Back to Journals » International Journal of Nanomedicine » Volume 18
Cancer Cell Membrane-Coated Gambogic Acid Nanoparticles for Effective Anticancer Vaccination by Activating Dendritic Cells
Authors Huang F ![]() , Zhang Q, Xiao J, Zhang X, Han X
, Zhang Q, Xiao J, Zhang X, Han X ![]() , Shi X, Hu J, Li L, Qian X
, Shi X, Hu J, Li L, Qian X ![]()
Received 14 February 2023
Accepted for publication 24 April 2023
Published 2 May 2023 Volume 2023:18 Pages 2261—2273
DOI https://doi.org/10.2147/IJN.S408521
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Israel Rubinstein
Fengli Huang,1,* Qun Zhang,2,* Jie Xiao,3,* Xin Zhang,1 Xingzhi Han,3 Xiao Shi,3 Jing Hu,2 Li Li,2 Xiaoping Qian1,2
1Department of Oncology, Nanjing Drum Tower Hospital Clinical College of Nanjing University of Chinese Medicine, Nanjing, People’s Republic of China; 2Department of Oncology, Nanjing Drum Tower Hospital, Nanjing, People’s Republic of China; 3Department of Oncology, Nanjing Drum Tower Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoping Qian, Department of Oncology, Nanjing Drum Tower Hospital Clinical College of Nanjing University of Chinese Medicine, 321 Zhongshan Road, Nanjing, Jiangsu, 210008, People’s Republic of China, Tel +86-13951743162, Fax +86-25-68182342, Email [email protected]
Purpose: Recent studies have shown that traditional Chinese medicine (TCM), such as gambogic acid (GA), is involved in the regulation of tumor immune microenvironment and can be combined with other anti-tumor treatment strategies. Here, we used GA as an adjuvant to construct a nano-vaccine to improve the anti-tumor immune response of colorectal cancer (CRC).
Materials and Methods: We used a previously reported two-step emulsification method to obtain poly (lactic-co-glycolic acid) /GA nanoparticles (PLGA/GA NPs), and then CT26 colon cancer cell membrane (CCM) was used to obtain CCM-PLGA/GA NPs. This novel nano-vaccine, CCM-PLGA/GA NPs, was co-synthesized with GA as an adjuvant and neoantigen provided by CT26 CCM. We further confirmed the stability, tumor targeting, and cytotoxicity of CCM-PLGA/GA NPs. The regulatory effect on the tumor immune microenvironment, the anti-tumor efficacy, and the combined anti-tumor efficacy with anti-PD-1 monoclonal Antibodies (mAbs) of this novel nano-vaccine was also detected in vivo.
Results: We successfully constructed the CCM-PLGA/GA NPs. In vitro and in vivo tests showed low biological toxicity, as well as the high tumor-targeting ability of the CCM-PLGA/GA NPs. Besides, we revealed a remarkable effect of CCM-PLGA/GA NPs to activate the maturation of dendritic cells (DCs) and the formation of a positive anti-tumor immune microenvironment.
Conclusion: This novel nano-vaccine constructed with GA as the adjuvant and CCM providing the tumor antigen can not only directly kill tumors by enhancing the ability of GA to target tumors, but also indirectly kill tumors by regulating tumor immune microenvironment, providing a new strategy for immunotherapy of CRC.
Keywords: cancer immunotherapy, gambogic acid, nano-vaccine, tumor immune microenvironment, colorectal cancer
Introduction
Colorectal cancer (CRC) is one of the most common malignancies with high incidence and mortality. Although the prognosis of CRC patients has been improved, the 5-year survival rate of patients with advanced CRC is only approximately 10%.1 Consequently, exploring new strategies for the treatment of advanced CRC remains significant and challenging. In recent years, immunotherapy has brought revolutionary changes to cancer therapy, including CRC.2–4 The key to the success of immunotherapy is to rely on remodeling the positive tumor immune microenvironment to enhance the activation and killing of CD8-positive T cells.5
According to recent studies, vascular endothelial growth factors (VEGF) signal is crucial to the tumor immune cycle and involved in tumor immune escape.6,7 VEGF signal participates in regulating the immune function of the tumor microenvironment through a variety of mechanisms,8,9 such as boosting the expression of IL-10, enhancing the differentiation towards M2-like macrophages, and inhibiting DC maturation, thereby reducing the activation of CD8+ T cells, which ultimately leads to the formation of tumor immunosuppressive microenvironment.10
Recent studies have revealed that traditional Chinese medicine (TCM) can also synergize the efficacy of immunotherapy by modulating the immune microenvironment of tumors.11 Gambogic acid (GA) is a TCM monomer extracted from the Garcinia hanburyi tree. It has been found that GA has strong anticancer activity against a variety of solid tumors including CRC.12,13 Our previous studies revealed that GA exerts antitumor effects by counteracting VEGF-mediated angiogenesis,14 suggesting its potential to modulate the tumor immune microenvironment.15 Nevertheless, the poor water solubility and toxic side effects of GA restricted its clinical application.12
Nowadays, cell membrane-based biomimetic nanosystems for drug delivery have obtained increasing attention. Cell membranes for biomimetic nanosystems are mainly derived from red blood cells, white blood cells, cancer cells, stem cells, and hybrid cells. The cell membranes-coated biomimetic nanoparticles could significantly prolong the circulation time and increase the stability of nanoparticles in vivo.16 Our previous study successfully constructed a GA-loaded biomimetic nanoparticle using the red blood cell membrane and demonstrated its anti-tumor effects in vitro and in vivo.17 Unlike other cell membranes, the cancer cell membrane could produce immune escape, and provide tumor antigen and tumor homotypic targeting capacities.18–20 Therefore, cancer cell membrane (CCM) is the preferred cell membrane for encapsulating nanoparticles and in combination with the adjuvant to the preparation of nano-vaccine. In this study, we used CT26 colon CCM to construct biomimetic nanoparticles loaded with GA and explored its anti-tumor efficacy and regulatory functions on the immune microenvironment of CRC (Figure 1).
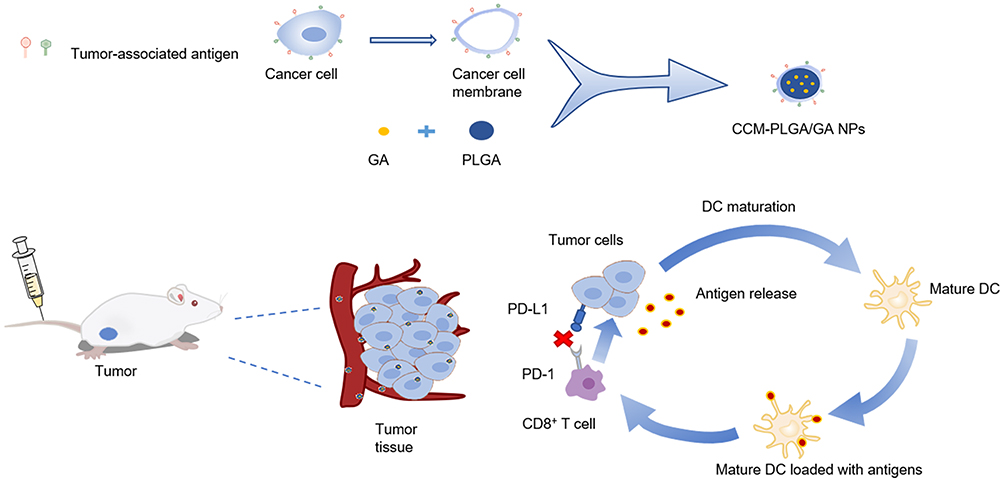 |
Figure 1 Schematic diagram of the preparation of CCM-PLGA/GA NPs and improving tumor immune microenvironment. |
Materials and Methods
Materials
GA (purity ≥99%) was purchased from Aladdin Industrial Corporation (Shanghai, China). Poly (lactic-co-glycolic acid) (PLGA) and polyvinyl alcohol (PVA) were purchased from Macklin (Shanghai, China). Roswell Park Memorial Institute (RPMI) 1640 medium, trypsin, Fetal bovine serum (FBS) and collagenase IV were acquired from Gibco. The antibodies include anti-CD11c-FITC, anti-CD86-PE, anti-CD80-APC, anti-CD279-PE, and anti-CD274-PC7 were purchased from Biolegend (USA).
Cells and Animals
Mouse colon cancer CT26 cells were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). B16-F10 melanoma cells, Mouse Forestomach Carcinoma cells, and Raw264.7 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured at 37°C with a 5% CO2 cell culture incubator. BALB/c male mice aged 5–6 weeks were purchased from Gempharmatech Co. Ltd. (Nanjing, China). All mice were kept in the specific pathogen-free Laboratory Animal Center of Nanjing Drum Tower Hospital (Nanjing, China). All animal procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Nanjing Drum Tower Hospital and approved by the Ethics Committee of Nanjing Drum Tower Hospital.
Preparation of CCM-PLGA/GA NPs
To obtain PLGA/GA NPs, a previously reported two-step emulsification method was used.17 After the sediment was resuspended in phosphate buffered saline (PBS) and centrifuged at 1500 rpm to remove unloaded GA, the PLGA/GA NPs were prepared.
The method for extracting the cancer cell membrane is as shown in the prior literature.21 In short, CT26 tumor cells were cultured with RPMI 1640 containing 10% FBS and then digested with trypsin. The entire cell solution was lysed by freeze-thaw cycling six times. The lysate was then centrifuged at 700g to remove the precipitation. After sonicating for 2 minutes, centrifuge the harvested supernatant at 14000g for 30 minutes, and collect the precipitate, which is the cancer cell membrane. The resulting vesicles were then repeatedly extracted via 400 nm and 200 nm polycarbonate porous membranes by a small extruder. (Avanti Polar Lipids, Alabaster, AL, USA). CT26 CCM was obtained after extrusion for 21 cycles.
The CT26 CCM solution obtained in the previous step was mixed with PLGA/GA NPs aqueous suspension. Then, the mixture was successively extruded through an extruder 11 times to acquire CCM-PLGA/GA NPs. The resulting NPs were re-dispersed in PBS to further characterize.
Characterization of CCM-PLGA/GA NPs
Then the particle size and polydispersity of the CCM-PLGA/GA NPs were measured using a DLS analyzer (Zetasizer, Malvern, UK). To evaluate the stability of NPs, the diameter of NPs determined by DLS for 14 days. The morphology of CCM-PLGA/GA NPs was characterized using a transmission electron microscopy (TEM). For confocal imaging, PLGA NPs loaded with fluorescent dye DiI (PLGA/DiI) were prepared by O/W ultrasonic emulsification method. Fluorescent nanoparticles were prepared in the above manner after staining CT26 cancer cell membranes with the fluorescent dye DiO, and the images were visualized by confocal laser scanning microscopy. (CLSM, FV1000, Olympus, Japan). The chemical interaction between GA and PLGA was evaluated by Fourier-transform IR (FTIR) spectra (Thermo Fisher Scientific, USA).
In vitro Cytotoxicity Assay
The CCK8 assay was used to measure the cytotoxicity of CT26 cells. The cells were cultured in a 96-well plate and incubated overnight. After cells had been exposed to gradient concentrations of CCM-PLGA/GA NPs and free GA for 24 hours, 10 µL of CCK8 was added to each well and incubated for 40 minutes. The absorbance was quantified at 450 nm using a spectrophotometer.
Characterization of Cancer Cell Membrane Proteins
A sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) method was used to characterize the cancer cell membrane protein of CCM-PLGA/GA NPs. The membrane proteins were extracted from the CT26 cells or CT26 CCM-PLGA/GA NPs by RIPA lysis buffer and further measured with the BCA assay kit (Beyotime, China). All samples were added into the wells with SDS-PAGE gel. Protein was stained by Coomassie Blue and imaged after water destaining for 12 hours.
In vitro Cellular Uptake
CT26, B16-F10 melanoma cells (B16), and Mouse Forestomach Carcinoma cells (MFC) were selected to assess the cellular uptake in vitro. A mouse monocyte macrophage leukemia cell line, Raw 264.7, was selected to evaluate uptake profiles in the phagocytosis study. All cells were incubated separately in confocal petri dishes overnight. PLGA NPs loaded with DiI dyes were prepared by the above method. Raw 264.7 cell membrane was dyed with DiO. The cells were incubated with PLGA/DiI or CCM-PLGA/DiI for 60 minutes, respectively. Then the cell samples were stained with DAPI to label nuclei. Then, the tumor cells were visualized by CLSM. Meanwhile, Raw 264.7 cells were observed by Fluorescent Inverted microscope.
Biodistribution Study
CT26 cells (5 × 105) were subcutaneously injected into the right wing of BALB/c mice. After two weeks, the mice were randomly assigned to two groups and intravenously injected with PLGA/DiR or CCM-PLGA/DiR. The mice were anesthetized at the designated time points after injection and scanned with the CRi Maestro In Vivo Imaging System (Cambridge Research & Instrumentation, Massachusetts, USA). The mice were sacrificed 24 hours later, while the tumors and other primary organ tissues (the heart, liver, spleen, lungs, and kidneys) were collected and washed with PBS.
In vitro BMDC Stimulation
Bone marrow-derived dendritic cells (BMDCs) were obtained from C57BL/6 male mice, cultured with rmGM-CSF (20 ng/mL, Peprotech, USA) and rmIL-4 (10 ng/mL, Peprotech, USA). During the incubation period, three-quarters of the medium was changed every 2–3 days until the 8th day when all supernatants were discarded and all adherent cells were collected simultaneously. The immature DCs were randomly assigned to four groups (n=3) and co-cultured with Normal saline (NS), PLGA/GA(PG), CCM-PLGA/GA(CPG) (4 ug/mL of GA), and lipopolysaccharide (LPS), for 24 hours, respectively. Finally, all the cells were collected and then incubated with the corresponding flow cytometry fluorescent antibody for 30 minutes before being evaluated and analyzed using the flow cytometric analysis (CytoFLEX, Beckman, USA).
Flow Cytometry Detection of the CCM-PLGA/GA NPs Induced Immune Response
The in vivo colon tumor model was established by subcutaneously injected with 1×106 CT26 cells into the right wing of BALB/c mice. When the tumor volume grew to about 100 mm3, the mice were randomly assigned to three groups (n=3) and intravenously injected with saline, PLGA/GA, and CCM-PLGA/GA (4 mg/kg of GA, iv), once a week for 2 weeks, respectively. The animal weight and tumor volume were measured every 2 or 3 days until the test end. Tumor volume (V) was calculated using the formula: V = (L × W × W)/2 (L: the longest diameter of tumor and W: the shortest diameter perpendicular to length). All the BALB/c mice were sacrificed on the third day after the last injection, and spleens and tumors were collected for flow cytometry. Tumors were cut into small pieces, incubated with collagenase type IV at 37 °C for 3 hours, filtered, and suspended in PBS. All samples were stained with specific antibodies for 30 minutes at 4 °C in the dark, and washed subsequently before being collected with the flow cytometric analysis and analyzed using FlowJo X (FlowJo). The following monoclonal antibodies (mAbs) were used for flow cytometry: CD11c-FITC, CD80-APC, CD86-PE, CD3-FITC, CD8a-PC5.5, anti-CD279-PE, CD274-PC7(Biolegend, USA).
In vivo Antitumor Effect
The previous animal experiments are roughly the same as the above-described process and will not be repeated here. The differences are briefly described below, the mice were randomly assigned to five groups (n = 4) and injected with Normal saline, PLGA/GA, anti-PD-1 monoclonal Antibodies (aPD-1), CCM-PLGA/GA and CCM-PLGA/GA with aPD-1 (4 mg/kg of GA, iv; 100 ug per mouse of anti-mouse PD-1 (CD279) monoclonal antibody, ip), once a week for 2 weeks, respectively. The tumor-bearing mice were sacrificed on the third day after the last administration.
Safety Studies
After execution of the mice, tumor tissues and major organs (including heart, lungs, liver, spleen and kidneys) were collected from each group of mice for hematoxylin and eosin (H&E). The blood from each group of mice was collected for blood biochemical analysis. The Ki67 immunohistochemistry was applied to assess the cell proliferation in the tumors. And systemic toxicity was evaluated via blood biochemistry and H&E staining of organ slices.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8.0.2 statistical software. Independent Student’s t-test or One-way analysis of variance (ANOVA) was used to determine the differences between various treatments. Data are presented as the mean ± S.D. unless otherwise noted. Significance levels were defined as ns (not significant, P > 0.05), *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Preparation and Characterization of CCM-PLGA/GA
A previously reported two-step emulsification method was used to acquire PLGA/GA NPs,17 and then the CT26 cancer cell membrane was coated on the surface of PLGA/GA NPs by mechanical extrusion. The encapsulation efficiency (EE) and drug loading capacity (DL) of PLGA/GA NPs were measured by Ultraviolet-visible Spectrophotometer, and the results showed that EE=85.5±4.6%, and DL=29.9±0.8% (mean±S.D.), respectively. The shell–core structure of the CCM-PLGA/GA NPs was observed by TEM (Figure 2A), indicating that NPs coated with cancer cell membrane were successfully prepared. The DLS results showed that, after the PLGA/GA NPs-vesicles fusion process, the resulting CCM-PLGA/GA NP was approximately 182 nm in diameter (Figure 2B). The obtained CCM-PLGA/GA NP has no apparent changes in particle size and polydispersity index (PDI) at 14 days in PBS (Figure 2C), which confirmed its long-term stability and guaranteed the feasibility of subsequent trials. In addition, DiI and DiO were labeled PLGA NPs and CCM, respectively. After co-extrusion, we observed the colocalization of PLGA NPs and CCM on CLSM (Figure 2D), indicating the successful preparation of CPG nanoparticles. Moreover, the characteristic absorption peaks of GA (1630 cm−1) and the carbonyl group of PLGA (1750 cm−1) were observed in the FTIR spectra of PLGA/GA NP (Figure S1), proving the successful binding of GA in PLGA microspheres.
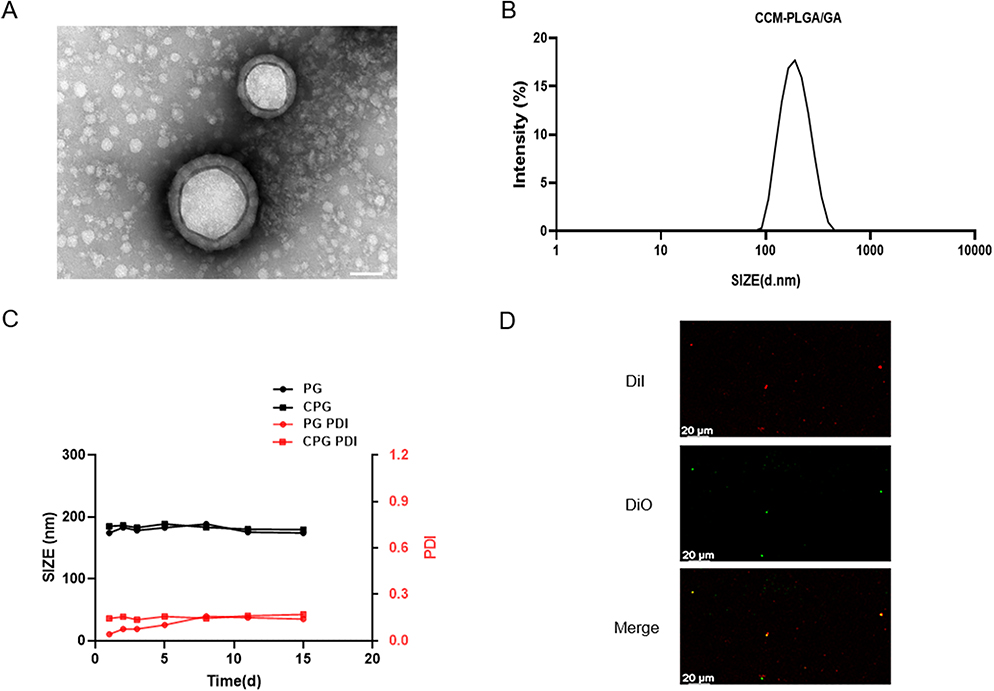 |
Figure 2 Characterization of CCM-PLGA/GA NPs. (A) TEM images of CCM-PLGA /GA NPs. (The scale bar is 50 nm). (B) Size distribution of CCM-PLGA/GA NPs determined via DLS. (C) Size and PDI of PG and CPG NPs in PBS over 14 days. (D) Confocal images of the colocalization between DiO-labeled CCM and DiI-labeled NPs. (The scale bar is 20 μm). |
In vitro Uptake and Cytotoxicity
CCM with endogenous molecules on its surface can produce prolonged blood circulation, immune escape, and homotypic targeting capacities in NPs.18,19 We performed the SDS-PAGE assay to confirm the protein profiles on the CCM-PLGA/GA NPs. CCM-PLGA/GA NPs have a protein distribution similar to that derived from CT26 cell membranes, showing that the fabrication process had not damaged the membrane proteins on NPs (Figure 3A). The homotypic tumor targeting of CCM-PLGA/GA NPs was investigated by CLSM after incubation of CT26, B16 and MFC cells with CCM-PLGA/DiI NPs, respectively (Figure 3B). Fluorescence images showed that red points (PLGA cores were labeled with DiI dye) were around the blue nuclei (labeled with DAPI dye) and overlap each other. Meanwhile, CT26 cells treated with NPs exhibited remarkably higher fluorescence intensity than those other cells at the same incubation periods. Immunoescape study of CCM-PLGA/GA NPs showed that the internalization of PLGA/DiI was significantly higher than CCM-PLGA/DiI by Raw264.7 macrophages (Figure 3C), indicating that cell membrane coated NPs could effectively evade recognition by the immune system to some extent. CCK8 assays were performed to assess whether the GA encapsulated in the CCM-PLGA/GA NPs was able to maintain its antitumor activity towards CT26 cells. The experimental results demonstrated that the free GA and CPG NPs exhibited no statistically significant on cell viability (P >0.05, n=4) against CT26 cells for 24 hours (Figure 3D).
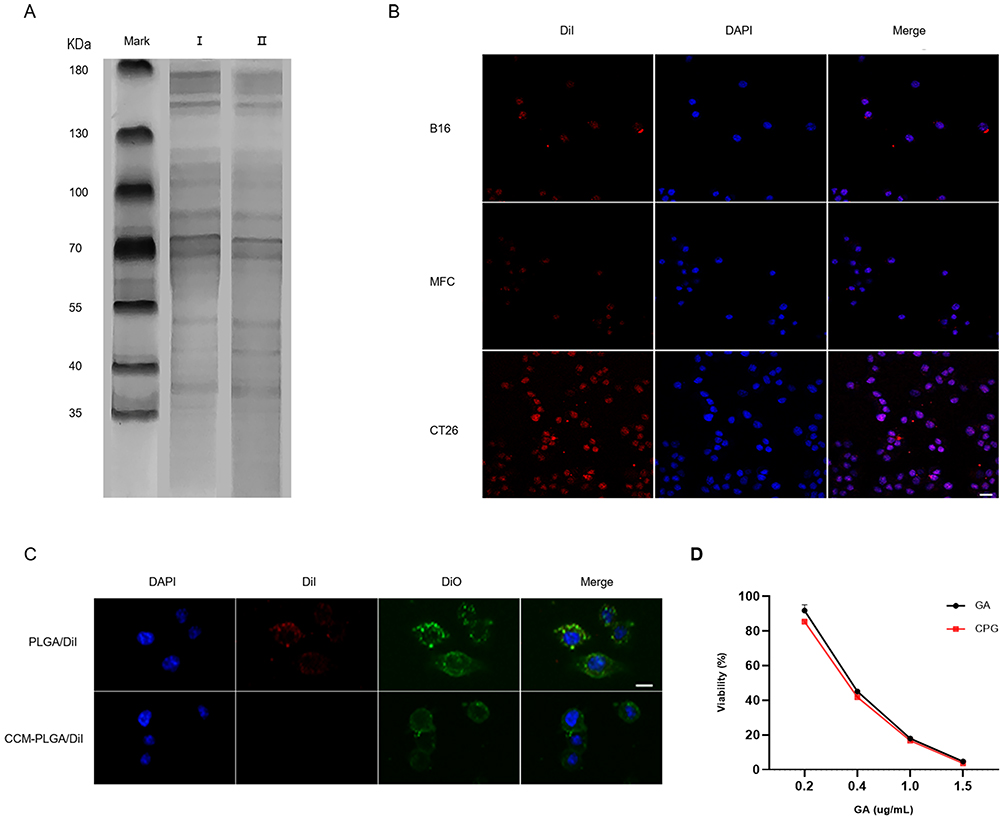 |
Figure 3 In vitro uptake and cytotoxicity of CCM-PLGA/GA NPs. (A) SDS-PAGE protein analysis. I: CT26 cancer cell lysate, II: CCM-PLGA/GA NPs. (B) Confocal images of various cancer cells after 1 hour co-incubation with CCM-PLGA/GA NPs. NPs were labeled with DiI and the cancer-cell nucleus were stained with DAPI. (The scale bar is 20 μm). (C) Fluorescent images of Raw264.7 cells incubated with DiI-labeled NPs for 1 hour via Fluorescent Inverted microscope. Raw 264.7 cell membrane was labeled with DiO and nucleus was stained with DAPI. (The scale bar is 10 μm). (D) In vitro cytotoxicity of CCM-PLGA/GA NPs in comparison to free GA on CT26 cells after 24 hours of incubation (n=4, P >0.05). |
In vivo Biodistribution and Targeting Ability
The biodistribution of two groups NPs (PLGA/DiR and CCM-PLGA/DiR) was detected via Near-infrared (NIR) fluorescence imaging (Figure 4A). We found that, compared to the PLGA/DiR NPs, CCM-PLGA/DiR NPs displayed superior tumor-targeting ability and a certain amount of fluorescence accumulation can be detected at the tumor site after 24 hours postinjection, which lasted until 48 hours. At 24 hours postinjection, tumors and other organs were collected to further investigate the biodistribution of NPs via ex vivo NIR fluorescence imaging (Figure 4B and C). The imaging exhibited that CCM-PLGA/DiR NPs showed a higher DiR signal at the tumor site and a lower fluorescent signal at the liver than PLGA/DiR NPs, supporting that CCM-PLGA/DiR NPs have strong tumor targeting efficacy. A lower amount of CCM-PLGA/DiR accumulated at the liver also demonstrated that because of cancer cell membrane coating, CCM-PLGA/DiR NPs could be disguised as cells to reduce liver interception to some extent.
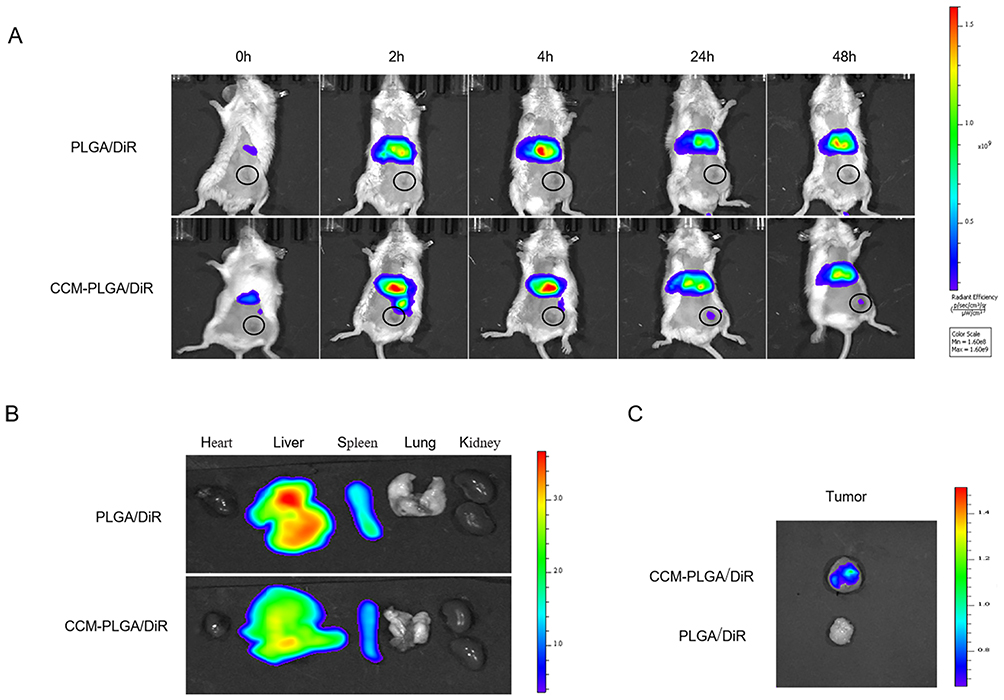 |
Figure 4 In Vivo Biodistribution and targeting ability. (A) Representative real-time NIR fluorescence imaging of CT26 tumor-bearing mice following intravenous injection of PLGA/DiR and CCM-PLGA/DiR NPs. The tumor of each mouse was circled. (B and C) Ex vivo NIR images of the various organs and tumor at 24 hours postadministration with two different NPs. |
Immune Response Induced by the CCM-PLGA/GA NPs
In order to verify the effect of CPG NPs on DCs, BMDCs were treated with NS, PG, CPG NPs and LPS in vitro for 24 hours. Meanwhile, NS and LPS were used as negative and positive controls. The fraction of mature DC (mDC) (CD11c+CD86+) was nearly 2-fold higher in the PG group than in the NS group, which is noteworthy. At the same time, it is surprising that CPG NPs play a more significant role in activating DCs which was about 4 times that of the NS group (Figure 5A and B). Subsequently, animal experiments were performed for further validation. Spleens and tumors were collected for flow cytometry on the third day after the last injection. The expression levels of both CD11c+CD86+ cells and CD11c+CD80+CD86+ cells in the CPG group were 1.3-fold upregulated compared to the NS group (Figure 5C-F). Meanwhile, the proportion of CD8+ T cells was examined, and the results revealed that in both the spleen and tumor, there was at least a 2-fold increase in CD8+ T cells in the CPG group compared with the NS (Figure 5G-J). In addition, the expression of PD-1 on CD8+ T cells and PD-L1 on tumor cells were slightly decreased, which was beneficial in reducing the immune escape of tumor cells (Figure 5K-N).22 These findings have proved that CPG NPs have a precise effect on improving the immune microenvironment. Based on the above findings, we hypothesized that there might be an excellent synergistic effect between the CPG NPs and anti-PD-1 monoclonal antibodies.
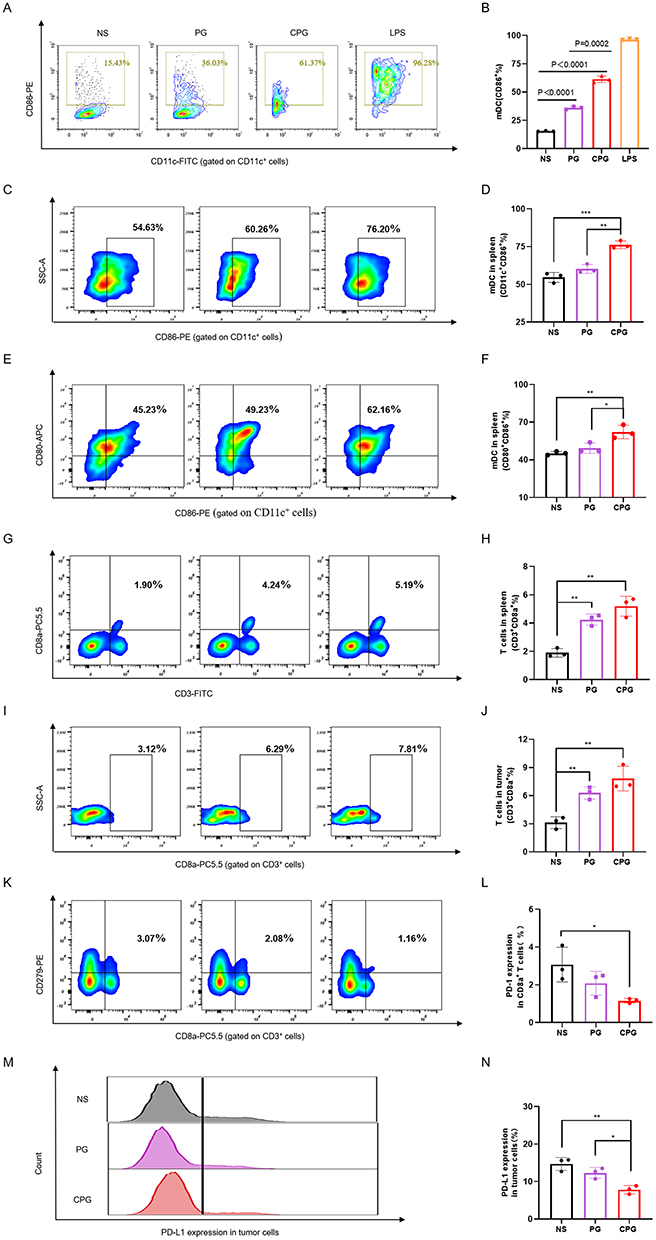 |
Figure 5 Immune Response Induced by the CCM-PLGA/GA NPs. (A) Flow cytometry images of mDC (CD11c+CD86+) after co-incubation with NS, PG, CPG or LPS in vitro for 24 hours. (B) The percentage of mDC in vitro co-incubation with different groups for 24 hours (n=3). (C and D) Representative flow cytometry images and the percentage of mDC (CD11c+CD86+) in the spleen of BALB/c mice in different groups on the third day after the last injection (n=3). (E and F) Representative flow cytometry images and the percentage of mDC (CD11c+CD80 + CD86+) in the spleen of BALB/c mice on the third day after the last injection (n=3). (G-J) Representative flow cytometry images and the percentage of CD3+CD8a + T cells in the spleen or tumor of BALB/c mice in different groups on the third day after the last injection (n=3). (K and L) Flow cytometry images and the percentage of the expression of PD-1 on CD8+ T cells in the spleen of BALB/c mice in different groups on the third day after the last administration (n=3). (M and N) Representative flow cytometry images and the percentage of the expression of PD-L1 on tumor cells of BALB/c mice on the third day after the last administration (n=3). *, **, and ***were used to represent p value of < 0.05, < 0.01, and < 0.001, respectively. |
In vivo Antitumor Effect
Inspired by the previous experimental results, we tried to combine the therapy of CPG NPs with anti-PD-1 monoclonal antibodies to obtain better anti-tumor effects. The mice were subjected to treatments as illustrated in Figure 6A. CT26 tumor-bearing BALB/C mice were randomly divided into five groups: the NS group, PG group, CPG group, aPD-1 group and CPG+aPD-1 group. In the NS group, the tumor volume rapidly increased with time, while the CPG+aPD-1 group had the most significant tumor inhibition effect (Figure 6B). Tumor growth of each mouse revealed that after 18 days of tumor inoculation, three-quarters of mice showed a slower-growing tendency in the dual drug group, whose volumes were all below 170mm3 (Figure 6D). The photographs of the tumors also verified the excellent anti-tumor ability of the dual drug group (Figure 6E). The body weight growth curves were similar in all groups of mice, revealing that the NPs were safe in vivo (Figure 6C). Moreover, immunohistochemistry staining demonstrated that the CPG+aPD-1 group remarkably inhibited the proliferation of CT26 tumors in vivo, as indicated by the decreased expression of Ki-67 (Figure 6F).
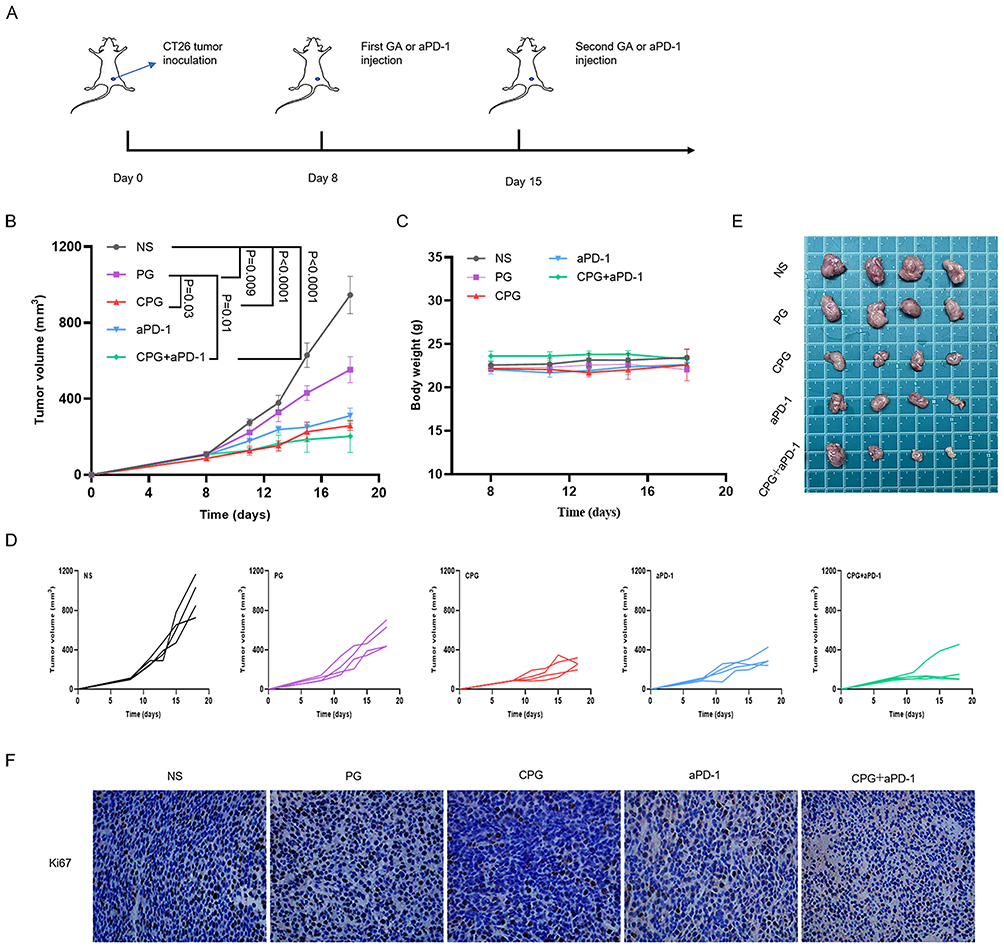 |
Figure 6 Combination therapy of NPs with PD-1 antibody. (A) Schematic diagram of administration route in tumor suppression Experiment. (B) Average tumor-growth curves of CT26 tumor-bearing mice with different treatments as indicated (n=4). The error bars shown as mean ± s.e.m. p-values were calculated by one-way ANOVA. (C) Body weights from five groups of CT26 tumor-bearing mice during treatment (n = 4). The error bars shown as mean ± s.e.m. (D) Tumor growth curves in five groups of each mouse (n=4). (E) Photographs of tumors from five groups of mice (n=4). (F) Ki67 staining images of tumor issues proliferation magnified 400×. |
Biosafety Assessment
When NPs was applied to treat mouse of different groups, all mice did not experience abnormal weight fluctuations (Figure 6C). H&E-stained images of the main organs (heart, liver, spleen, lung, kidney) in five groups showed no detectable signal of organ damage (Figure 7A). Besides, we performed serum biochemistry assays (Figure 7B). The serum biochemical indexes of mice in all groups had no significant differences and were within the normal range, suggesting the good biocompatibility and safety of CPG NPs in vivo.
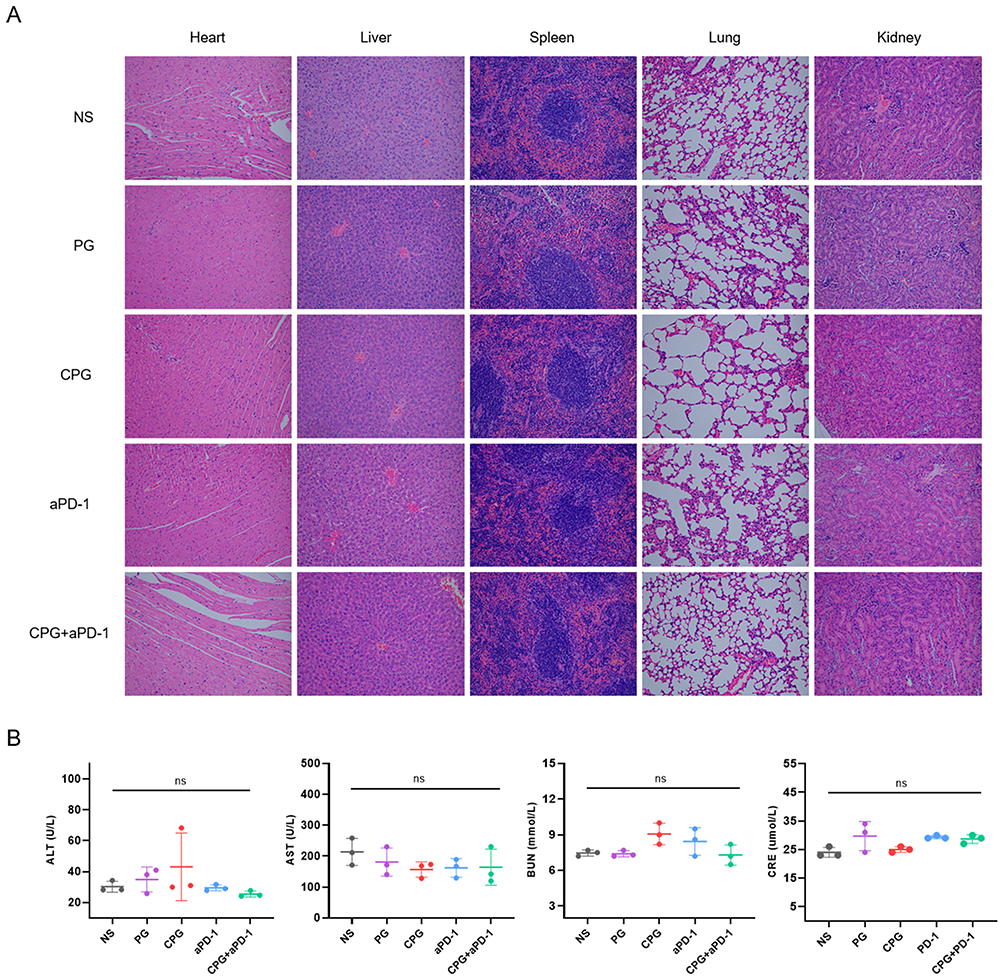 |
Figure 7 Biosafety assessment of CPG NPs in vivo. (A) Representative images of H&E-stained sections of the primary organs in each group were magnified 200x. (B) Blood biochemistry data of mice on the third day after the last treatment (n=3). p-values were calculated by one-way ANOVA. Abbreviations: ALT, glutamic-pyruvic transaminase; AST, aspartate aminotransferase; BUN, Blood Urea Nitrogen; CRE, Blood creatinine. |
Discussion
The past decade has witnessed significant antitumor effects of immunotherapy, represented by immune checkpoint inhibitors, in a variety of solid tumors. However, this efficacy is limited to a small subset of microsatellite stability-high (MSI-H) CRC patients. It is reported that the formation of immunosuppressive microenvironment is the main reason for the failure of immunotherapy. Nano-vaccines activate T cell-mediated anti-tumor immune responses by delivering tumor-associated antigens and immune-activating adjuvants to target organs.23 Therefore, it has become an essential strategy for anti-tumor immunotherapy.
Immunotherapy kills tumors by mobilizing the body’s immune system, which is similar to the TCM theory of “nourishing the positive and dispelling the evil”. The advantage of TCM lies in regulating the tumor-host microenvironment, so that the immune cells can perform their duties and cause apoptosis or autophagy of tumor cells.24 Thus, TCM is also a candidate adjuvant for the preparation of nano-vaccine. Nowadays, there are four broad categories of herbal medicines used as adjuvants: saponins, polysaccharides, propolis and other natural active ingredients.25 Among them, polysaccharides are generally used as an adjuvant.26,27 Our previous study focused on the evaluation of the anti-tumor effect of GA in CRC, and found that in addition to directly killing tumors through cytotoxic effects,14 it may be involved in immune regulation by counteracting VEGF-mediated angiogenesis. The discoveries suggest that GA can also be used as an adjuvant to prepare the nano-vaccine.
In the present study, we used CT26 colon cancer cell membrane to further optimize the nanoparticle in order to improve its homologous targeting ability. Based on this, we successfully constructed a novel nano-vaccine using GA as an adjuvant. In vitro and in vivo tests confirmed its high tumor targeting ability as well as low biological toxicity. Additionally, we found that CPG NPs could stimulate the maturation of DC, increase the expression of CD8+ T cells in the spleen, and reduce the expression of PD-L1 on tumor cells and PD-1 on CD8+ T cells, indicating that CPG NPs can improve the formation of a positive tumor immune microenvironment to some extent. Moreover, it is surprising that the effect of CPG was more remarkable than PG, even though the fluorescence intensity of the spleen was higher in the PG group on the NIR ex vivo organ images. As a result, we speculate that part of the reason is that the vaccine has been formed, as the CCM provides the tumor antigen and the GA acts as an adjuvant for DC activation, which could amplify the anti-tumor immune response.20 In addition, in vivo experiments, CCM on the surface of CPG NPs may also play a role by causing higher concentrations of NPs to escape phagocytosis and clearance by the immune system and enter the circulation successfully to exert anti-tumor effects.28 Especially, the low intensity of CPG NPs could not only kill tumor cells in targeted lesions, but also activate DCs maturation, showing the superiority of CPG nanoparticles in cancer immunotherapy.
Based on the experimental results of improving immune microenvironment by CPG NPs, we hypothesized that the combination of aPD-1 and nanoparticles might have synergistic anti-tumor effects.29 Our studies showed that the combined group had the best tumor-suppressive effect, while the protein expression of Ki67 was the lowest in the tumor tissues of this group. It was seen that the dual drug group had the best anti-tumor effect with the best trend of tumor suppression. These findings enhance our confidence in the future clinical translation of this nano-vaccine. However, there are corresponding shortcomings in our study, for example, the specific mechanisms of immune microenvironment regulation need to be studied in depth subsequently.
Conclusion
In conclusion, we developed a novel nano-vaccine using CCM as the antigen and GA as an adjuvant, and confirmed its excellent tumor targeting and anti-tumor efficacy. In addition, it also reduces the side effects of GA and ensures the safety of the drug in vivo. Thus, this nano-vaccine with GA as an adjuvant enhances the release of GA at the tumor site through homologous targeting, and exerts immune regulatory function. This nano-vaccine has the potential for clinical translation and will provide a new strategy for colorectal cancer.
Ethical Approval
This study was conducted with the approval of the Ethics Committee of Nanjing Drum Tower Hospital.
Acknowledgments
This study was supported by grants from Nanjing health science and technology development key program (no ZKX21028), Provincial Natural Science Foundation of Jiangsu (no BK20211007), Jiangsu scientific and technological development of traditional Chinese medicine Key projects (no ZD202227).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Horn L, Mansfield AS, Szczesna A, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med. 2018;379(23):2220–2229. doi:10.1056/NEJMoa1809064
3. Xie Y, Xie F, Zhang L, et al. Targeted anti-tumor immunotherapy using tumor infiltrating cells. Adv Sci. 2021;8(22):e2101672. doi:10.1002/advs.202101672
4. Mo F, Duan S, Jiang X, et al. Nanobody-based chimeric antigen receptor T cells designed by CRISPR/Cas9 technology for solid tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1):80. doi:10.1038/s41392-021-00462-1
5. Deng G, Zhou L, Wang B, et al. Targeting cathepsin B by cycloastragenol enhances antitumor immunity of CD8 T cells via inhibiting MHC-I degradation. J Immunother Cancer. 2022;Oct(10):10. doi:10.1136/jitc-2022-004874
6. Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol. 2018;52(Pt 2):117–124. doi:10.1016/j.semcancer.2017.12.002
7. Huang Y, Kim BYS, Chan CK, Hahn SM, Weissman IL, Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat Rev Immunol. 2018;18(3):195–203. doi:10.1038/nri.2017.145
8. Ren S, Xiong X, You H, Shen J, Zhou P. The combination of immune checkpoint blockade and angiogenesis inhibitors in the treatment of advanced non-small cell lung cancer. Front Immunol. 2021;12:689132. doi:10.3389/fimmu.2021.689132
9. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med. 2020;52(9):1475–1485. doi:10.1038/s12276-020-00500-y
10. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022;21(1):28. doi:10.1186/s12943-021-01489-2
11. Cao M, Yan H, Han X, et al. Ginseng-derived nanoparticles alter macrophage polarization to inhibit melanoma growth. J Immunother Cancer. 2019;7(1):326. doi:10.1186/s40425-019-0817-4
12. Yang M, Yang Y, Cui H, et al. The natural compound gambogic acid radiosensitizes nasopharyngeal carcinoma cells under hypoxic conditions. Tumori. 2016;102(2):135–143. doi:10.5301/tj.5000411
13. Wen C, Huang L, Chen J, et al. Gambogic acid inhibits growth, induces apoptosis, and overcomes drug resistance in human colorectal cancer cells. Int J Oncol. 2015;47(5):1663–1671. doi:10.3892/ijo.2015.3166
14. Wan L, Zhang Q, Wang S, et al. Gambogic acid impairs tumor angiogenesis by targeting YAP/STAT3 signaling axis. Phytother Res. 2019;33(5):1579–1591. doi:10.1002/ptr.6350
15. Chen X, Chen DR, Liu H, et al. Local delivery of gambogic acid to improve anti-tumor immunity against oral squamous cell carcinoma. J Control Release. 2022;351:381–393. doi:10.1016/j.jconrel.2022.09.010
16. Luk BT, Zhang L. Cell membrane-camouflaged nanoparticles for drug delivery. J Control Release. 2015;220(Pt B):600–607. doi:10.1016/j.jconrel.2015.07.019
17. Zhang Z, Qian H, Yang M, et al. Gambogic acid-loaded biomimetic nanoparticles in colorectal cancer treatment. Int J Nanomedicine. 2017;12:1593–1605. doi:10.2147/IJN.S127256
18. Feng S, Ren Y, Li H, et al. Cancer cell-membrane biomimetic boron nitride nanospheres for targeted cancer therapy. Int J Nanomedicine. 2021;16:2123–2136. doi:10.2147/IJN.S266948
19. Zhang L, Zhang W, Peng H, et al. Bioactive cytomembrane@poly(citrate-peptide)-miRNA365 nanoplatform with immune escape and homologous targeting for colon cancer therapy. Mater Today Bio. 2022;15:100294. doi:10.1016/j.mtbio.2022.100294
20. Fang RH, Hu CM, Luk BT, et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano Lett. 2014;14(4):2181–2188. doi:10.1021/nl500618u
21. Ren X, Yang S, Yu N, et al. Cell membrane camouflaged bismuth nanoparticles for targeted photothermal therapy of homotypic tumors. J Colloid Interface Sci. 2021;591:229–238. doi:10.1016/j.jcis.2021.02.006
22. Jiang X, Wang J, Deng X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):10. doi:10.1186/s12943-018-0928-4
23. Jiang X, Wang J, Zheng X, et al. Intratumoral administration of STING-activating nanovaccine enhances T cell immunotherapy. J Immunother Cancer. 2022;10(5):May. doi:10.1136/jitc-2021-003960
24. Yang DD, Wang ZY, Li Q, et al. Regulating immune function in patients with colorectal carcinoma by traditional Chinese medicine. Jilin J Chine Med. 2015;35(07):753–756. doi:10.13463/j.cnki.jlzyy.2015.07.032
25. Liu HY, Wang JY, Cui ZZ, et al. Research progress on immune adjuvant effect and related mechanism of active ingredients of traditional Chinese medicine on animal vaccines. China Animal Husbandry Veterinary Med. 2023;50(1):390–397. doi:10.16431/j.cnki.1671-7236.2023.01.039
26. Chang WT, Lai TH, Chyan YJ, et al. Specific medicinal plant polysaccharides effectively enhance the potency of a DC-based vaccine against mouse mammary tumor metastasis. PLoS One. 2015;10(3):e0122374. doi:10.1371/journal.pone.0122374
27. Gu P, Liu Z, Sun Y, et al. Angelica sinensis polysaccharide encapsulated into PLGA nanoparticles as a vaccine delivery and adjuvant system for ovalbumin to promote immune responses. Int J Pharm. 2019;554:72–80. doi:10.1016/j.ijpharm.2018.11.008
28. Nie D, Dai Z, Li J, et al. Cancer-cell-membrane-coated nanoparticles with a yolk-shell structure augment cancer chemotherapy. Nano Lett. 2020;20(2):936–946. doi:10.1021/acs.nanolett.9b03817
29. Yi C, Chen L, Lin Z, et al. Lenvatinib Targets FGF Receptor 4 to Enhance Antitumor Immune Response of Anti-Programmed Cell Death-1 in HCC. Hepatology. 2021;74(5):2544–2560. doi:10.1002/hep.31921
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.