Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Calcifying nanoparticles promote mineralization in vascular smooth muscle cells: implications for atherosclerosis
Authors Hunter L, Charlesworth J, Yu S, Lieske J, Miller V
Received 27 February 2014
Accepted for publication 29 March 2014
Published 27 May 2014 Volume 2014:9(1) Pages 2689—2698
DOI https://doi.org/10.2147/IJN.S63189
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Larry W Hunter,1 Jon E Charlesworth,2 Sam Yu,3,4 John C Lieske,5,6 Virginia M Miller1,7
1Department of Surgery, 2Department of Biochemistry and Molecular Biology, Mayo Clinic, Rochester, MN, USA; 3Lincoln University, Christchurch, New Zealand; 4Izon Science Ltd., Christchurch, New Zealand; 5Department of Internal Medicine, Division of Nephrology and Hypertension; 6Department of Laboratory Medicine and Pathology; 7Department of Physiology
and Biomedical Engineering, Mayo Clinic, Rochester, MN, USA
Background: Nano-sized complexes of calcium phosphate mineral and proteins (calcifying nanoparticles [CNPs]) serve as mineral chaperones. Thus, CNPs may be both a result and cause of soft tissue calcification processes. This study determined if CNPs could augment calcification of arterial vascular smooth muscle cells in vitro.
Methods: CNPs 210 nm in diameter were propagated in vitro from human serum. Porcine aortic smooth muscle cells were cultured for up to 28 days in medium in the absence (control) or presence of 2 mM phosphate ([P] positive calcification control) or after a single 3-day exposure to CNPs. Transmission electron-microscopy was used to characterize CNPs and to examine their cellular uptake. Calcium deposits were visualized by light microscopy and von Kossa staining and were quantified by colorimetry. Cell viability was quantified by confocal microscopy of live-/dead-stained cells and apoptosis was examined concurrently by fluorescent labeling of exposed phosphatidylserine.
Results: CNPs, as well as smaller calcium crystals, were observed by transmission electron-microscopy on day 3 in CNP-treated but not P-treated cells. By day 28, calcium deposits were visible in similar amounts within multicellular nodules of both CNP- and P-treated cells. Apoptosis increased with cell density under all treatments. CNP treatment augmented the density of apoptotic bodies and cellular debris in association with mineralized multicellular nodules.
Conclusion: Exogenous CNPs are taken up by aortic smooth muscle cells in vitro and potentiate accumulation of smooth-muscle-derived apoptotic bodies at sites of mineralization. Thus, CNPs may accelerate vascular calcification.
Keywords: biomineralization, hydroxyapatite, vascular calcification, protein-mineral complex
Introduction
Cardiovascular disease is the leading cause of mortality worldwide.1 Calcification of the arterial wall and heart valves is a common characteristic of advanced atherosclerosis,2 Mönckeberg’s sclerosis,3 and aortic valvular stenosis.4 Under physiological conditions, the concentrations of calcium (Ca) and phosphate (P) in the extracellular milieu of healthy arteries and serum are approximately 1.8 mM and 0.9 mM, respectively,5 which would be sufficient to induce their precipitation spontaneously if it were not for endogenous inhibitors that bind these minerals, ie, fetuin-A (also known as human α-2-HS-glycoprotein), matrix gla protein (MGP), and inorganic pyrophosphate (PPi). These inhibitors are sequestered6 or produced7 by vascular smooth muscle cells and packaged into matrix vesicles – membranous structures in the nm to micron size-range that are shed from the cell membrane. In healthy arteries, inhibitors within the released vesicles serve as mineral chaperones, maintaining calcium solubility and allowing excess calcium to be cleared from the cells and the extracellular environment. However, under pathological conditions, vesicles deficient in these inhibitory proteins are released, becoming nidi for crystal growth.
Other structures similar in size to matrix vesicles are now being investigated for their possible function in ectopic calcification, including calcification that is associated with cardiovascular disease. However, lack of uniformity in nomenclature has led to some degree of duplication or overlap in the identification of these structures. In general, cell-derived membrane-bound structures, including those originating from smooth muscle cells, may be divided into three major groups based upon morphology: microvesicles, microparticles, and exosomes, which, like matrix vesicles, have been linked to calcification.8–10 Another group of structures includes mineral/protein complexes which can form independently of cellular activity; termed calciprotein particles,11 fetuin-mineral complexes,12 microcalcifications,13 nanocrystals,14 nanons,15 or calcifying nanoparticles (CNPs).16 This second group includes particles initially considered to be a putative life-form, termed nanobacteria.17 However, current evidence shows that they are most likely biochemical entities, which are complexes of calcium phosphate and protein with a high binding affinity for charged molecules.18–21 Indeed, nm-sized structures composed of apatite mineral and the proteins fetuin-A and MGP13,20,22,23 have been identified at sites of calcification in human aortic aneurysms,24,25 cardiac valves,26 calcium oxalate kidney stones,23,25,27 renal papillary Randall’s plaques,27 calcific tissue that encapsulates implanted silicone prosthetic devices,16 and serum.28,29
Although deposition of extracellular calcium in vascular smooth muscle cells cultured in a hyperphosphatemic environment is coincident with upregulation of osteogenic proteins, including bone morphogenic protein-2 (BMP-2) and osteopontin, these effects are not due directly to P, but to calcium phosphate nanocrystals formed subsequent to elevation of extracellular P levels.14 High extracellular P levels also augment formation of CNPs, and propagation of CNPs derived from calcific vascular tissue is inhibited by PPi, an inhibitor of apatite crystal growth, and is increased by alkaline phosphatase, which hydrolyzes PPi, producing P.25 Thus it is possible that nano-sized protein/mineral complexes in the blood and extracellular fluid may influence pathological calcification associated with human disease. Therefore, the aim of this study was to determine if human-derived CNPs induce mineralization of vascular smooth cells.
Materials and methods
General experimental design
To model arterial calcification in vitro, porcine aortic smooth muscle cells were cultured to post-confluency, which favors the formation of multicellular nodules containing a core of necrotic and apoptotic cells together with extensive matrix material; this model mimics sites of plaque formation within the vascular wall.30,31
Materials
Human serum and gamma-irradiated fetal bovine serum were purchased from Atlanta Biologicals (Lawrenceville, GA, USA). M-199 medium was from Life Technologies (Carlsbad, CA, USA). The primary antibodies against α-smooth muscle actin (mouse monoclonal) and smooth muscle myosin heavy-chain 2 (rabbit polyclonal) were from Abcam PLC (Cambridge, MA, USA). Radiolabeled calcium (45Ca) was from Perkin-Elmer, Inc. (Waltham, MA, USA). 4-well glass slides and reagents were from Thermo Fisher Scientific.
Isolation and characterization of CNPs
CNPs were isolated from human serum as described previously,25,32 with slight modification. Briefly, 15% serum in M-199 was filtered (0.2 μm), placed into vented culture flasks, and maintained in a humidified incubator at 37°C 5% CO2 for 4–6 weeks. Adherent CNPs that had developed during incubation were then scraped from the flask bottoms into the medium, which itself contained CNPs that were nonadherent. The medium was then centrifuged at 20,000 ×g for 60 minutes at 4°C (Model J2-MC centrifuge; Beckman Coulter Inc., Brea, CA, USA). The pelleted CNPs were resuspended in phosphate-buffered saline (PBS) and quantified turbidimetrically (in nephelometric turbidity units) using a Hach model 2100N turbidimeter (Hach Co., Loveland, CO, USA). CNPs were then inoculated into fresh medium for subculture in order to increase amounts for experimentation. The resulting pelleted stocks were stored in PBS at −80°C. The morphological and mineral characteristics of the CNPs were examined using scanning-electron microscopy and transmission-electron microscopy (TEM) with energy-dispersive elemental analysis,16,32 and the protein-containing components that remained after CNP demineralization with 0.6N HCl were examined by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).25,32 CNP diameters and size distribution were determined with a particle-analyzer employing tunable nanopores together with resistive pulse sensing (qNANO; Izon Science Ltd., Oxford, UK).33,34 CNP plating density (ie, nephelometric turbidity units per cm2) was used to quantify the amount CNPs applied to cells in culture.
Cell culture
Vascular smooth muscle cells were isolated from medial layer explants of thoracic aorta from 3-month old female pigs and were cultured in standard growth medium ([GM] M-199/15% fetal bovine serum with penicillin [100 U/mL], streptomycin [100 U/mL], and amphotericin [0.27 μg/mL]).35 Medium was replaced every 2–3 days. The cellular phenotype of all cells examined was confirmed by positive immunofluorescence staining with antibodies against α-smooth muscle actin as well as smooth muscle myosin heavy chain-2. Stocks of cells were stored in liquid nitrogen, and cells between passages three and six were used for study. Each experiment was initiated by seeding cells in GM into 24-well culture plates and in 4-chamber slides at 1.3×104 cells/cm2. Cells were cultured until nearly confluent (day 0), then divided into three experimental groups differing only in treatment: 1) media alone, no treatment (control); 2) CNPs, ten nephelometric turbidity units per cm2; or 3) 2 mM P as a positive control for calcification.36 Cells were cultured for up to an additional 28 days before harvesting. CNPs, when used, were applied from days 0–3; the cells were then rinsed thoroughly in medium to remove extracellular particles and further incubated in medium without addition of CNPs for the remainder of the study.
Additional experiments were carried out to assess calcium accumulation by CNPs themselves. CNPs in GM were first allowed to adhere to collagen-coated glass cover-slips overnight in the absence of cells. Each cover-slip was then rinsed in GM, transferred to a culture plate well containing GM augmented with 45Ca (0.5 μCi/well), and cultured for an additional 15 days. At 3-day intervals, a subgroup of cover-slips was removed and rinsed in PBS. The adhered particles were then demineralized in 0.6 N HCl for 16 hours at 4°C. Radioactivity in each HCl extract was measured by liquid-scintillation counting and used as an index of Ca accumulation.
CNP effects on cell viability
For each determination, cells cultured on 4-well slides were rinsed in HEPES buffer then exposed to fresh buffer containing calcein AM (2 μM), ethidium homodimer-1 (EthD-1; 2 μM), and annexin V–AlexaFluor® 647 conjugate (Life Technologies) (20 μL/mL) for 20 minutes to label live, dead, and apoptotic cells, respectively. After rinsing in HEPES, the cells were imaged by confocal laser-scanning microscopy, employing a C-apochromat 40× objective,16 with excitation/emission wavelengths of 495/515 nm, 528/615 nm, and 650/665 nm for calcein AM, EthD-1, and annexin V conjugate, respectively. For each well, five randomly chosen fields, each encompassing 325 μm2, were imaged; live and dead cells in each field were counted and averaged, and annexin V-positive staining, if present, was noted.
Assessment of CNP-induced cellular mineralization
Mineral deposits were visualized using von Kossa staining. Cells cultured on 12 mm round glass cover-slips were rinsed in PBS after experimentation then fixed in 4% paraformaldehyde in PBS for 20 minutes at 4°C. Thereafter, water was used to dissolve agents and for rinsing (three times) between each step. Next, cells were exposed to 3% silver nitrate under a 75-watt lamp for 30 minutes, then to 5% sodium thiosulfate for 3 minutes to remove unreduced silver. After rinsing, cells were stained with eosin-Y for 10 seconds to label cytoplasm pink, then cleared in xylene and mounted on a glass slide. Stained cells were examined by light microscopy for black areas indicating deposited mineral, and the number of mineral-positive multicellular nodules were counted and expressed as percent of total nodules. Ca deposition was quantified in a separate set of cells treated identically except that, after experimentation, they were decalcified with HCl. The Ca content of the HCl supernate was measured using the o-cresolphthalein complexone method (Calcium Colorimetric Assay Kit; BioVision Inc., Milpitas, CA, USA), and normalized to cellular protein.36
CNP cellular uptake
Cells were cultured and treated as described above, except that cover-slips were composed of Aclar® polymer (Electron Microscopy Sciences, Hatfield, PA, USA). Cells were then fixed overnight in Trumps’ solution at 4°C then surface-embedded in epoxy resin. Transverse thin-sections were cut, transferred to a carbon-coated grid, and examined by TEM (Tecnai 12; FEI, Hillsboro, OR, USA). Elemental analysis of intracellular components was carried out using an energy pulse processor (EDAX, Inc., Mahwah, NJ, USA). In addition, an aliquot of CNPs from the original stock was pelleted by centrifugation, sectioned, and examined by TEM23,25 for comparison with intracellular CNP-like structures.
Statistics
Quantified data are presented as mean ± standard error of the mean. Analysis of variance was used to determine differences among treatment groups; significance was accepted at P<0.05. For each data-point tested, values of two to five replicates were averaged.
Results
Characterization of CNPs
CNP isolates were spheroid structures less than 1.2 μm in diameter with Ca and P components (Figures 1A and B). Their sizes ranged from 20 nm to about 1 μm. The average size was 210 nm (Figure 1C) and the largest proportion of particles, relative to the total population, was found in a size range less than 220 nm (Figure 1D). Demineralization of CNPs with HCl revealed smaller structures (Figure 1E) that were comprised of numerous proteins (Figure 1F), consistent with previous published work by our group.27 CNPs adhered to collagen-coated cover-slips in the absence of cells and accumulated 45Ca from the media over the study period of 15 days (Figure 1G).
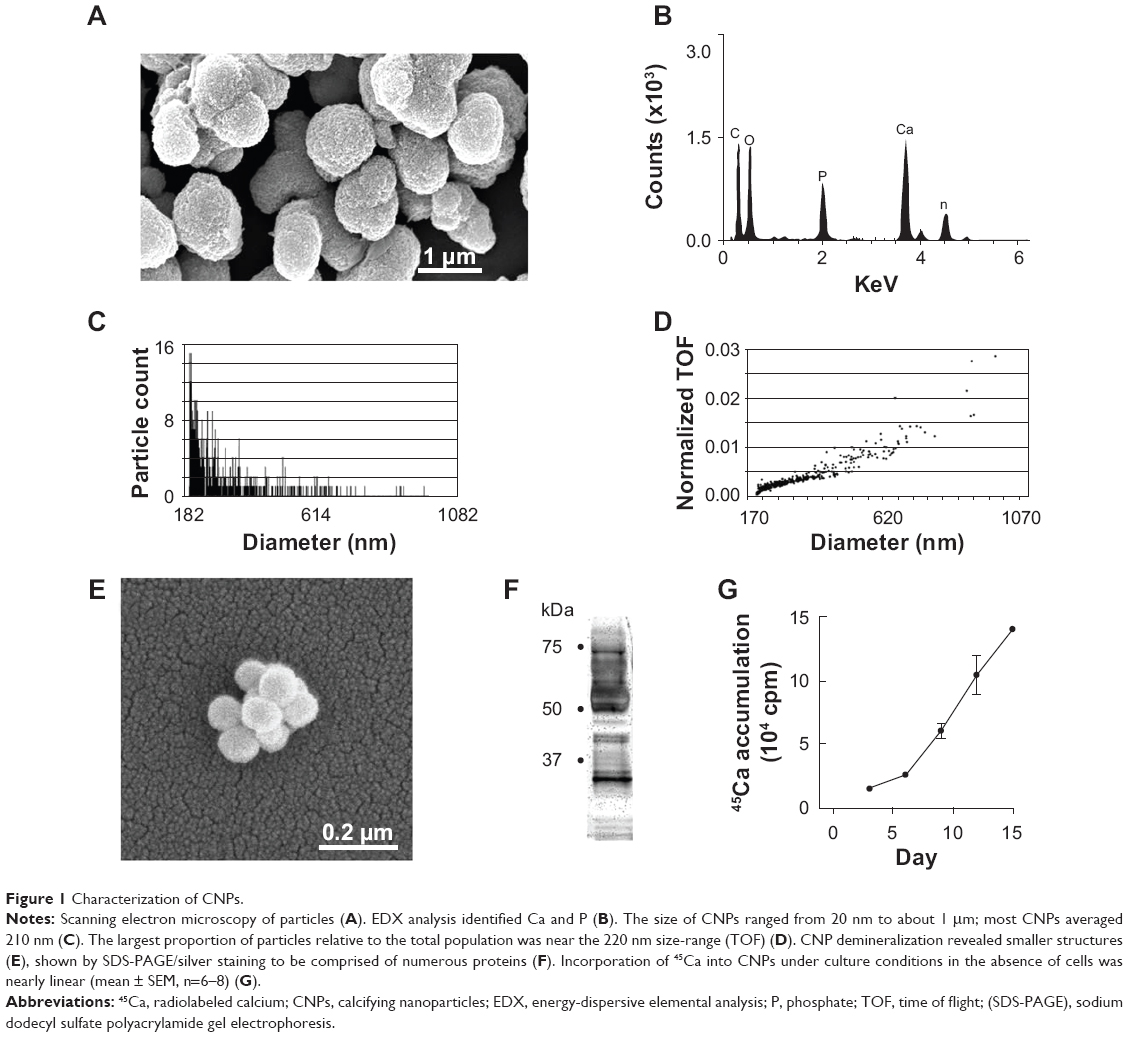 | Figure 1 Characterization of CNPs. |
CNP-induced calcification of vascular smooth muscle cells
After 3 days in culture, cells formed a monolayer of nearly-confluent cells (Figures 2A–C). Subsequently, groups of cells formed ridges, which ultimately developed into multicellular nodules (Figures 2D–F). Under all culture conditions, Ca deposition was not detected at day 3 by von Kossa staining. However, by day 28, von Kossa-positive crystals were present only in nodules of CNP-treated and 2 mM P-treated cultures. Crystals in CNP-treated and P-treated cells averaged about 1.7 μm and 8.4 μm in diameter, respectively (Figures 2E and F). Cells cultured in control media lacked crystals, even though there were no significant differences in the number of nodules per coverslip between control, CNP-, and P-treated cells (61.5±1.3, 48.8±6.2, and 57.5±4.6, respectively; n=4 each; Figure 3A). Among CNP- and P-treated cells, 81.0%±2.0% and 37.6%±4.2% of nodules were mineralized, respectively (Figure 3B). Ca was not detected in the HCl demineralizing solution from any of the control cultures at any time-point (Figure 3C), or in that of CNP-treated or P-treated cells after 3 days. However, it was present at 20.03±3.41 and 27.45±4.77 μg/mg protein in CNP-treated and P-treated cells, respectively, at experiment completion (Figure 3C).
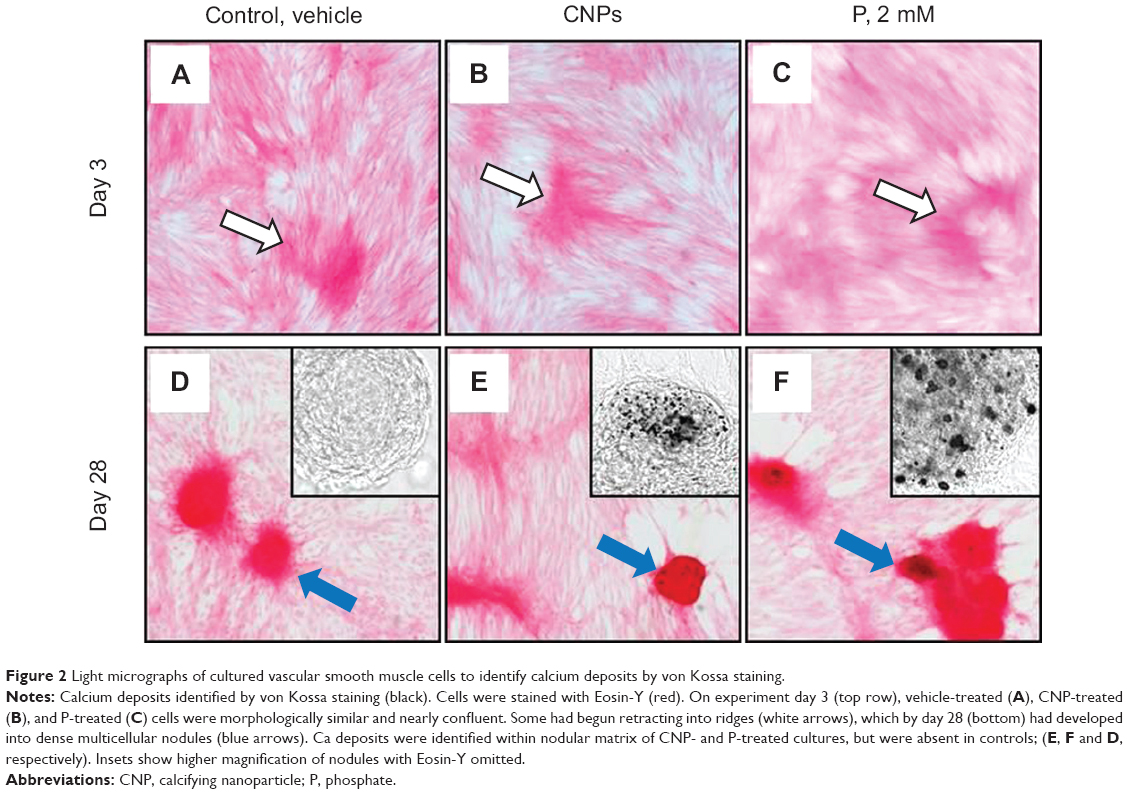 | Figure 2 Light micrographs of cultured vascular smooth muscle cells to identify calcium deposits by von Kossa staining. |
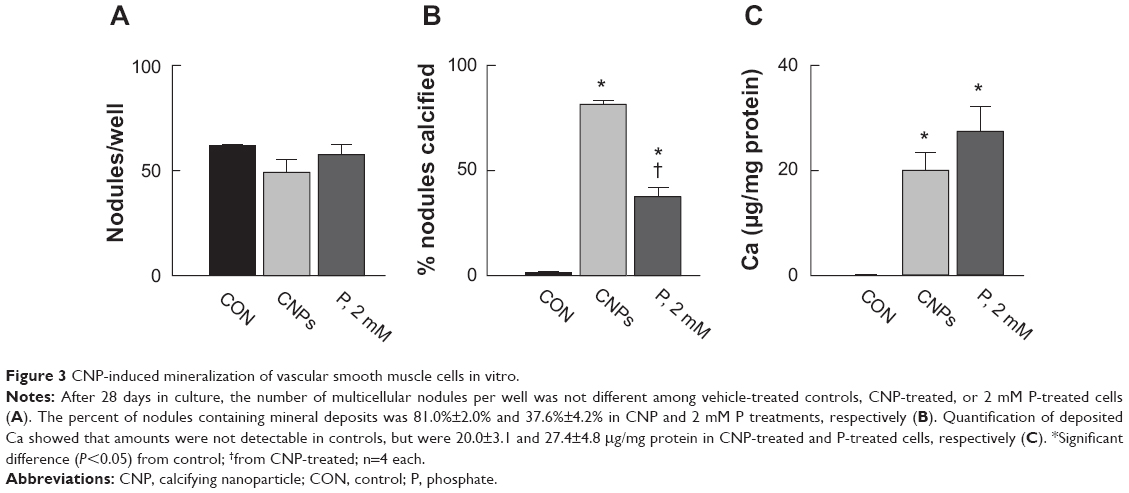 | Figure 3 CNP-induced mineralization of vascular smooth muscle cells in vitro. |
CNP uptake into vascular smooth muscle cells
The serum-derived CNPs applied to smooth muscle cells appeared, in cross-section, to consist of concentric layers that alternated in electron density (Figures 4A and B). Structures similar in size and appearance were present within 19.6%±3.7% (n=8) of CNP-treated cells (Figures 4C and D), but not in control or P-treated cells after 3 days of treatment. At this time point, highly electron-dense crystalline structures less than 50 nm in diameter were also observed in a small proportion of CNP-treated cells (Figure 4E). After 28 days in culture, CNPs were only observed within multicellular nodules of CNP-treated cells, either incorporated into the extracellular matrix (Figures 4F and G) or, less commonly, localized within intracellular vacuoles of nodular peripheral cells (Figure 4H). Although these particles were similar in morphology to those observed after 3 days, they were typically more electron-dense. Energy-dispersive elemental analysis of intra- and extracellular particles revealed Ca and phosphorus components (Figure 4I). Cells treated with 2 mM P displayed large crystals up to several μm in size within the nodules. However, it was not possible to confirm their original localization since it appeared that the particles may have been displaced by the sectioning procedure (Figure 4J).
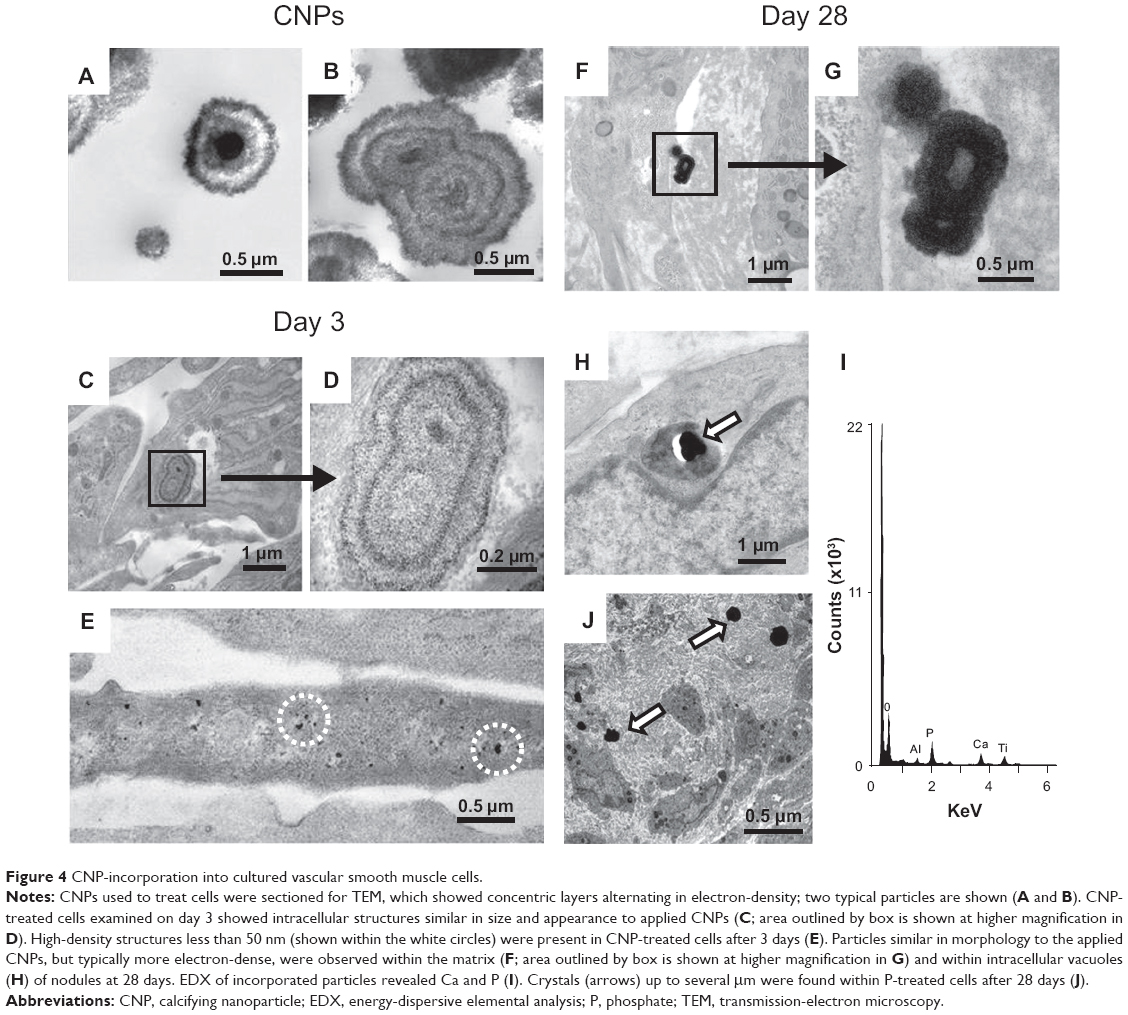 | Figure 4 CNP-incorporation into cultured vascular smooth muscle cells. |
CNP effects on cell viability
Annexin V labeling, indicating exposed phosphatidylserine, was negative in monolayer cells of all cultures examined. However, annexin V-labeled structures up to about 300 nm in size, most likely representing apoptotic bodies, were typically distributed sparsely among the cells. Numerous apoptotic bodies, as well as an increased proportion of dead cells, colocalized within ridges formed by retracted monolayer cells (Figures 5A and B). Multicellular nodules that subsequently formed from the ridge cells contained a necrotic core of EthD-1-labeled nuclear fragments surrounded by a layer of live cells with associated apoptotic bodies (Figures 5C–F). It was not possible to quantify cell viability within multicellular nodules using this methodology. However, nodules from CNP-treated cells typically displayed more intense nucleic-acid staining of fragmented cells and greater numbers of apoptotic bodies than was observed in control cells (Figures 5G and H). After 21 days, monolayer cell viability, determined by calcein AM/EthD-1 labeling, was 97.9%±0.5% in control cultures, and was slightly but significantly reduced to 96.2%±1.0% and 95.6%±0.7% in CNP-treated and 2 mM P-treated cultures, respectively; n=9–10 each (Figure 5I).
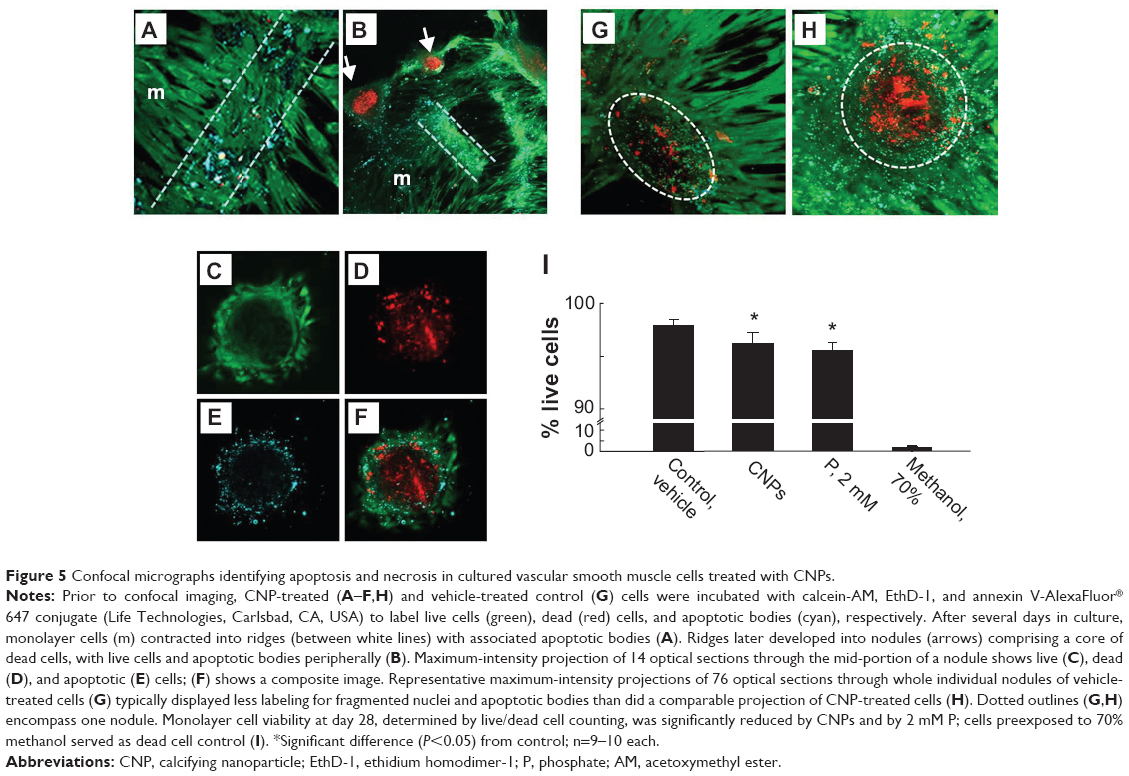 | Figure 5 Confocal micrographs identifying apoptosis and necrosis in cultured vascular smooth muscle cells treated with CNPs. |
Discussion
This study demonstrates that human-derived CNPs are taken up by vascular smooth muscle cells in culture, promoting accumulation of apoptotic bodies at sites that are colocalized with extracellular mineral deposits. The findings suggest that CNPs modify cellular processes that favor vascular calcification.
The CNPs that localized within the monolayer of the smooth muscle cells after 3 days most likely originated from those applied to the cells, since they were similar in morphology and elemental composition. In addition, similar structures were not observed in cells cultured in control media. The CNPs used in these experiments were within the size range of synthetic basic calcium phosphate crystals shown to be endocytosed by cultured vascular smooth muscle cells, resulting in rapid cell death.37 The cell-death effect is attributed to a rise in intracellular Ca following crystal dissolution within lysosomes, and is diminished by preincubating the crystals in serum. In the present investigation, the CNPs used were derived from human serum and were comprised of calcium phosphate and proteins; thus, their protein components (fetuin-A and MGP, for example) could have had a protective effect in this regard by limiting particle endocytosis or intracellular dissolution.
Confocal microscopy, using fluoroconjugated annexin V and EthD-1 to label apoptotic bodies and necrotic cells, respectively, indicated that monolayer cells were highly viable with low levels of apoptosis, regardless of treatment. Typical of this in vitro model of vascular calcification, retraction of the cells into dense multicellular nodules was associated with increased apoptosis within the nodule cores. The increased density of apoptotic bodies, as well as fragmented nuclei and cellular debris within the nodules of CNP-treated cells, compared to those of untreated cells, and together with the finding of colocalized mineral deposits within nodules of the CNP-treated cells, suggests that CNPs induced mineralization in vascular smooth muscle cells by promoting apoptosis. This effect would likely reflect modulation of both pro- and antiapoptotic regulatory processes, for example, activation of BMP-2/smad-induced apoptosis through phosphorylation of Smad1 would result in loss of expression of Bcl-2, an important anti-apoptotic protein.38 Another possibility is through downregulation of the vitamin K-dependent growth arrest-specific gene 6 (Gas6) and its receptor, Ax1, a tyrosine kinase. Under physiological conditions, the Gas6–Ax1 survival pathway functions to protect vascular smooth muscle cells from apoptotic death, partly by inhibiting caspase 3 activation.39,40 However, hyperphosphatemia inhibits this pathway by downregulating Gas6 as well as Ax1, and this effect is concurrent with calcium phosphate deposition and increased apoptosis.41 Thus, CNPs might have a similar effect in this regard, and these mechanisms could be a focus of future experiments.
Several possible mechanisms may account for the CNP-induced mineralization observed in this study. Firstly, since the deposits ranged up to several μm in size, which is larger than the individual CNPs that were added, and they were not present after 3 days of cell culture, CNPs that were associated with or taken up into the cells and not directed to lysosomes could have become nidi for mineral deposition. The applied CNPs did in fact have the capacity to accumulate environmental Ca as indicated by the 45Ca-binding experiments. Secondly, the applied particles could have initiated a series of cellular events that induced or accelerated conversion of the smooth muscle cells to osteogenic bone-forming cells. In vitro studies indicate that the osteogenic markers osteopontin, BMP-2, and runt-related transcription factor 2 (Runx2) are upregulated in vascular smooth muscle cells under hyperphosphatemic conditions. Notably, the initiating factor for this activation appears to be uptake of calcium phosphate nanocrystals that precipitate into the medium independently of cell activity – not soluble phosphate.14,42 Within the context of the present study, BMP-2 is particularly significant since it initiates a program that enhances the expression of multiple osteogenic transcription factors including Msx-2, Osterix, and runt-related transcription factor 2.43 In healthy smooth muscle cells, the osteoinductive properties of BMP-2 are neutralized by its formation of a complex with γ-carboxylated MGP, a potent mineralization inhibitor produced by smooth muscle cells, which itself is downregulated in vascular calcification.44 Thus, if CNPs induce BMP-2 upregulation, the increased levels might overwhelm the capacity of γ-carboxylated MGP for binding, which would favor mineralization.
Clinical implications
The present findings are consistent with the concept that protein/mineral complexes, CNPs in particular, influence atherosclerotic disease processes, and they are in accord with our previous study in which CNPs injected intravenously into rabbits induced calcification and occlusion at sites of arterial injury in vivo.45 Taken together, these results suggest that, under certain circumstances, CNPs are pathological agents. Apoptotic bodies are efficient nucleators of calcium phosphate30 and have been strongly linked to cardiovascular disease. Increased density of apoptotic bodies is a characteristic feature of arteries of patients on hemodialysis with end-stage renal failure,44 aortic plaque,46 and calcific aortic valves.4 Increased apoptosis localized to shoulder regions of arterial plaque may also contribute to plaque instability by thinning of the fibrous cap.47,48
Under physiological conditions, CNPs function to remove excess Ca and P from soft tissues by entering the circulation, which is followed by clearance by the reticuloendothelial system via scavenger receptor-A-mediated endocytosis, primarily in the liver and spleen.49 In diseases characterized by hyperphosphatemia, their formation increases and clearance mechanisms could be overwhelmed, favoring mineral deposition within the vascular extracellular matrix. Consistent with this concept are our previous studies, which demonstrated that CNPs were detectable in calcific but not healthy human arteries, and that they could be propagated in vitro from calcific vessel isolates, provided sufficient P was available.24,25
Effects of CNPs are likely to be modulated partly by their protein components. In vitro studies show that CNPs are pleomorphic, ie, their composition changes reflective of the constituents within their local environment.20 Thus, the proteinaceous makeup of the CNPs would be expected to vary in vivo depending upon the relative availability of specific mineral-binding proteins in the extracellular milieu, which would influence pathological events such as apoptosis. For example, levels of fetuin-A and MGP in the circulation are altered in chronic kidney disease and diabetes,29,50,51 which would likely affect the composition of circulating CNPs and, ultimately, contribute to vascular mineral deposition. Furthermore, the proportion and amount of fetuin-A complexed with mineral relative to free fetuin-A appears to be dynamic, since the complexes are undetectable in normal human serum but present in serum from patients with chronic kidney disease, and they increase in quantity in parallel with disease severity.29
A possible contribution for CNPs in progression of atherosclerosis should be viewed within the context of other similar-sized entities, such as smooth-muscle-derived membrane-bound matrix vesicles, which share certain functional features in promoting calcification. Both are localized to vascular plaque in vivo and increase in response to pathological elevations in extracellular mineral.16,24,25,52,53 Smooth-muscle-derived matrix vesicles may also serve as nucleation foci for Ca deposition.6,53 However, it is yet unknown whether CNPs act in combination with matrix vesicles to promote calcification. Additionally, membrane-bound microvesicles, primarily shed from platelets, are present in the circulation, and microvesicles of leukocyte, endothelial, and smooth muscle origin accumulate within atherosclerotic plaques.9,52 Evidence that these microvesicles participate directly in vascular calcification are lacking; however, circulating endothelial microvesicles impair endothelial function,54,55 and plaque microvesicles, which are more concentrated than those in the circulation, are more thrombogenic,9 thus contributing to atherosclerosis through proinflammatory and procoagulant mechanisms.
Improved methodologies are needed for isolation and analysis of protein/mineral complexes, such as CNPs used in this study, in order to more fully understand their character and to provide uniformity in classification. Since these complexes have the capacity to induce osteogene expression and apoptosis – two processes which precede calcification within the vasculature – the ability to identify and quantitate possible diagnostic markers of cardiovascular disease, such as using CNPs isolated from serum, would be beneficial.
Conclusion
CNPs are internalized by vascular smooth muscle cells in culture, resulting in accumulation of apoptotic bodies at sites correspondent with extracellular matrix mineralization. Because apoptotic bodies are localized to sites of calcification in the vasculature, and apoptosis is an important regulator of calcification, CNPs may augment vascular calcification under circumstances in which normal physiological processes for CNP clearance from the local environment are disrupted.
Acknowledgments
This work was supported in part by a developmental research grant from the Department of Surgery, Mayo Clinic, NIH R21 Grant HL88988, and the John E Fetzer Memorial Trust. We thank Ryan L Barness, Mayo Clinic, Rochester, MN, USA for expert technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization – The top 10 causes of death. 2011; Fact Sheet N310. Available from: http://www.who.int/mediacentre/factsheets/fs310_2008.pdf. Accessed on 3 March 2014. | ||
Bobryshev YV, Lord RS, Warren BA. Calcified deposit formation in intimal thickenings of the human aorta. Atherosclerosis. 1995;118:9–21. | ||
Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization-regulating proteins in association with Monckeberg’s sclerosis: Evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999;100: 2168–2176. | ||
Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465; discussion 465–466. | ||
American College of Physicians. Laboratory Values. Philadelphia, PA: American College of Physicians; 2014. Available from: http://im2014.acponline.org/assets/public/normal-lab-values.pdf. Accessed on 3 March 2014. | ||
Reynolds JL, Skepper JN, McNair R, et al. Multifunctional roles for serum protein fetuin-a in inhibition of human vascular smooth muscle cell calcification. J Am Soc Nephrol. 2005;16:2920–2930. | ||
Shanahan CM, Weissberg PL, Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res. 1993;73:193–204. | ||
Liu ML, Scalia R, Mehta JL, Williams KJ. Cholesterol-induced membrane microvesicles as novel carriers of damage-associated molecular patterns: mechanisms of formation, action, and detoxification. Arterioscler Thromb Vac Biol. 2012;32:2113–2121. | ||
Leroyer AS, Isobe H, Leseche G, et al. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007;49:772–777. | ||
Coppinger JA, Cagney G, Toomey S, et al. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood. 2004;103:2096–2104. | ||
Heiss A, DuChesne A, Denecke B, et al. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J Biol Chem. 2003;278:13333–13341. | ||
Price PA, Lim JE. The inhibition of calcium phosphate precipitation by fetuin is accompanied by the formation of a fetuin-mineral complex. J Biol Chem. 2003;278:22144–22152. | ||
Schlieper G, Aretz A, Verberckmoes SC, et al. Ultrastructural analysis of vascular calcifications in uremia. J Am Soc Nephrol. 2010;21:689–696. | ||
Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422. | ||
Raoult D, Drancourt M, Azza S, et al. Nanobacteria are mineralo fetuin complexes. PLoS Pathog. 2008;4:e41. | ||
Hunter LW, Lieske JC, Tran NV, Miller VM. The association of matrix Gla protein isomers with calcification in capsules surrounding silicone breast implants. Biomaterials. 2011;32:8364–8373. | ||
Kajander EO, Ciftcioglu N. Nanobacteria: An alternative mechanism for pathogenic intra- and extracellular calcification and stone formation. Proc Natl Acad Sci U S A. 1998;95:8274–8279. | ||
Cisar JO, Xu DQ, Thompson J, Swaim W, Hu L, Kopecko DJ. An alternative interpretation of nanobacteria-induced biomineralization. Proc Natl Acad Sci U S A. 2000;97:11511–11515. | ||
Barba I, Villacorta E, Bratos-Perez MA, et al. Aortic Valve-derived calcifyng nanoparticles: no evidence of life. Rev Esp Cardiol. 2012;65: 813–818. | ||
Young JD, Martel J, Young D, et al. Characterization of granulations of calcium and apatite in serum as pleomorphic mineralo-protein complexes and as precursors of putative nanobacteria. PLoS One. 2009;4:e5421. | ||
Young JD, Martel J, Young L, Wu CY, Young A, Young D. Putative nanobacteria represent physiological remnants and culture by-products of normal calcium homeostasis. PLoS One. 2009;4:e4417. | ||
Price PA, Thomas GR, Pardini AW, Figueira WF, Caputo JM, Williamson MK. Discovery of a high molecular weight complex of calcium, phosphate, fetuin, and matrix gamma-carboxyglutamic acid protein in the serum of etidronate-treated rats. J Biol Chem. 2002;277:3926–3934. | ||
Shiekh FA, Charlesworth JA, Sung-Hoon K, et al. Proteomic evaluation of biological nanoparticles isolated from human kidney stones and calcified arteries. Acta Biomater. 2010;6:4065–4072. | ||
Miller VM, Rodgers G, Charlesworth JA, et al. Evidence of nanobacterial-like structures in human calcified arteries and cardiac valves. Am J Physiol Heart Circ Physiol. 2004;287:H1115–H1124. | ||
Hunter LW, Shiekh FA, Pisimisis GT, et al. Key role of alkaline phosphatase in the development of human-derived nanoparticles in vitro. Acta Biomater. 2011;7:1339–1345. | ||
Bratos-Perez MA, Sanchez PL, Garcia de Cruz S, et al. Association between self-replicating calcifying nanoparticles and aortic stenosis: a possible link to valve calcification. Eur Heart J. 2008;29:371–376. | ||
Kumar V, Farell G, Yu S, et al. Cell biology of pathologic renal calcification: contribution of crystal transcytosis, cell-mediated calcification, and nanoparticles. J Investig Med. 2006;54:412–424. | ||
Martel J, Young D, Young A, et al. Comprehensive proteomic analysis of mineral nanoparticles derived from human body fluids and analyzed by liquid chromatography-tandem mass spectrometry. Anal Biochem. 2011;418:111–125. | ||
Hamano T, Matsui I, Mikami S, et al. Fetuin-mineral complex reflects extraosseous calcification stress in CKD. J Am Soc Nephrol. 2010;21:1998–2007. | ||
Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062. | ||
Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler Thromb Vac Biol. 1998;18:379–388. | ||
Linnes MP, Shiekh FA, Hunter LW, Miller VM, Lieske JC. Isolation, propagation, and analysis of biological nanoparticles. Methods Mol Biol. 2011;790:263–275. | ||
van der Pol E, Coumans F, Varga Z, Krumrey M, Nieuwland R. Innovation in detection of microparticles and exosomes. J Thromb Haemost. 2013;11 Suppl 1:36–45. | ||
Roberts GS, Yu S, Zeng Q, et al. Tunable pores for measuring concentrations of synthetic and biological nanoparticle dispersions. Biosens Bioelectron. 2012;31:17–25. | ||
Rzewuska-Lech E, Jayachandran M, Fitzpatrick LA, Miller VM. Differential effects of 17beta-estradiol and raloxifene on VSMC phenotype and expression of osteoblast-associated proteins. Am J Physiol Endocrinol Metab. 2005;289:E105–E112. | ||
Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–E17. | ||
Ewence AE, Bootman M, Roderick HL, et al. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008;103:e28–e34. | ||
Zhang S, Fantozzi I, Tigno DD, et al. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003;285:L740–L754. | ||
Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287:H1207–H1213. | ||
Son BK, Kozaki K, Iijima K, et al. Gas6/Axl-PI3K/Akt pathway plays a central role in the effect of statins on inorganic phosphate-induced calcification of vascular smooth muscle cells. Eur J Pharmacol. 2007;556:1–8. | ||
Son BK, Kozaki K, Iijima K, et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ Res. 2006;98:1024–1031. | ||
Pazar B, Ea HK, Narayan S, et al. Basic calcium phosphate crystals induce monocyte/macrophage IL-1beta secretion through the NLRP3 inflammasome in vitro. J Immunol. 2011;186:2495–2502. | ||
Matsubara T, Kida K, Yamaguchi A, et al. BMP2 regulates Osterix through Msx2 and Runx2 during osteoblast differentiation. J Biol Chem. 2008;283:29119–29125. | ||
Shroff RC, McNair R, Figg N, et al. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. | ||
Schwartz MK, Lieske JC, Hunter LW, Miller VM. Systemic injection of planktonic forms of mammalian-derived nanoparticles alters arterial response to injury in rabbits. Am J Physiol Heart Circ Physiol. 2009;296: 1434–1441. | ||
Koleganova N, Piecha G, Ritz E, et al. Arterial calcification in patients with chronic kidney disease. Nephrol Dial Transplant. 2009;24:2488–2496. | ||
Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998;97:2307–2315. | ||
Dhume AS, Soundararajan K, Hunter WJ 3rd, Agrawal DK. Comparison of vascular smooth muscle cell apoptosis and fibrous cap morphology in symptomatic and asymptomatic carotid artery disease. Ann Vasc Surg. 2003;17:1–8. | ||
Herrmann M, Schafer C, Heiss A, et al. Clearance of fetuin-A – containing calciprotein particles is mediated by scavenger receptor-A. Circ Res. 2012;111:575–584. | ||
Laughlin GA, Cummins KM, Wassel CL, Daniels LB, Ix JH. The association of fetuin-A with cardiovascular disease mortality in older community-dwelling adults: the Rancho Bernardo study. J Am Coll Cardiol. 2012;59:1688–1696. | ||
Cranenburg EC, Brandenburg VM, Vermeer C, et al. Uncarboxylated matrix Gla protein (ucMGP) is associated with coronary artery calcification in haemodialysis patients. Thromb Haemost. 2009;101:359–366. | ||
Bobryshev YV, Killingsworth MC, Huynh TG, Lord RS, Grabs AJ, Valenzuela SM. Are calcifying matrix vesicles in atherosclerotic lesions of cellular origin? Basic Res Cardiol. 2007;102:133–143. | ||
Reynolds JL, Joannides AJ, Skepper JN, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. | ||
Boulanger CM, Scoazec A, Ebrahimian T, et al. Circulating microparticles from patients with myocardial infarction cause endothelial dysfunction. Circulation. 2001;104:2649–2652. | ||
Amabile N, Guérin AP, Leroyer A, et al. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J Am Soc Nephrol. 2005;16:3381–3388. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.