novel CRYAB mutation resulting in multisystemic disease. Neuromuscul Disord. 2012;22(1):66–72. doi:10.1016/j.nmd.2011.07.004
45. van der Smagt JJ, Vink A, Kirkels JH, et al. Congenital posterior pole cataract and adult onset dilating cardiomyopathy: expanding the phenotype of αB-crystallinopathies. Clin Genet. 2014;85(4):381–385. doi:10.1111/cge.12169
46. Fichna JP, Potulska-Chromik A, Miszta P, et al. A novel dominant D109A CRYAB mutation in a family with myofibrillar myopathy affects αB-crystallin structure. BBA Clin. 2017;7:1–7. doi:10.1016/j.bbacli.2016.11.004
47. Jiao X, Khan SY, Irum B, et al. Missense mutations in CRYAB are liable for recessive congenital cataracts. PLoS One. 2015;10(9):e0137973. doi:10.1371/journal.pone.0137973
48. Safieh LA, Khan AO, Alkuraya FS. Identification of a novel CRYAB mutation associated with autosomal recessive juvenile cataract in a Saudi family. Mol Vis. 2009;15:980–984.
49. Muranova LK, Strelkov SV, Gusev NB. Effect of cataract-associated mutations in the N-terminal domain of αB-crystallin (HspB5). Exp Eye Res. 2020;197:108091. doi:10.1016/j.exer.2020.108091
50. Rossen JL, Williams AL, Bohnsack BL. Zebrafish as a model for crystallin-associated congenital cataracts in humans. Front Cell Dev Biol. 2025;13:1552988. doi:10.3389/fcell.2025.1552988
51. Slingsby C, Clout NJ. Structure of the crystallins. Eye. 1999;13(Pt 3b):395–402. doi:10.1038/eye.1999.113
52. Gillespie RL, O’Sullivan J, Ashworth J, et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology. 2014;121(11):2124–37.e1–2. doi:10.1016/j.ophtha.2014.06.006
53. Zhang J, ZHANG Y, FANG F, et al. Congenital cataracts due to a novel 2‑bp deletion in CRYBA1/A3. Mol Med Rep. 2014;10(3):1614–1618. doi:10.3892/mmr.2014.2324
54. Liu HL, Zhang D-W, Hu F-Y, et al. Mutational spectrum in a Chinese cohort with congenital cataracts. Mol Genet Genomic Med. 2023;11(9):e2196. doi:10.1002/mgg3.2196
55. Qi YH, Jia H-Y, Huang S-Z, et al. [Autosomal dominant congenital nuclear cataract caused by a deletion mutation in the beta A1-crystallin gene]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2003;20(6):486–489. Chinese
56. Zhai Y, Li J, Yu W, et al. Targeted exome sequencing of congenital cataracts related genes: broadening the mutation spectrum and genotype-phenotype correlations in 27 chinese han families. Sci Rep. 2017;7(1):1219. doi:10.1038/s41598-017-01182-9
57. Patel A, Hayward JD, Tailor V, et al. The oculome panel test: next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology. 2019;126(6):888–907. doi:10.1016/j.ophtha.2018.12.050
58. Ni SH, Zhang JM, Zhao J. A novel missense mutation of CRYBA1 in a northern Chinese family with inherited coronary cataract with blue punctate opacities. Eur J Ophthalmol. 2022;32(1):193–199. doi:10.1177/11206721211008355
59. Bateman JB, Geyer DD, Flodman P, et al. A new betaA1-crystallin splice junction mutation in autosomal dominant cataract. Invest Ophthalmol Vis Sci. 2000;41(11):3278–3285.
60. Yang Z, Li Q, Ma Z, et al. A G→T splice site mutation of CRYBA1/A3 associated with autosomal dominant suture cataracts in a Chinese family. Mol Vis. 2011;17:2065–2071.
61. Yang Z, Su D, Li Q, et al. A novel T→G splice site mutation of CRYBA1/A3 associated with autosomal dominant nuclear cataracts in a Chinese family. Mol Vis. 2012;18:1283–1288.
62. Wu MH, Yu Y-H, Hao Q-L, et al. A novel splice site mutation of CRYBA3/A1 gene associated with congenital cataract in a Chinese family. Int J Ophthalmol. 2017;10(1):1–5. doi:10.18240/ijo.2017.01.01
63. Kannabiran C, Rogan PK, Olmos L, et al. Autosomal dominant zonular cataract with sutural opacities is associated with a splice mutation in the betaA3/A1-crystallin gene. Mol Vis. 1998;4:21.
64. Li J, Leng Y, Han S, et al. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet J Rare Dis. 2018;13(1):94. doi:10.1186/s13023-018-0828-0
65. Li D, Jing Q, Jiang Y. The identification and characterization of the p.G91 deletion in CRYBA1 in a Chinese family with congenital cataracts. BMC Med Genet. 2019;20(1):153. doi:10.1186/s12881-019-0882-z
66. Khan AO, Aldahmesh MA, Alkuraya FS. Phenotypes of recessive pediatric cataract in a cohort of children with identified homozygous gene mutations (An American Ophthalmological Society Thesis). Trans Am Ophthalmol Soc. 2015;113:T7.
67. Graw J, Jung M, Löster J, et al. Mutation in the betaA3/A1-crystallin encoding gene Cryba1 causes a dominant cataract in the mouse. Genomics. 1999;62(1):67–73. doi:10.1006/geno.1999.5974
68. Hegde S, Kesterson RA, Srivastava OP. CRYβA3/A1-crystallin knockout develops nuclear cataract and causes impaired lysosomal cargo clearance and calpain activation. PLoS One. 2016;11(2):e0149027. doi:10.1371/journal.pone.0149027
69. Ma Z, Yao W, Chan -C-C, et al. Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells. Biochim Biophys Acta. 2016;1862(6):1214–1227. doi:10.1016/j.bbadis.2016.02.003
70. Reddy MA, Bateman OA, Chakarova C, et al. Characterization of the G91del CRYBA1/3-crystallin protein: a cause of human inherited cataract. Human Mol Genet. 2004;13(9):945–953. doi:10.1093/hmg/ddh110
71. Joseph R, Robinson ML, Lambert L, et al. Lens-specific βA3/A1-conditional knockout mice: phenotypic characteristics and calpain activation causing protein degradation and insolubilization. PLoS One. 2023;18(3):e0281386. doi:10.1371/journal.pone.0281386
72. Andley UP, Hamilton PD, Ravi N, et al. A knock-in mouse model for the R120G mutation of αB-crystallin recapitulates human hereditary myopathy and cataracts. PLoS One. 2011;6(3):e17671. doi:10.1371/journal.pone.0017671
73. Gerasimovich ES, Strelkov SV, Gusev NB. Some properties of three αB-crystallin mutants carrying point substitutions in the C-terminal domain and associated with congenital diseases. Biochimie. 2017;142:168–178. doi:10.1016/j.biochi.2017.09.008
74. Bova MP, Yaron O, Huang Q, et al. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A. 1999;96(11):6137–6142. doi:10.1073/pnas.96.11.6137
75. Andley UP, Tycksen E, McGlasson-Naumann BN, et al. Probing the changes in gene expression due to α-crystallin mutations in mouse models of hereditary human cataract. PLoS One. 2018;13(1):e0190817. doi:10.1371/journal.pone.0190817
76. Frankfater C, Bozeman SL, Hsu -F-F, et al. Alpha-crystallin mutations alter lens metabolites in mouse models of human cataracts. PLoS One. 2020;15(8):e0238081. doi:10.1371/journal.pone.0238081
77. Salehi A, Bahrami Z, Shahsavani MB, et al. Structural characterization and functional analysis of human αB-crystallin with the p.R11G mutation: insights into cataractogenesis and cardiomyopathy. Int J Biol Macromol. 2025;307(Pt 3):141895. doi:10.1016/j.ijbiomac.2025.141895
78. Wang Z, Huang C, Sun Y, et al. Novel mutations associated with autosomal-dominant congenital cataract identified in Chinese families. Exp Ther Med. 2019;18(4):2701–2710. doi:10.3892/etm.2019.7865
79. Yu Y, Qiao Y, Ye Y, et al. Identification and characterization of six β-crystallin gene mutations associated with congenital cataract in Chinese families. Mol Genet Genomic Med. 2021;9(3):e1617. doi:10.1002/mgg3.1617
80. Caswell RC, Gunning AC, Owens MM, et al. Assessing the clinical utility of protein structural analysis in genomic variant classification: experiences from a diagnostic laboratory. Genome Med. 2022;14(1):77. doi:10.1186/s13073-022-01082-2
81. Zhang X, Liang C, Liu M, et al. A novel missense variant c.71G > T (p.Gly24Val) of the CRYBA4 gene contributes to autosomal-dominant congenital cataract in a Chinese family. Int Ophthalmol. 2023;43(1):43–50. doi:10.1007/s10792-022-02386-3
82. AlAbdi L, Maddirevula S, Shamseldin HE, et al. Diagnostic implications of pitfalls in causal variant identification based on 4577 molecularly characterized families. Nat Commun. 2023;14(1):5269. doi:10.1038/s41467-023-40909-3
83. Chen J, Wang Q, Cabrera PE, et al. Molecular genetic analysis of pakistani families with autosomal recessive congenital cataracts by homozygosity screening. Invest Ophthalmol Vis Sci. 2017;58(4):2207–2217. doi:10.1167/iovs.17-21469
84. Cohen D, Bar-Yosef U, Levy J, et al. Homozygous CRYBB1 deletion mutation underlies autosomal recessive congenital cataract. Invest Ophthalmol Vis Sci. 2007;48(5):2208–2213. doi:10.1167/iovs.06-1019
85. Meyer E, Rahman F, Owens J, et al. Initiation codon mutation in betaB1-crystallin (CRYBB1) associated with autosomal recessive nuclear pulverulent cataract. Mol Vis. 2009;15:1014–1019.
86. Lenfant C, Baz P, Degavre A, et al. Juvenile-onset diabetes and congenital cataract: “double-gene” mutations mimicking a syndromic diabetes presentation. Genes. 2017;8(11):309. doi:10.3390/genes8110309
87. Aksay S, Bildirici İ, Coşar CB, et al. Intrauterine cataract diagnosis and follow-up. Turk J Ophthalmol. 2020;50(4):245–247. doi:10.4274/tjo.galenos.2020.05014
88. Mackay DS, Boskovska OB, Knopf HLS, et al. A nonsense mutation in CRYBB1 associated with autosomal dominant cataract linked to human chromosome 22q. Am J Hum Genet. 2002;71(5):1216–1221. doi:10.1086/344212
89. Wang KJ, Wang S, Cao N-Q, et al. A novel mutation in CRYBB1 associated with congenital cataract-microcornea syndrome: the p.Ser129Arg mutation destabilizes the βB1/βA3-crystallin heteromer but not the βB1-crystallin homomer. Hum Mutat. 2011;32(3):E2050–60. doi:10.1002/humu.21436
90. Reis LM, Tyler RC, Muheisen S, et al. Whole exome sequencing in dominant cataract identifies a new causative factor, CRYBA2, and a variety of novel alleles in known genes. Hum Genet. 2013;132(7):761–770. doi:10.1007/s00439-013-1289-0
91. Li D, Wang S, Ye H, et al. Distribution of gene mutations in sporadic congenital cataract in a Han Chinese population. Mol Vis. 2016;22:589–598.
92. Sun Y, Man J, Wan Y, et al. Targeted next-generation sequencing as a comprehensive test for Mendelian diseases: a cohort diagnostic study. Sci Rep. 2018;8(1):11646. doi:10.1038/s41598-018-30151-z
93. Rechsteiner D, Issler L, Koller S, et al. Genetic Analysis in a Swiss Cohort of Bilateral Congenital Cataract. JAMA Ophthalmol. 2021;139(7):691–700. doi:10.1001/jamaophthalmol.2021.0385
94. Sergouniotis PI, Barton SJ, Waller S, et al. The role of small in-frame insertions/deletions in inherited eye disorders and how structural modelling can help estimate their pathogenicity. Orphanet J Rare Dis. 2016;11(1):125. doi:10.1186/s13023-016-0505-0
95. Billingsley G, Santhiya ST, Paterson AD, et al. CRYBA4, a novel human cataract gene, is also involved in microphthalmia. Am J Hum Genet. 2006;79(4):702–709. doi:10.1086/507712
96. Zhou G, Zhou N, Hu S, et al. A missense mutation in CRYBA4 associated with congenital cataract and microcornea. Mol Vis. 2010;16:1019–1024.
97. Kumar M, Agarwal T, Kaur P, et al. Molecular and structural analysis of genetic variations in congenital cataract. Mol Vis. 2013;19:2436–2450.
98. Sun W, Xiao X, Li S, et al. Exome sequencing of 18 Chinese families with congenital cataracts: a new sight of the NHS gene. PLoS One. 2014;9(6):e100455. doi:10.1371/journal.pone.0100455
99. Zin OA, Neves LM, Motta FL, et al. Novel mutation in CRYBB3 causing pediatric cataract and microphthalmia. Genes. 2021;12(7):1069. doi:10.3390/genes12071069
100. Wang KJ, Liao X-Y, Lin K, et al. A novel F30S mutation in γS-crystallin causes autosomal dominant congenital nuclear cataract by increasing susceptibility to stresses. Int J Biol Macromol. 2021;172:475–482. doi:10.1016/j.ijbiomac.2021.01.079
101. Sinha D, Wyatt MK, Sarra R, et al. A temperature-sensitive mutation of Crygs in the murine Opj cataract. J Biol Chem. 2001;276(12):9308–9315. doi:10.1074/jbc.M010583200
102. Lee S, Mahler B, Toward J, et al. A single destabilizing mutation (F9S) promotes concerted unfolding of an entire globular domain in gammaS-crystallin. J Mol Biol. 2010;399(2):320–330. doi:10.1016/j.jmb.2010.04.003
103. Mahler B, Doddapaneni K, Kleckner I, et al. Characterization of a transient unfolding intermediate in a core mutant of γS-crystallin. J Mol Biol. 2011;405(3):840–850. doi:10.1016/j.jmb.2010.11.005
104. Bateman A, Martin M-J, Orchard S. UniProt: the universal protein knowledgebase in 2025. Nucleic Acids Res. 2025;53(D1):D609–d617. doi:10.1093/nar/gkae1010
105. Dave A, Laurie K, Staffieri SE, et al. Mutations in the EPHA2 gene are a major contributor to inherited cataracts in South-Eastern Australia. PLoS One. 2013;8(8):e72518. doi:10.1371/journal.pone.0072518
106. Zhai Y, Zhu S, Li J, et al. A novel human congenital cataract mutation in EPHA2 kinase domain (p.G668D) alters receptor stability and function. Invest Ophthalmol Vis Sci. 2019;60(14):4717–4726. doi:10.1167/iovs.19-27370
107. Dave A, Martin S, Kumar R, et al. EPHA2 mutations contribute to congenital cataract through diverse mechanisms. Mol Vis. 2016;22:18–30.
108. Zhou Y, Bennett TM, Shiels A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation. 2019;109:16–27. doi:10.1016/j.diff.2019.07.003
109. Shiels A, Bennett TM, Knopf HLS, et al. CHMP4B, a novel gene for autosomal dominant cataracts linked to chromosome 20q. Am J Hum Genet. 2007;81(3):596–606. doi:10.1086/519980
110. Jackson D, Malka S, Harding P, et al. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am J Med Genet C Semin Med Genet. 2020;184(3):578–589. doi:10.1002/ajmg.c.31837
111. Wang X, Wang D, Wang Q, et al. Broadening the mutation spectrum in GJA8 and CHMP4B: novel missense variants and the associated phenotypes in six chinese han congenital cataracts families. Front Med Lausanne. 2021;8:713284. doi:10.3389/fmed.2021.713284
112. Berry V, Pontikos N, Moore A, et al. A novel missense mutation in HSF4 causes autosomal-dominant congenital lamellar cataract in a British family. Eye. 2018;32(4):806–812. doi:10.1038/eye.2017.268
113. Lv H, Huang C, Zhang J, et al. A novel HSF4 gene mutation causes autosomal-dominant cataracts in a Chinese family. G3. 2014;4(5):823–828. doi:10.1534/g3.113.009860
114. Rao Y, Dong S, Li Z, et al. A novel truncation mutation in CRYBB1 associated with autosomal dominant congenital cataract with nystagmus. Mol Vis. 2017;23:624–637.
115. Jing X, Zhu M, Lu X, et al. Cataract-causing Y204X mutation of crystallin protein CRYβB1 promotes its C-terminal degradation and higher-order oligomerization. J Biol Chem. 2023;299(8):104953. doi:10.1016/j.jbc.2023.104953
116. Graw J, Neuha¨user-Klaus A, Klopp N, et al. Genetic and allelic heterogeneity of cryg mutations in eight distinct forms of dominant cataract in the mouse. Invest Ophthalmol Vis Sci. 2004;45(4):1202–1213. doi:10.1167/iovs.03-0811
117. Pei R, Liang PF, Ye W, et al. A novel mutation of LIM2 causes autosomal dominant membranous cataract in a Chinese family. Int J Ophthalmol. 2020;13(10):1512–1520. doi:10.18240/ijo.2020.10.02
118. Pras E, Levy-Nissenbaum E, Bakhan T, et al. A missense mutation in the LIM2 gene is associated with autosomal recessive presenile cataract in an inbred Iraqi Jewish family. Am J Hum Genet. 2002;70(5):1363–1367. doi:10.1086/340318
119. Ponnam SP, Ramesha K, Tejwani S, et al. A missense mutation in LIM2 causes autosomal recessive congenital cataract. Mol Vis. 2008;14:1204–1208.
120. Irum B, Khan SY, Ali M, et al. Mutation in LIM2 is responsible for autosomal recessive congenital cataracts. PLoS One. 2016;11(11):e0162620. doi:10.1371/journal.pone.0162620
121. Long X, Huang Y, Tan H, et al. Identification of a novel MIP frameshift mutation associated with congenital cataract in a Chinese family by whole-exome sequencing and functional analysis. Eye. 2018;32(8):1359–1364. doi:10.1038/s41433-018-0084-5
122. Wang H, Zhang T, Wu D, et al. A novel beaded filament structural protein 1 (BFSP1) gene mutation associated with autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2013;19:2590–2595.
123. Ramachandran RD, Perumalsamy V, Hejtmancik JF. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum Genet. 2007;121(3–4):475–482. doi:10.1007/s00439-006-0319-6
124. Wang H, Ouyang G, Zhu Y. D348N mutation of BFSP1 gene in congenital cataract: it does matter. Cell Biochem Biophys. 2023;81(4):757–763. doi:10.1007/s12013-023-01169-6
125. Cui X, Gao L, Jin Y, et al. The E233del mutation in BFSP2 causes a progressive autosomal dominant congenital cataract in a Chinese family. Mol Vis. 2007;13:2023–2029.
126. Jakobs PM, Hess JF, FitzGerald PG, et al. Autosomal-dominant congenital cataract associated with a deletion mutation in the human beaded filament protein gene BFSP2. Am J Hum Genet. 2000;66(4):1432–1436. doi:10.1086/302872
127. Ma X, Li -F-F, Wang S-Z, et al. A new mutation in BFSP2 (G1091A) causes autosomal dominant congenital lamellar cataracts. Mol Vis. 2008;14:1906–1911.
128. Irum B, Kabir F, Shoshany N, et al. A genomic deletion encompassing CRYBB2-CRYBB2P1 is responsible for autosomal recessive congenital cataracts. Hum Genome Var. 2022;9(1):31. doi:10.1038/s41439-022-00208-7
129. Garnai SJ, Huyghe JR, Reed DM, et al. Congenital cataracts: de novo gene conversion event in CRYBB2. Mol Vis. 2014;20:1579–1593.
130. Vanita V, Sarhadi V, Reis A, et al. A unique form of autosomal dominant cataract explained by gene conversion between beta-crystallin B2 and its pseudogene. J Med Genet. 2001;38(6):392–396. doi:10.1136/jmg.38.6.392
131. Hansen L, Mikkelsen A, Nu¨rnberg P, et al. Comprehensive mutational screening in a cohort of Danish families with hereditary congenital cataract. Invest Ophthalmol Vis Sci. 2009;50(7):3291–3303. doi:10.1167/iovs.08-3149
132. Fernández-Alcalde C, Nieves-Moreno M, Noval S, et al. Molecular and genetic mechanism of non-syndromic congenital cataracts. mutation screening in Spanish families. Genes. 2021;12(4):580. doi:10.3390/genes12040580
133. Riazuddin SA, Yasmeen A, Yao W, et al. Mutations in betaB3-crystallin associated with autosomal recessive cataract in two Pakistani families. Invest Ophthalmol Vis Sci. 2005;46(6):2100–2106. doi:10.1167/iovs.04-1481
134. Astiazarán MC, García‐Montaño LA, Sánchez‐Moreno F, et al. Next generation sequencing-based molecular diagnosis in familial congenital cataract expands the mutational spectrum in known congenital cataract genes. Am J Med Genet A. 2018;176(12):2637–2645. doi:10.1002/ajmg.a.40524
135. Jiao X, Viswanathan M, Bobrova NF, et al. Molecular genetic analysis of Ukrainian families with congenital cataracts. Children. 2022;10(1). doi:10.3390/children10010051.
136. Xu W, Xu J, Shi C, et al. A novel cataract-causing mutation Ile82Met of γA crystallin trends to aggregate with unfolding intermediate. Int J Biol Macromol. 2022;211:357–367. doi:10.1016/j.ijbiomac.2022.04.205
137. Cheng MH, Tam CN, Choy KW, et al. A γA-crystallin mouse mutant secc with small eye, cataract and closed eyelid. PLoS One. 2016;11(8):e0160691. doi:10.1371/journal.pone.0160691
138. Klopp N, Favor J, Löster J, et al. Three murine cataract mutants (Cat2) are defective in different gamma-crystallin genes. Genomics. 1998;52(2):152–158. doi:10.1006/geno.1998.5417
139. AlFadhli S, Abdelmoaty S, Al-Hajeri A, et al. Novel crystallin gamma B mutations in a Kuwaiti family with autosomal dominant congenital cataracts reveal genetic and clinical heterogeneity. Mol Vis. 2012;18:2931–2936.
140. Mehra S, Kapur S, Vasavada AR. Polymorphisms of the gamma crystallin A and B genes among Indian patients with pediatric cataract. J Postgrad Med. 2011;57(3):201–205. doi:10.4103/0022-3859.85205
141. Sandilands A, Prescott AR, Wegener A, et al. Knockout of the intermediate filament protein CP49 destabilises the lens fibre cell cytoskeleton and decreases lens optical quality, but does not induce cataract. Exp Eye Res. 2003;76(3):385–391. doi:10.1016/S0014-4835(02)00330-5
142. Müller M, Bhattacharya SS, Moore T, et al. Dominant cataract formation in association with a vimentin assembly disrupting mutation. Hum Mol Genet. 2009;18(6):1052–1057. doi:10.1093/hmg/ddn440
143. Aldahmesh MA, Khan AO, Mohamed JY, et al. Genomic analysis of pediatric cataract in Saudi Arabia reveals novel candidate disease genes. Genet Med. 2012;14(12):955–962. doi:10.1038/gim.2012.86
144. Lachke SA, Ho JWK, Kryukov GV, et al. iSyTE: integrated Sy stems T ool for E ye Gene Discovery. Invest Ophthalmol Vis Sci. 2012;53(3):1617–1627. doi:10.1167/iovs.11-8839
145. Knöpfel EB, Vilches C, Camargo SMR, et al. Dysfunctional LAT2 amino acid transporter is associated with cataract in mouse and humans. Front Physiol. 2019;10:688. doi:10.3389/fphys.2019.00688
146. Patel N, Anand D, Monies D, et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum Genet. 2017;136(2):205–225. doi:10.1007/s00439-016-1747-6
147. Kloss BA, Reis LM, Brémond-Gignac D, et al. Analysis of FOXD3 sequence variation in human ocular disease. Mol Vis. 2012;18:1740–1749.
148. Wu Z, Meng D, Fang C, et al. PITX3 mutations associated with autosomal dominant congenital cataract in the Chinese population. Mol Med Rep. 2019;19(4):3123–3131. doi:10.3892/mmr.2019.9989
149. Tang Y, Xu J, Lu Y, et al. Three novel mutations of microphthalmos identified in two Chinese families. Phenomics. 2022;2(4):254–260. doi:10.1007/s43657-022-00053-2
150. Khan K, Rudkin A, Parry D, et al. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am J Hum Genet. 2011;89(3):464–473. doi:10.1016/j.ajhg.2011.08.005
151. Lin ZB, Li J, Ye L, et al. Novel SOX2 mutation in autosomal dominant cataract-microcornea syndrome. BMC Ophthalmol. 2022;22(1):70. doi:10.1186/s12886-022-02291-4
152. Cheong SS, Hentschel L, Davidson A, et al. Mutations in CPAMD8 cause a unique form of autosomal-recessive anterior segment dysgenesis. Am J Hum Genet. 2016;99(6):1338–1352. doi:10.1016/j.ajhg.2016.09.022
153. Siggs OM, Souzeau E, Taranath DA, et al. Biallelic CPAMD8 variants are a frequent cause of childhood and juvenile open-angle glaucoma. Ophthalmology. 2020;127(6):758–766. doi:10.1016/j.ophtha.2019.12.024
154. Sharma R, Grover A. Myocilin-associated glaucoma: a historical perspective and recent research progress. Mol Vis. 2021;27:480–493.
155. Skarie JM, Link BA. The primary open-angle glaucoma gene WDR36 functions in ribosomal RNA processing and interacts with the p53 stress-response pathway. Hum Mol Genet. 2008;17(16):2474–2485. doi:10.1093/hmg/ddn147
156. Hughes AE, Bradley D, Campbell M, et al. Mutation altering the miR-184 seed region causes familial keratoconus with cataract. Am J Hum Genet. 2011;89(5):628–633. doi:10.1016/j.ajhg.2011.09.014
157. Mei S, Wu Y, Wang Y, et al. Disruption of PIKFYVE causes congenital cataract in human and zebrafish. Elife. 2022;11:e71256.
158. Chen P, Hao X, Li W, et al. Mutations in the TMCO3 gene are associated with cornea guttata and anterior polar cataract. Sci Rep. 2016;6:31021. doi:10.1038/srep31021
159. Tan MH, Mackay DS, Cowing J, et al. Leber congenital amaurosis associated with AIPL1: challenges in ascribing disease causation, clinical findings, and implications for gene therapy. PLoS One. 2012;7(3):e32330. doi:10.1371/journal.pone.0032330
160. Gradstein L, Zolotushko J, Sergeev YV, et al. Novel GUCY2D mutation causes phenotypic variability of Leber congenital amaurosis in a large kindred. BMC Med Genet. 2016;17(1):52. doi:10.1186/s12881-016-0314-2
161. Ahmad A, Daud S, Kakar N, et al. Identification of a novel LCA5 mutation in a Pakistani family with Leber congenital amaurosis and cataracts. Mol Vis. 2011;17:1940–1945.
162. Cai XB, Wu K-C, Zhang X, et al. Whole-exome sequencing identified ARL2 as a novel candidate gene for MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma) syndrome. Clin Genet. 2019;96(1):61–71. doi:10.1111/cge.13541
163. Boon CJ, Klevering BJ, Leroy BP, et al. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009;28(3):187–205. doi:10.1016/j.preteyeres.2009.04.002
164. El-Haig WM, Jakobsson C, Favez T, et al. Novel ADAM9 homozygous mutation in a consanguineous Egyptian family with severe cone-rod dystrophy and cataract. Br J Ophthalmol. 2014;98(12):1718–1723. doi:10.1136/bjophthalmol-2014-305231
165. Lim JM, Lee J-H, Wee W-R, et al. Downregulated expression of ADAM9 in anterior polar cataracts. J Cataract Refract Surg. 2002;28(4):697–702. doi:10.1016/S0886-3350(01)01236-6
166. Parry DA, Toomes C, Bida L, et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am J Hum Genet. 2009;84(5):683–691. doi:10.1016/j.ajhg.2009.04.005
167. Khan K, Logan CV, McKibbin M, et al. Next generation sequencing identifies mutations in Atonal homolog 7 (ATOH7) in families with global eye developmental defects. Hum Mol Genet. 2012;21(4):776–783. doi:10.1093/hmg/ddr509
168. Khan AO, Bergmann C, Neuhaus C, et al. A distinct vitreo-retinal dystrophy with early-onset cataract from recessive KCNJ13 mutations. Ophthalmic Genet. 2015;36(1):79–84. doi:10.3109/13816810.2014.985846
169. Tang S, Di G, Hu S, et al. AQP5 regulates vimentin expression via miR-124-3p.1 to protect lens transparency. Exp Eye Res. 2021;205:108485. doi:10.1016/j.exer.2021.108485
170. Zhong J, Shi J, Zhang X, et al. A novel splicing variant of VCAN identified in a Chinese family initially diagnosed with familial exudative vitreoretinopathy. Mol Genet Genomic Med. 2023;11(2):e2083. doi:10.1002/mgg3.2083
171. Elhusseiny AM, Jabroun M, Rajabi F, et al. A novel variant in the TSPAN12 gene-presenting as unilateral myopia, pediatric cataract, and heterochromia in a patient with familial exudative vitreoretinopathy. Eur J Ophthalmol. 2022;32(6):Np6–np9. doi:10.1177/11206721211027415
172. Poulter JA, Ali M, Gilmour DF, et al. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86(2):248–253. doi:10.1016/j.ajhg.2010.01.012
173. Conte I, Hadfield KD, Barbato S, et al. MiR-204 is responsible for inherited retinal dystrophy associated with ocular coloboma. Proc Natl Acad Sci U S A. 2015;112(25):E3236–45. doi:10.1073/pnas.1401464112
174. Wang L, He F, Bu J, et al. ABCB6 mutations cause ocular coloboma. Am J Hum Genet. 2012;90(1):40–48. doi:10.1016/j.ajhg.2011.11.026
175. Sun H, Ma Z, Li Y, et al. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans. J Med Genet. 2005;42(9):706–710. doi:10.1136/jmg.2004.028274
176. Vanita V, Singh JR, Singh D, et al. Novel mutation in the gamma-S crystallin gene causing autosomal dominant cataract. Mol Vis. 2009;15:476–481.
177. Yang Z, Li Q, Zhu S, et al. A G57W mutation of CRYGS associated with autosomal dominant pulverulent cataracts in a Chinese family. Ophthalmic Genet. 2015;36(3):281–283. doi:10.3109/13816810.2013.865761
178. Zhang T, Yan L, Leng Y, et al. A novel missense mutation of CRYGS underlies congenital cataract in a Chinese family. Gene. 2018;675:9–14. doi:10.1016/j.gene.2018.06.100
179. Gao M, Huang Y, Wang L, et al. HSF4 regulates lens fiber cell differentiation by activating p53 and its downstream regulators. Cell Death Dis. 2017;8(10):e3082. doi:10.1038/cddis.2017.478
180. Hassan AY, Yousaf S, Levin MR, et al. Novel homozygous missense variant in GJA3 connexin domain causing congenital nuclear and cortical cataracts. Int J Mol Sci. 2021;23(1):240. doi:10.3390/ijms23010240
181. Beyer EC, Ebihara L, Berthoud VM. Connexin mutants and cataracts. Front Pharmacol. 2013;4:43. doi:10.3389/fphar.2013.00043
182. Ding N, Chen Z, Song X, et al. Novel mutation of GJA8 in autosomal dominant congenital cataracts. Ann Transl Med. 2020;8(18):1127. doi:10.21037/atm-20-4663
183. Alizadeh A, Clark JI, Seeberger T, et al. Targeted genomic deletion of the lens-specific intermediate filament protein CP49. Invest Ophthalmol Vis Sci. 2002;43(12):3722–3727.
184. Alizadeh A, Clark J, Seeberger T, et al. Targeted deletion of the lens fiber cell-specific intermediate filament protein filensin. Invest Ophthalmol Vis Sci. 2003;44(12):5252–5258. doi:10.1167/iovs.03-0224
185. Evers C, Paramasivam N, Hinderhofer K, et al. SIPA1L3 identified by linkage analysis and whole-exome sequencing as a novel gene for autosomal recessive congenital cataract. Eur J Hum Genet. 2015;23(12):1627–1633. doi:10.1038/ejhg.2015.46
186. Yang D, Zhou H, Lin J, et al. Case report: a novel missense variant in the sipa1l3 gene associated with cataracts in a Chinese family. Front Genet. 2021;12:715599. doi:10.3389/fgene.2021.715599
187. Ansar M, Chung H-L, Taylor RL, et al. Bi-allelic loss-of-function variants in DNMBP cause infantile cataracts. Am J Hum Genet. 2018;103(4):568–578. doi:10.1016/j.ajhg.2018.09.004
188. Iqbal H, Khan SY, Zhou L, et al. Mutations in FYCO1 identified in families with congenital cataracts. Mol Vis. 2020;26:334–344.
189. Khaliq S, Hameed A, Ismail M, et al. A novel locus for autosomal dominant nuclear cataract mapped to chromosome 2p12 in a Pakistani family. Invest Ophthalmol Vis Sci. 2002;43(7):2083–2087.
190. Sun L, Song F, Liu H, et al. The novel mutation P36R in LRP5L contributes to congenital membranous cataract via inhibition of laminin γ1 and c-MAF. Graefes Arch Clin Exp Ophthalmol. 2020;258(12):2737–2751. doi:10.1007/s00417-020-04846-x
191. Sun M, Chen C, Hou S, et al. A novel mutation of PANK4 causes autosomal dominant congenital posterior cataract. Hum Mutat. 2019;40(4):380–391. doi:10.1002/humu.23696
192. Chen JH, Huang C, Zhang B, et al. Mutations of RagA GTPase in mTORC1 pathway are associated with autosomal dominant cataracts. PLoS Genet. 2016;12(6):e1006090. doi:10.1371/journal.pgen.1006090
193. Yu YJ, Qiu F, Zhang XA. Studies of congenital cataract-related TSR1 mutation and its expression in the lens. Yi Chuan. 2020;42(2):161–171. doi:10.16288/j.yczz.19-166
194. Khan AO, Aldahmesh MA, Alsharif H, et al. Recessive mutations in LEPREL1 underlie a recognizable lens subluxation phenotype. Ophthalmic Genet. 2015;36(1):58–63. doi:10.3109/13816810.2014.985847
195. Guo H, Tong P, Peng Y, et al. Homozygous loss-of-function mutation of the LEPREL1 gene causes severe non-syndromic high myopia with early-onset cataract. Clin Genet. 2014;86(6):575–579. doi:10.1111/cge.12309
196. Yuan L, Yi J, Lin Q, et al. Identification of a PRX variant in a Chinese family with congenital cataract by exome sequencing. Qjm. 2016;109(11):731–735. doi:10.1093/qjmed/hcw058
197. Maddala R, Skiba NP, Lalane R, et al. Periaxin is required for hexagonal geometry and membrane organization of mature lens fibers. Dev Biol. 2011;357(1):179–190. doi:10.1016/j.ydbio.2011.06.036
198. Kloeckener-Gruissem B, Vandekerckhove K, Nürnberg G, et al. Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet. 2008;82(3):772–779. doi:10.1016/j.ajhg.2007.12.013
199. Tan YQ, Tu C, Meng L, et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med. 2019;21(5):1209–1217. doi:10.1038/gim.2017.130
200. Lachke SA, Alkuraya FS, Kneeland SC, et al. Mutations in the RNA granule component TDRD7 cause cataract and glaucoma. Science. 2011;331(6024):1571–1576. doi:10.1126/science.1195970
201. Barnum CE, Al Saai S, Patel SD, et al. The Tudor-domain protein TDRD7, mutated in congenital cataract, controls the heat shock protein HSPB1 (HSP27) and lens fiber cell morphology. Hum Mol Genet. 2020;29(12):2076–2097. doi:10.1093/hmg/ddaa096
202. Jones JL, McComish BJ, Staffieri SE, et al. Pathogenic genetic variants identified in Australian families with paediatric cataract. BMJ Open Ophthalmol. 2022;7(1):e001064. doi:10.1136/bmjophth-2022-001064
203. Vona B, Maroofian R, Bellacchio E, et al. Expanding the clinical phenotype of IARS2-related mitochondrial disease. BMC Med Genet. 2018;19(1):196. doi:10.1186/s12881-018-0709-3
204. Jones JL, Corbett MA, Yeaman E, et al. A 127 kb truncating deletion of PGRMC1 is a novel cause of X-linked isolated paediatric cataract. Eur J Hum Genet. 2021;29(8):1206–1215. doi:10.1038/s41431-021-00889-8
205. Bennett TM, Mackay DS, Siegfried CJ, et al. Mutation of the melastatin-related cation channel, TRPM3, underlies inherited cataract and glaucoma. PLoS One. 2014;9(8):e104000. doi:10.1371/journal.pone.0104000
206. Zhang J, Li Y, Fan Y, et al. Compound heterozygous mutations in SMO associated with anterior segment dysgenesis and morning glory syndrome. Gene. 2019;713:143973. doi:10.1016/j.gene.2019.143973
207. Tan Y, Tian H, Mai J, et al. A case of congenital cataracts with hypotrichosis caused by compound heterozygous variants in the LSS gene. Mol Genet Genomic Med. 2024;12(1):e2320. doi:10.1002/mgg3.2320
208. Zhao SH, Yap KL, Allegretti V, et al. A case of non-syndromic congenital cataracts caused by a novel MAF variant in the C-Terminal DNA-binding domain-case report and literature review. Genes. 2024;15(6):686. doi:10.3390/genes15060686
209. Dafsari HS, Kocaturk NM, Daimagüler H-S, et al. Bi-allelic mutations in uncoordinated mutant number-45 myosin chaperone B are a cause for congenital myopathy. Acta Neuropatholog Commun. 2019;7(1):211. doi:10.1186/s40478-019-0869-1
210. Hansen L, Comyn S, Mang Y, et al. The myosin chaperone UNC45B is involved in lens development and autosomal dominant juvenile cataract. Eur J Hum Genet. 2014;22(11):1290–1297. doi:10.1038/ejhg.2014.21
211. Chen P, Dai Y, Wu X, et al. Mutations in the ABCA3 gene are associated with cataract-microcornea syndrome. Invest Ophthalmol Vis Sci. 2014;55(12):8031–8043. doi:10.1167/iovs.14-14098
212. Berry V, Pontikos N, Ionides A, et al. Pathogenic variants in the CYP21A2 gene cause isolated autosomal dominant congenital posterior polar cataracts. Ophthalmic Genet. 2022;43(2):218–223. doi:10.1080/13816810.2021.1998556
213. Nieves-Moreno M, Noval S, Peralta J, et al. Expanding the phenotypic spectrum of PAX6 mutations: from congenital cataracts to nystagmus. Genes. 2021;12(5):707. doi:10.3390/genes12050707
214. Lima Cunha D, Arno G, Corton M, et al. The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes. 2019;10(12):1050. doi:10.3390/genes10121050
215. Landsend ECS, Lagali N, Utheim TP. Congenital aniridia – a comprehensive review of clinical features and therapeutic approaches. Survey Ophthalmol. 2021;66(6):1031–1050. doi:10.1016/j.survophthal.2021.02.011
216. Zhang X, Zhang Q, Tong Y, et al. Large novel deletions detected in Chinese families with aniridia: correlation between genotype and phenotype. Mol Vis. 2011;17:548–557.
217. Plaisancié J, Ragge NK, Dollfus H, et al. FOXE3 mutations: genotype-phenotype correlations. Clin Genet. 2018;93(4):837–845. doi:10.1111/cge.13177
218. Young TL, Whisenhunt KN, LaMartina SM, et al. Sonic hedgehog intron variant associated with an unusual pediatric cortical cataract. Invest Ophthalmol Vis Sci. 2022;63(6):25. doi:10.1167/iovs.63.6.25
219. Schimmenti LA, de la Cruz J, Lewis RA, et al. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A. 2003;116a(3):215–221. doi:10.1002/ajmg.a.10884
220. Azuma N, Tadokoro K, Yamada M, et al. Sonic hedgehog determines early retinal development and adjusts eyeball Architecture. Int J Mol Sci. 2025;26(2):496. doi:10.3390/ijms26020496
221. Elnahry AG, Tripathy K. Gyrate atrophy of the choroid and retina. STATPearls. 2023.
222. Khan AO. Microcornea with myopic chorioretinal atrophy, telecanthus and posteriorly-rotated ears: a distinct clinical syndrome. Ophthalmic Genet. 2012;33(4):196–199. doi:10.3109/13816810.2012.681097
223. Chandra A, Arno G, Williamson K, et al. Expansion of ocular phenotypic features associated with mutations in ADAMTS18. JAMA Ophthalmol. 2014;132(8):996–1001. doi:10.1001/jamaophthalmol.2014.940
224. Knight LSW, Mullany S, Taranath DA, et al. The phenotypic spectrum of ADAMTSL4-associated ectopia lentis: additional cases, complications, and review of literature. Mol Vis. 2022;28:257–268.
225. Urano F. Wolfram syndrome: diagnosis, management, and treatment. Curr Diab Rep. 2016;16(1):6. doi:10.1007/s11892-015-0702-6
226. Reynier P, Amati-Bonneau P, Verny C, et al. OPA3 gene mutations responsible for autosomal dominant optic atrophy and cataract. J Med Genet. 2004;41(9):e110. doi:10.1136/jmg.2003.016576
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.