Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
C5 Variant rs10985126 is Associated with Mortality in Patients with Symptomatic Coronary Artery Disease
Authors Henes JK, Groga-Bada P ![]() , Schaeffeler E, Winter S, Hack L
, Schaeffeler E, Winter S, Hack L ![]() , Zdanyte M, Mueller K
, Zdanyte M, Mueller K ![]() , Droppa M
, Droppa M ![]() , Stimpfle F, Gawaz M, Langer H, Schwab M, Geisler T, Rath D
, Stimpfle F, Gawaz M, Langer H, Schwab M, Geisler T, Rath D
Received 24 April 2021
Accepted for publication 22 June 2021
Published 21 July 2021 Volume 2021:14 Pages 893—903
DOI https://doi.org/10.2147/PGPM.S307827
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jessica Kristin Henes,1,* Patrick Groga-Bada,1,* Elke Schaeffeler,2,3 Stefan Winter,2,3 Luis Hack,1 Monika Zdanyte,1 Karin Mueller,1 Michal Droppa,1 Fabian Stimpfle,1 Meinrad Gawaz,1 Harald Langer,4 Matthias Schwab,2,3,5,6 Tobias Geisler,1 Dominik Rath1
1Department of Cardiology, University Hospital Tuebingen, Tuebingen, Germany; 2Dr. Margarete Fischer-Bosch Institute of Clinical Pharmacology, Stuttgart, Germany; 3University of Tuebingen, Tuebingen, Germany; 4Department of Cardiology, Angiology and Intensive Care, University Hospital Luebeck, Luebeck, Germany; 5Department of Clinical Pharmacology, University Hospital Tuebingen, Tuebingen, Germany; 6Department of Pharmacy and Biochemistry, University of Tuebingen, Tuebingen, Germany
*These authors contributed equally to this work
Correspondence: Tobias Geisler
Medizinische Klinik III, Department Cardiology and Cardiovascular Research, Otfried-Müller-Strasse 10, Tübingen, 72076 Tel +49 7071/29-8 36 88
Fax +49 7071/29-57 49
Email [email protected]
Background: Complement component 5a (C5a) is a highly potent anaphylatoxin with a variety of pro-inflammatory effects. C5a contributes to progression of atherosclerosis and inhibition of the receptor (C5aR) might offer a therapeutic strategy in this regard. Single nucleotide polymorphisms (SNPs) of the C5 gene may modify protein expression levels and therefore function of C5a and C5aR. This study aimed to examine associations between clinically relevant C5a SNPs and the prognosis of patients with symptomatic coronary artery disease (CAD). Furthermore, we sought to investigate the influence of C5 SNPs on C5aR platelet surface expression and circulating C5a levels.
Methods: C5 variants (rs25681, rs17611, rs17216529, rs12237774, rs41258306, and rs10985126) were analyzed in a consecutive cohort of 833 patients suffering from symptomatic coronary artery disease (CAD). Circulating C5a levels were determined in 116 patients whereas C5aR platelet surface expression was measured in 473 CAD patients. Endpoints included all-cause mortality, myocardial infarction (MI), and ischemic stroke (IS). Homozygous carriers (HC) of the minor allele (rs10985126) showed significantly higher all-cause mortality than major allele carriers. While we could not find significant associations between rs10985126 allele frequency and C5aR platelet surfazl ce expression, significantly elevated levels of circulating C5a were found in HC of the minor allele of the respective genotype. rs17216529 allele frequency correlated with the composite combined endpoint and bleeding events. However, since the number of HC of the minor allele of this genotype was low, we cannot draw a robust conclusion about the observed associations.
Conclusion: In this study, we provide evidence for the prognostic relevance of rs10985126 in CAD patients. C5 rs10985126 may serve as a prognostic biomarker for risk stratification in high-risk CAD patients and consequently promote tailored therapies.
Keywords: coronary artery disease, SNPs, complement C5, prognostic factors
Introduction
The complement system is part of the innate immunity and represents a first defense line against microbial pathogens.1 Consequently, complement components are critically involved in infectious (eg, meningitis) and autoimmune disease (eg, systemic lupus erythematosus).2 This system consists of over 20 proteins and may eliminate cells by forming a membrane attack complex in order to induce cytolysis or by opsonization for macrophages.3 Furthermore, complement component 5a (C5a), a cleavage product of C5, is a highly potent anaphylatoxin and possesses a variety of pro-inflammatory effects, facilitating chemotaxis of leukocytes,4 vasodilatation,5 vascular permeability6 and the release of histamine by mast cells.7 As inflammation contributes to the pathogenesis of atherosclerosis,8 several studies suggest C5a to be a promoter of atherosclerosis. CD88 (C5aR) is highly present in aortas of ApoE−/- mice, coinciding with atherosclerotic lesion development and inhibition of C5aR reduces lesion size.9 Moreover, in human coronary artery lesions, a higher expression of C5a was found in lipid-rich inflammatory plaques with cholesterol clefts and necrotic cells, while lower C5a expression was found in stable plaques.10 The complement system might be activated by cholesterol crystals (CC) in plaques, leading to the release of cytokines in whole blood, including TNFα and IL-1β.11,12 Consequently, TNFα mediates endothelial cell activation, which is essential for inflammatory cell recruitment to the lesion site.13 Since CC-induced cytokine production is reduced after inhibiting C5aR in whole blood, C5a may represent a key player in this process.11 Plaque rupture represents a serious complication in atherosclerotic vessels leading to eg, myocardial or cerebral infarction. C5 induced smooth muscle and endothelial cell degradation via apoptosis may be associated with atherosclerotic plaque disruption.14 In lipid-rich plaques, a co-localization of C5a and matrix metalloproteinases (MMPs) has been demonstrated. C5a induces the release of MMPs in vitro which might be of importance since MMPs contribute to plaque rupture.10
C5aR blockage may serve a therapeutic purpose to inhibit progress of atherosclerosis.9 Furthermore, circulating C5a correlates with future risk for adverse cardiovascular events in patients with advanced atherosclerosis.15 C5aR platelet surface exposure levels are elevated in patients suffering from coronary artery disease (CAD) when compared to healthy controls.16 Platelet C5aR is associated with platelet activation levels.16 C5 single nucleotide polymorphisms (SNPs) on chromosome 9q34.117 may alter protein expression levels and therefore function of C5a and eventually also C5aR. SNPs of the C5 gene have previously been investigated regarding a possible influence on common and/or inflammatory diseases. rs17611 has already been reported to be associated with cardiovascular outcome in patients with asymptomatic carotid atherosclerosis,18 as well as ischemic stroke19 or diabetic retinopathy.20 Furthermore, rs25681 and rs17611 are associated with the outcome of renal allografts.21 In diabetic retinopathy and renal allografts, rs10985126 and rs12237774 however failed to yield significant results.20,21 In addition, rs25681, rs17611, rs10985126 and rs12237774 are used to identify a protective haplotype against bronchial asthma.22 Significant associations could neither be demonstrated for rs17216529 and age-related macular degeneration23 nor rs41258306 in glioma patients, respectively.24 Hence, this study aimed to examine (1) associations between clinically relevant and previously investigated C5a SNPs (rs25681, rs17611, rs17216529, rs12237774, rs41258306 and rs10985126) and the prognosis of patients with symptomatic CAD. Furthermore, we investigated (2) C5 SNPs’ association with both platelet surface expression of C5aR and circulating C5a.
Materials and Methods
Subjects
The study was conducted from November of 2011 until March of 2016. Clinically significant C5 SNPs were analyzed in 833 consecutive CAD patients (chronic coronary syndrome (CCS) and acute coronary syndrome (ACS)) that were admitted to the department of cardiology of the university of Tuebingen, Germany. CAD was diagnosed in case of a stenosis greater than 50% in at least one main coronary artery.25 CCS and ACS were defined according to current guidelines.26,27
Patients suffered from a significant amount of cardiovascular risk factors, including arterial hypertension (83%), diabetes mellitus type 2 (33%), hyperlipidemia (58%) and smoking (41%). The average body mass index (BMI) was 27.8 kg/m2. Harmful use of alcohol was registered in 3% of the overall cohort. Medication on admission was distributed as follows: Acetylsalicylic acid (55%), Clopidogrel (12%), Prasugrel (2%), Ticagrelor (2%), Oral anticoagulants (8%), ACE-inhibitors (43%), Angiotensin-II-receptor-antagonists (18%), Ca-channel-blockers (20%), Beta-blockers (57%) and Statins (48%). Further information on the baseline characteristics of the overall cohort is depicted in Tables 1 and 2.
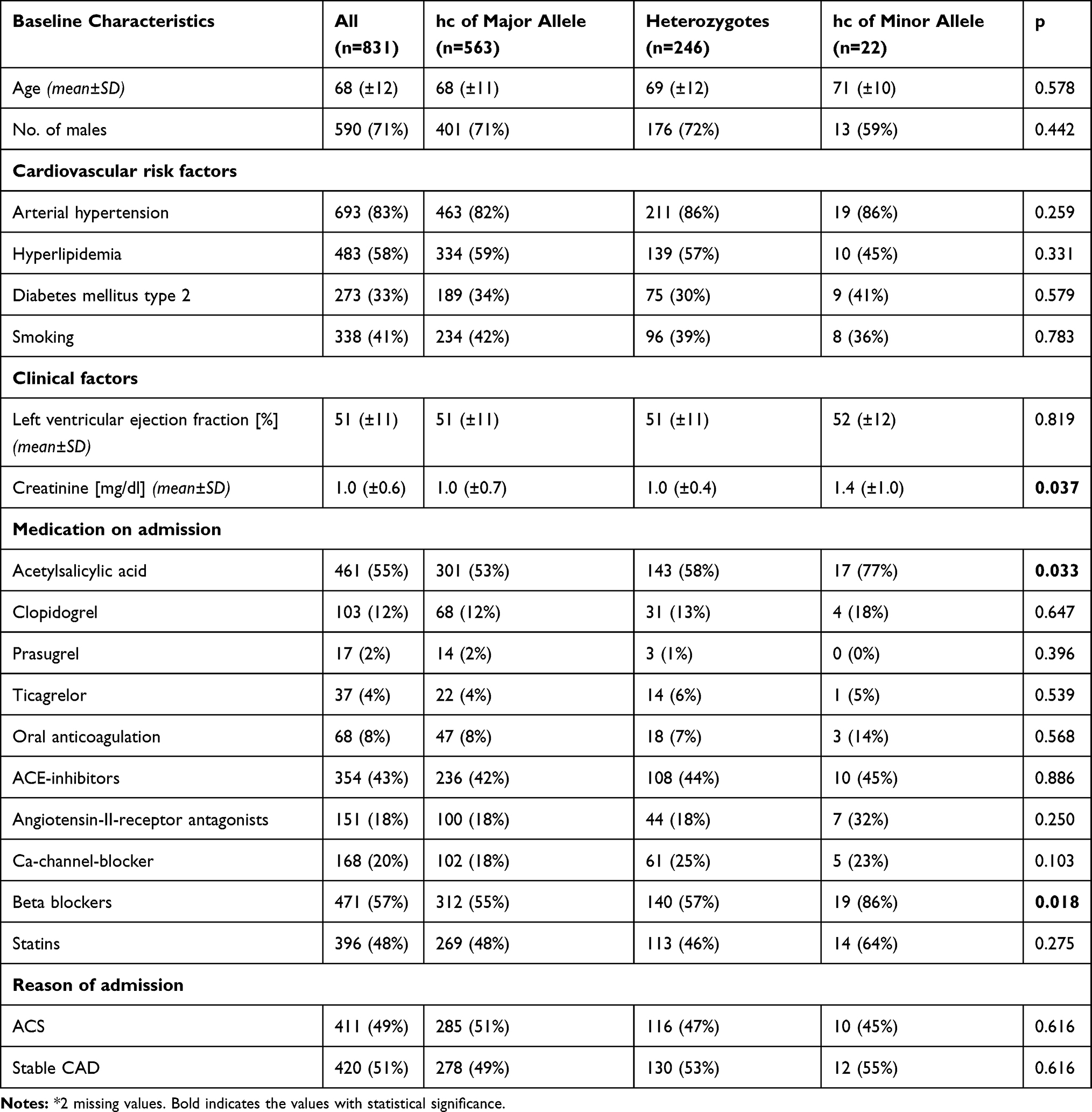 |
Table 1 Baseline Characteristics for the Overall Cohort Stratified According to Rs10985126 (n=831) * |
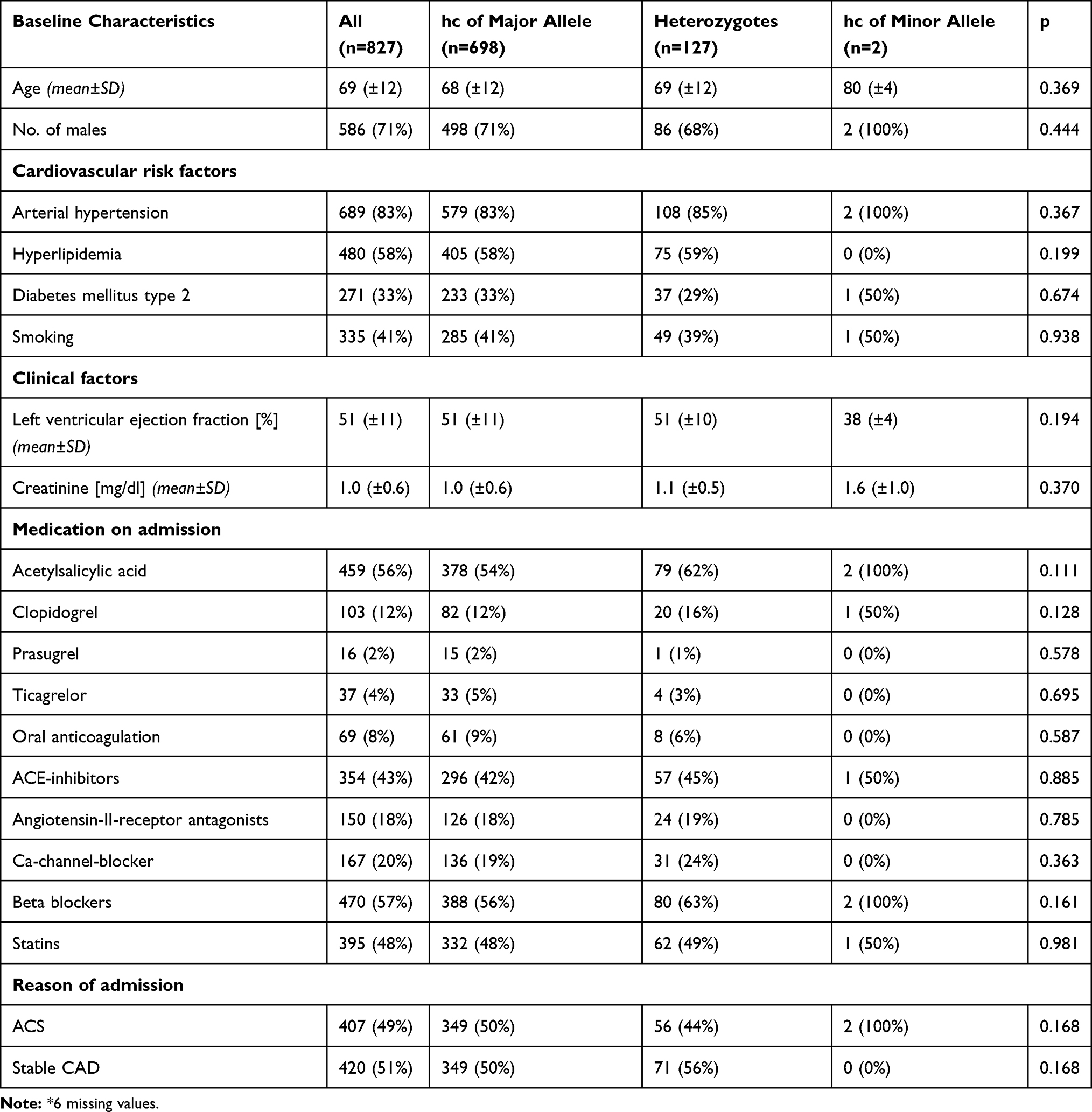 |
Table 2 Baseline Characteristics for the Overall Cohort Stratified According to Rs17216529 (n=827) * |
Ethics Statement
Hereby, we assure that for the current manuscript the following is fulfilled:
- This material is the authors’ own original work, which has not been previously published elsewhere.
- The paper is not currently being considered for publication elsewhere.
- The paper reflects the authors’ own research and analysis in a truthful and complete manner.
- The paper properly credits the meaningful contributions of co-authors and co-researchers.
- The results are appropriately placed in the context of prior and existing research.
- All sources used are properly disclosed.
- All authors have been personally and actively involved in substantial work leading to the paper and will take public responsibility for its content.
The violation of the Ethical Statement rules may result in severe consequences.
The ethics committee of the Eberhard Karls University Tuebingen approved this study (270/2011BO1 and 238/2018BO2). Finally, this investigation is in line with the Helsinki declaration as well as the good clinical practice guidelines.28–30
Inclusion and Exclusion Criteria
We enrolled all patients hospitalized due to either CCS (51%) or ACS (49%). Exclusion criteria were defined as age <18 years and absence of CAD. We confirm that the patients provided informed consent.
Genotyping of C5 Variants
Genomic DNA was isolated from ethylenediaminetetraacetic acid (EDTA) blood samples using the QIAmp® DNA Blood Mini Kit System (Qiagen, Hilden, Germany). C5 variants (rs25681, rs17611, rs17216529, rs12237774, rs41258306 and rs10985126) were selected according to PubMed records and reported associations with clinical parameters and functional consequences.18–24 Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) using the MassARRAY® Compact system (Sequenom, CA, USA) was applied to genotype for C5 variants. Details of assays and primers are provided upon request. Approximately 10% of samples within each assay served as quality control. Personnel investigating the study endpoints was blinded to the allele frequency of all participants during the whole genotyping process. Minor allele frequencies of the analyzed C5 variants are provided in Table 3.31,32
 |
Table 3 Allele Frequencies of the Investigated C5 Variants |
Enzyme-Linked Immunosorbent Assay (ELISA)
Circulating C5a levels were determined in plasma of 116 consecutive patients via a commercially available ELISA kit (R&D Systems).16 Arterial blood was collected during catheter procedure. Within 30 minutes of collection, ethylenediamine tetraacetic acid plasma (EDTA) probes were centrifuged for 15 minutes and subsequently aliquoted and stored at −80 degrees until analysis.
C5aR Platelet Surface Exposure Analyzed by Whole Blood Flow Cytometry (FACS)
C5aR platelet surface exposure was determined in 473 consecutive CAD patients. Platelets in whole blood were analyzed for C5aR surface exposure gating for the platelet specific marker CD42b. Blood collected in CPDA was diluted 1:50 with PBS (Gibco) and incubated with mouse monoclonal anti-human C5aR-FITC (AbD Serotec) and mouse anti-human CD42b PE (Becton Dickinson) antibodies or their respective isotype controls (R&D systems) for 30 min at room temperature. Thereafter, cells were fixed with 0.5% paraformaldehyde and analyzed by flow cytometry (FACS-Calibur flow cytometer Becton–Dickinson, Heidelberg, Germany).16
Follow-Up
Patients were followed-up over a period of 1080 days after study enrollment for clinical events consisting of all-cause mortality, myocardial infarction (MI), ischemic stroke (IS) and bleeding. Acute MI was defined as follows: detection of a rise and/or fall of cardiac troponin I with at least one of the following criteria: angina pectoris, significant ST-segment and/or T wave changes, new complete left bundle branch block and/or pathological Q waves in the electrocardiogram, loss of viable myocardium and/or new regional wall motion abnormality in cardiac imaging and/or a thrombus in coronary angiography.33 Ischemic stroke was defined as neurological dysfunction due to ischemic brain, spinal cord, or retinal cell death, based on pathological imaging, or other objective evidence of cerebral, spinal cord, or retinal focal ischemic injury in a defined vascular distribution.34 The Bleeding Academic Research Consortium (BARC) bleeding definition was used to detect clinically significant bleeding.35 The primary composite endpoint (CE) included first occurrence of all-cause mortality, MI, and/or IS. Secondary endpoints were defined as all-cause mortality, MI and bleeding. We aimed to follow-up all patients of the SNP cohort (n=833). Out of this cohort, we managed to track 758 patients (91.0%). Follow-up remained incomplete in 75 patients (9.0%). Follow-up was performed by telephone interview (patients and patients’ physicians) and/or review of patients’ files in case of readmission by independent examiners.
Statistical Analysis
All statistical analyses were conducted with SPSS version 25.0 (SPSS Inc., Chicago IL). Normally distributed and categorical values in the baseline characteristics are presented as mean ± standard deviation and percentage of total, respectively. Normally distributed data were compared using Welch’s t-test. Mean fluorescence intensities (MFIs) and circulating C5 values are presented as median and 25th/75th percentiles. We applied Kruskal–Wallis-H-tests with Dunn-Bonferroni correction for comparison of both circulating C5a and C5aR platelet surface expression levels between different genotypes. Incidence rates are given per 100-person years. Chi-squared tests were used to compare event rates between genotype groups. For survival data, Kaplan–Meier estimates and corresponding log rank tests were determined. Multiple Cox regression analysis was performed to analyse independent associations between C5 SNPs and the pre-defined endpoints after adjustment for epidemiological factors that might have influenced patients’ outcome. Bonferroni’s procedure was applied to adjust the obtained p-values for multiple testing, considering the 5 investigated C5 variants. All statistical tests were two-sided, and the significance level was set to 5%.
Results
Baseline characteristics for the overall cohort, stratified according to rs10985126 and rs17216529 allele distribution are shown in Tables 1 and 2.
Numbers and incident rates of events stratified according to C5 SNPs are provided in Table 4
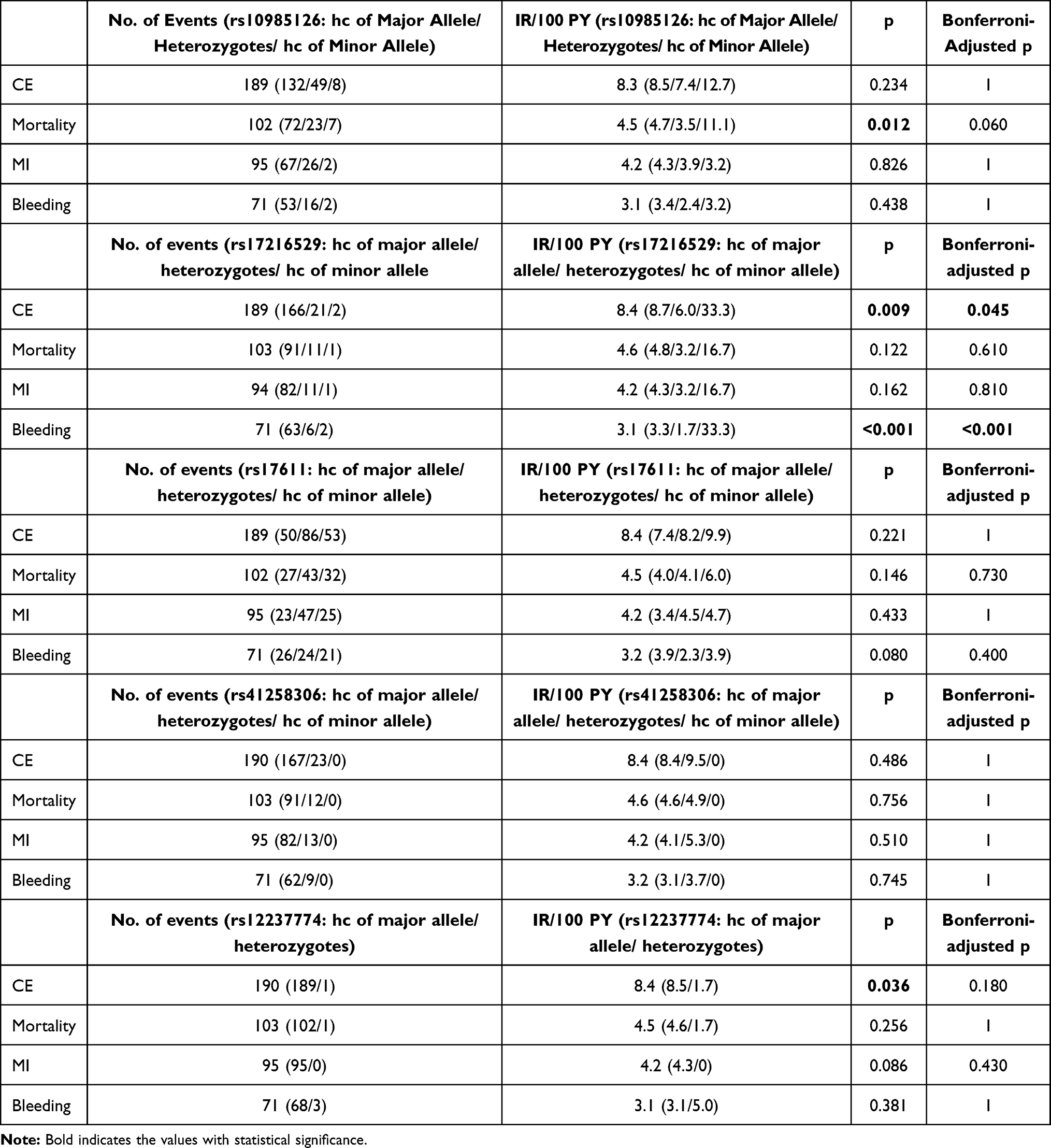 |
Table 4 Events and Incident Rates (IR) per 100-Person Years (PY) Within the Overall Cohort |
We observed significantly worse event-free survival in homozygous carriers (hc) of the minor allele (rs10985126) in comparison to heterozygotes and hc of the major allele in univariate analysis (log rank =0.008 for all-cause mortality, Bonferroni-adjusted p-value =0.040) (Figure 1). Furthermore, univariate analysis revealed significantly worse event-free survival in hc of the minor allele (rs17216529) in comparison to heterozygotes and hc of the major allele (log rank <0.001 for CE, Bonferroni-adjusted p-value <0.001). However, number of homozygous minor allele carriers was too low to conclude clinical implications for this SNP. rs10985126 was independently associated with all-cause mortality (p =0.003, Bonferroni-adjusted p-value =0.015) (Table 5).
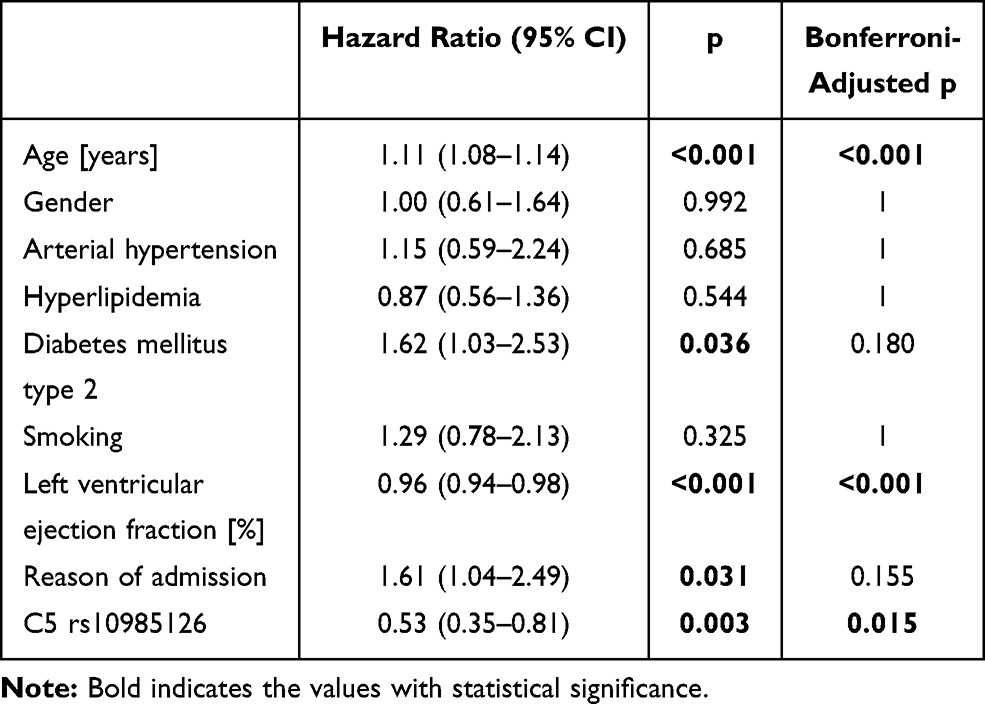 |
Table 5 Cox Regression Analysis with All-Cause Mortality as Dependent Variable, C5 Rs10985126 as Well as Epidemiological Factors (Age, Gender, Arterial Hypertension, Hyperlipidemia, Diabetes Mellitus Type 2, Smoking, Left Ventricular Ejection Fraction and Reason of Admission) as Covariates |
 |
Figure 1 Cumulative event-free survival for all-cause mortality stratified according to rs10985126 allele frequency. Red: Hc major allele; Green: Heterozygotes; Blue: HC minor allele. |
When we analyzed associations between rs10985126 and circulating C5a levels, we found that circulating C5a levels were significantly higher in hc of minor allele compared to heterozygotes and hc of major allele (median plasma C5a (pg/mL) 24715; 25th/75thpercentile 18504/NA, n=3 vs 20956; 25th/75th percentile 19834/24185, n=35 vs 19264; 25th/75th percentile 16813/21991, n=78; p=0.016). Post-hoc analysis revealed a significant difference between hc of major allele and heterozygotes (adjusted p=0.026) (Figure 2). There was no statistically significant association between rs10985126 allele frequency and C5aR platelet surface expression levels (median platelet C5aR (MFI) 13.8; 25th/75th percentile 9.6/17.8, n=15 vs 13.4; 25th/75th percentile 11.8/17.0, n=142 vs 13.8; 25th/75th percentile 11.7/17.3, n=316; p=0.753).
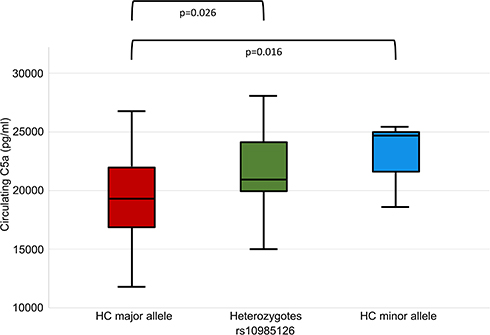 |
Figure 2 C5a plasma levels stratified according to rs10985126 allele frequency. |
Discussion
The major findings of the present study are (1) C5 rs10985126 is associated with mortality among patients suffering from symptomatic coronary artery disease. (2) Homozygous carriers of minor allele (rs10985126) show increased levels of circulating C5a when compared to major allele carriers.
As key elements of the complement cascade, C5a as well as C5aR offer a variety of pro-inflammatory effects. SNPs of the C5 gene might alter C5a and C5aR concentrations in human circulation and consequently affect inflammatory processes. Here, we could demonstrate an association between rs10985126 and all-cause death but failed to show an effect on myocardial infarction. Pre-existing evidence suggests that C5 rs17611 correlates with major adverse cardiovascular events in patients with asymptomatic atherosclerosis of the carotid arteries and that the allele at risk (rs17611) shows increased levels of circulating C5a.18 In the current study, hc of minor allele (rs10985126) display increased levels of circulating C5a and show increased mortality. C5 rs2269067, which is linked to C5 rs10985126 (r2≥0.86), is associated with proliferative diabetic retinopathy and elevated expression of C5a as well as production of IL-6.36 Interestingly, inhibition of IL-6 reduces C5aR expression in patients with NSTEMI correlating with attenuated troponin T release.37 Previously, a large number of SNPs including rs10985126 was used to draft a genetic model to estimate the risk for rheumatoid arthritis.38 Since rheumatoid arthritis is considered to be a risk factor for cardiovascular diseases,39 identifying an association between SNPs of the C5 gene and rheumatoid arthritis would be an interesting attempt to find similarities between different inflammatory diseases and to further the understanding of the underlying pathways. In the univariate survival analysis, rs10985126 was associated with a significantly worse cumulative event-free survival for homozygous carriers of the minor allele when compared to major allele carriers. This effect remained statistically significant after adjustment for multiple testing. When we applied Cox-regression analysis, we could demonstrate independent associations for rs10985126 on all-cause mortality after adjustment for epidemiological factors and multiple testing. In the current study neither rs17611 nor rs12237774 nor rs41258306 were significantly associated with prognosis. rs17216529 was associated with the combined endpoint and bleeding events. However, the number of hc of the minor allele was too low to draw a robust conclusion on these findings. Hoke et al previously showed a significant association of rs17611 with cardiovascular outcome in patients with asymptomatic atherosclerosis.18 Conversely, we could not obtain significant results for rs17611 in patients with symptomatic CAD. These different results may result due to distinct clinical circumstances upon admission: While Hoke et al tracked patients with asymptomatic carotid atherosclerosis for the occurrence of a first major adverse cardiovascular event,18 the patients in our study already suffered from CAD on the day of study enrollment. Furthermore, rs17611 was found to be over-expressed in patients with ischemic stroke compared to healthy controls.40 These observations notwithstanding, there might be well-preserved functional differences between SNPs of the same gene. rs17611 represents a missense mutation according to the NCBI SNP database (https://www.ncbi.nlm.nih.gov/snp/), whereas rs10985126 is a synonymous mutation. Synonymous mutations do not alter the protein sequence due to the degeneration of the genetic code, yet these SNPs might also have functional implications, albeit these are hard to predict.41 However, the current results suggest a functional relevance for rs10985126 due to associations with all-cause mortality in patients with symptomatic CAD.
Prior observations suggest that C5aR mediates tissue damage after myocardial ischemia.42 Previously, we have demonstrated elevated platelet C5aR and C3aR surface exposure in CAD patients compared to healthy controls.16 Currently, we failed to establish any correlation between C5 rs10985126 with C5aR platelet surface expression levels. C5 rs10985126 was however significantly associated with circulating C5a levels. Higher concentrations of circulating C5a correlate with adverse outcomes in patients suffering from advanced atherosclerosis.15 Furthermore, increased serum levels of C5a and C3a are associated with late re-stenosis of drug eluting stents after percutaneous transluminal coronary angioplasty.43 C5 levels are elevated in patients with generalized subclinical atherosclerosis and considered as a novel biomarker for subclinical atherosclerosis.44 In addition to contributing directly to the emergence and progress of CAD, C5a and C5aR signaling might also influence cardiovascular risk factors, as it has already been described for obesity45 and arterial hypertension.46 Nevertheless, other components of the complement cascade have already been established as prognostic markers since an increased C3/C4-ratio in patients with ACS was an independent risk factor to predict another ACS in the observation period.47 To conclude, C5a and C5aR seem to be involved in the pathophysiology of CAD in a way that might eventually be influenced by SNPs of the C5 gene.
Future Implications
As other SNPs have already been associated to prognosis in patients with cardiovascular disease,48 C5 rs10985126 might also be considered as a possible prognostic marker in these patients. Multi-locus genetic risk scores (GRS) consist of various gene variants associated with cardiovascular disease. This approach might be of use for identifying patients at an increased risk for suffering thromboischemic and/or bleeding events independently from classical cardiovascular risk factors.49,50
Furthermore, development of C5a receptor agonists may serve as a treatment strategy for thromboinflammatory processes fostering CAD. Previously, the antibody eculizumab, which binds to and blocks proteolysis of C5, has been developed and approved for treatment of paroxysmal nocturnal hemoglobinuria by the US Food and Drug Administration.51 Biologicals for chronic inflammatory diseases are expensive, offer low bioavailability and metabolic instability and need for repeated injections. The development of effective small molecule antagonists for C5aR, including peptides and peptidomimetics, as well as non-peptidic small molecules as ligands, represents an attractive alternative. While effects have mainly been studied in rodents, these studies have shown significant immunoregulatory effects of C5aR antagonists in vivo, which may offer major implications for cardiovascular disease.52 Consequently, preclinical pharmacokinetics of C5aR1 antagonists PMX53 and PMX205 targeting cyclic peptide compounds that act in a pseudo-irreversible and insurmountable manner at nanomolar concentrations are currently investigated in mice.53
Strengths and Limitations
The current study offers several strengths including the prospective, consecutive cohort of CAD patients. Furthermore, we analyzed clearly defined, “hard” endpoints. Finally, we present a reasonably long follow-up period with an acceptable number of patients who were lost to follow-up.
Major limitations of the current study lie within the observational character and moderate sample size. Furthermore, circulating C5a as well as platelet C5aR levels were determined only at study inclusion and were not available for the complete SNP cohort. Moreover, the current study lacks a validation cohort.
Highlights
- C5 rs10985126 is associated with all-cause mortality among patients suffering from symptomatic coronary artery disease
- C5 rs10985126 influences circulating C5a levels
- C5 rs10985126 might serve as a novel biomarker for risk stratification in patients with coronary artery disease and consequently promote individualized therapies
Acknowledgment
We thank the German Ministry of Education and Research, the “Robert Bosch Stiftung” Stuttgart, the Deutsche Forschungsgemeinschaft (DFG) (Grant number BO3786/1-1 and SCHW858/1-1/2) and Project number 374031971 – TRR 240 and the Klinische Forschergruppe KFO274 (Grant number 2133-0-0)” Platelets-Basic Mechanisms and Translational Implications” as well as the Open Access Publishing Fund of the University of Tuebingen for supporting this study.
Disclosure
Dr Elke Schaeffeler report grants from DFG (grant number SCHW858/1-1/2), grants from Robert Bosch Stiftung, Stuttgart (Germany), during the conduct of the study; Dr Stefan Winter report grants from Robert Bosch Stiftung (Stuttgart, Germany), grants from DFG Germany (grant number SCHW858/1-2), during the conduct of the study; Professor Matthias Schwab report grants from DFG SCHW858/1-1/2, grants from Robert Bosch Stiftung, during the conduct of the study; grants from Green Cross WellBeing Co. Ltd., grants from Gilead Sciences Inc., grants from Agena Bioscience GmbH, grants from University Hospital Tuebingen, grants, non-financial support from Robert Bosch GmbH, outside the submitted work; and Pharmacogenetics and Genomics, Editor in Chief Drug Research, Editor in Chief Genome Medicine, Section Editor Honoraria for oral presentations at academically organised congresses and meetings, External Reviewer for Research Impact Fund Hong Kong, External Reviewer for EU Horizon 2020, External Reviewer for the Science Council of the Federal Government of Germany. Professor Tobias Geisler report grants from German Research Foundation (DFG), during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Zipfel PF. Complement and immune defense: from innate immunity to human diseases. Immunol Lett. 2009;126(1–2):1–7. doi:10.1016/j.imlet.2009.07.005
2. Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine. 1984;63(5):243–273. doi:10.1097/00005792-198409000-00001
3. Müller-Eberhard HJ. Molecular organization and function of the complement system. Annu Rev Biochem. 1988;57(1):321–347. doi:10.1146/annurev.bi.57.070188.001541
4. Marder SR, Chenoweth DE, Goldstein IM, Perez HD. Chemotactic responses of human peripheral blood monocytes to the complement-derived peptides C5a and C5a des Arg. J Immunol. 1985;134(5):3325–3331.
5. Schumacher WA, Fantone JC, Kunkel SE, Webb RC, Lucchesi BR. The anaphylatoxins C3a and C5a are vasodilators in the canine coronary vasculature in vitroand in vivo. Agents Actions. 1991;34(3):345–349. doi:10.1007/BF01988727
6. Williams TJ, Jose PJ. Mediation of increased vascular permeability after complement activation. Histamine-independent action of rabbit C5a. J Exp Med. 1981;153(1):136–153. doi:10.1084/jem.153.1.136
7. Johnson AR, Hugli TE, Müller-Eberhard HJ. Release of histamine from rat mast cells by the complement peptides C3a and C5a. Immunology. 1975;28(6):1067.
8. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105(9):1135–1143. doi:10.1161/hc0902.104353
9. Manthey HD, Thomas AC, Shiels IA, et al. Complement C5a inhibition reduces atherosclerosis in ApoE−/− mice. FASEB J. 2011;25(7):2447–2455. doi:10.1096/fj.10-174284
10. Speidl WS, Kastl SP, Hutter R, et al. The complement component C5a is present in human coronary lesions in vivo and induces the expression of MMP-1 and MMP-9 in human macrophages in vitro. FASEB J. 2011;25(1):35–44. doi:10.1096/fj.10-156083
11. Samstad EO, Niyonzima N, Nymo S, et al. Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release. J Immunol. 2014;192(6):2837–2845. doi:10.4049/jimmunol.1302484
12. Okusawa S, Yancey KB, van der Meer JW, et al. C5a stimulates secretion of tumor necrosis factor from human mononuclear cells in vitro. Comparison with secretion of interleukin 1 beta and interleukin 1 alpha. J Exp Med. 1988;168(1):443–448. doi:10.1084/jem.168.1.443
13. Nymo S, Niyonzima N, Espevik T, Mollnes TE. Cholesterol crystal-induced endothelial cell activation is complement-dependent and mediated by TNF. Immunobiology. 2014;219(10):786–792. doi:10.1016/j.imbio.2014.06.006
14. Wezel A, de Vries MR, Lagraauw HM, et al. Complement factor C5a induces atherosclerotic plaque disruptions. J Cell Mol Med. 2014;18(10):2020–2030. doi:10.1111/jcmm.12357
15. Speidl WS, Exner M, Amighi J, et al. Complement component C5a predicts future cardiovascular events in patients with advanced atherosclerosis. Eur Heart J. 2005;26(21):2294–2299. doi:10.1093/eurheartj/ehi339
16. Patzelt J, Mueller KA, Breuning S, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis. 2015;238(2):289–295. doi:10.1016/j.atherosclerosis.2014.12.002
17. Wetsel RA, Lemons RS, Le Beau MM, Barnum SR, Noack D, Tack BF. Molecular analysis of human complement component C5: localization of the structural gene to chromosome 9. Biochemistry. 1988;27(5):1474–1482. doi:10.1021/bi00405a012
18. Hoke M, Speidl W, Schillinger M, et al. Polymorphism of the complement 5 gene and cardiovascular outcome in patients with atherosclerosis. Eur J Clin Invest. 2012;42(9):921–926. doi:10.1111/j.1365-2362.2012.02669.x
19. Guo L, Zheng L, Guo X, Chang Y, Zhou X. Single-Nucleotide Polymorphism rs17611 of complement component 5 shows association with ischemic stroke in Northeast Chinese population. Genet Test Mol Biomarkers. 2016;20(12):766–770. doi:10.1089/gtmb.2016.0125
20. Yang MM, Wang J, Ren H, et al. Genetic Investigation of Complement Pathway Genes in Type 2 Diabetic Retinopathy: an Inflammatory Perspective. Mediators Inflamm. 2016;2016:1313027. doi:10.1155/2016/1313027
21. Jeong JC, Hwang YH, Kim H, et al. Association of complement 5 genetic polymorphism with renal allograft outcomes in Korea. Nephrol, Dial, Transpl. 2011;26(10):3378–3385. doi:10.1093/ndt/gfr025
22. Hasegawa K, Tamari M, Shao C, et al. Variations in the C3, C3a receptor, and C5 genes affect susceptibility to bronchial asthma. Hum Genet. 2004;115(4):295–301. doi:10.1007/s00439-004-1157-z
23. Chen Y, Zhao C, Li B, Sun X, Yang Z, Zhang K. Evaluation on the Association of the Variants in Complement Component 5 Gene and Age Related Macular Degeneration. Invest. Ophthalmol Vis Sci. 2009;50(13):3440.
24. Michaud DS, Siddiq A, Cox DG, et al. Mannose-binding lectin 2 gene and risk of adult glioma. PLoS One. 2013;8(4):e61117. doi:10.1371/journal.pone.0061117
25. Burggraf GW, Parker JO. Prognosis in coronary artery disease. Angiographic, hemodynamic, and clinical factors. Circulation. 1975;51(1):146–156. doi:10.1161/01.CIR.51.1.146
26. Knuuti J, Wijns W, Saraste A, et al. ESC Guidelines for the diagnosis and management of chronic coronary syndromes: the Task Force for the diagnosis and management of chronic coronary syndromes of the European Society of Cardiology (ESC). Eur Heart J. 2019;41(3):407–477.
27. Thygesen K, Alpert JS, Jaffe AS, et al. Fourth universal definition of myocardial infarction. Eur Heart J. 2018;40(3):237–269.
28. World Medical Association Declaration of Helsinki. Recommendations guiding physicians in biomedical research involving human subjects. Cardiovasc Res. 1997;35(1):2–3.
29. ICH Harmonised Tripartite Guideline. Guideline for good clinical practice. J Postgrad Med. 2001;47(2):121–130.
30. Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the member states relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Med Etika Bioet. 2002;9(1–2):12–19.
31. Rath D, Schaeffeler E, Winter S. SDF1 Polymorphisms Influence Outcome in Patients with Symptomatic Cardiovascular Disease. PLoS One. 2016;11(9):e0161933.
32. Rath D, Schaeffeler E, Winter S, et al. GPla Polymorphisms Are Associated with Outcomes in Patients at High Cardiovascular Risk. Front Cardiovasc Med. 2017;4:52. doi:10.3389/fcvm.2017.00052
33. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD. Third universal definition of myocardial infarction. Eur Heart J. 2012;33(20):2551–2567. doi:10.1093/eurheartj/ehs184
34. Sacco RL, Kasner SE, Broderick JP, American Heart Association Stroke Council, Council on Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular and Stroke Nursing; Council on Epidemiology and Prevention; Council on Peripheral Vascular Disease; Council on Nutrition, Physical Activity and Metabolism. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(7):2064–2089. doi:10.1161/STR.0b013e318296aeca
35. Mehran R, Rao SV, Bhatt DL. Standardized bleeding definitions for cardiovascular clinical trials: a consensus report from the Bleeding Academic Research Consortium. Circulation. 2011;123(23):2736–2747. doi:10.1161/CIRCULATIONAHA.110.009449
36. Xu D, Yi H, Yu S. Association of Complement C5 Gene Polymorphisms with Proliferative Diabetic Retinopathy of Type 2 Diabetes in a Chinese Han Population. PLoS One. 2016;11(3):e0149704. doi:10.1371/journal.pone.0149704
37. Orrem HL, Nilsson PH, Pischke SE. IL-6 Receptor Inhibition by Tocilizumab Attenuated Expression of C5a Receptor 1 and 2 in Non-ST-Elevation Myocardial Infarction. Front Immunol. 2018;9:2035. doi:10.3389/fimmu.2018.02035
38. Chang M, Rowland CM, Garcia VE, et al. A large-scale rheumatoid arthritis genetic study identifies association at chromosome 9q33.2. PLoS Genet. 2008;4(6):e1000107. doi:10.1371/journal.pgen.1000107
39. Solomon DH, Karlson EW, Rimm EB, et al. Cardiovascular morbidity and mortality in women diagnosed with rheumatoid arthritis. Circulation. 2003;107(9):1303–1307. doi:10.1161/01.CIR.0000054612.26458.B2
40. Greisenegger S, Zehetmayer S, Bauer P, et al. Polymorphisms in inflammatory genes and the risk of ischemic stroke and transient ischemic attack: results of a multilocus genotyping assay. Clin Chem. 2009;55(1):134–138. doi:10.1373/clinchem.2008.112151
41. Zeng Z, Bromberg Y. Predicting Functional Effects of Synonymous Variants: a Systematic Review and Perspectives. Front Genet. 2019;10:914. doi:10.3389/fgene.2019.00914
42. Mueller M, Herzog C, Larmann J, et al. The receptor for activated complement factor 5 (C5aR) conveys myocardial ischemic damage by mediating neutrophil transmigration. Immunobiology. 2013;218(9):1131–1138. doi:10.1016/j.imbio.2013.03.006
43. Speidl WS, Katsaros KM, Kastl SP, et al. Coronary late lumen loss of drug eluting stents is associated with increased serum levels of the complement components C3a and C5a. Atherosclerosis. 2010;208(1):285–289. doi:10.1016/j.atherosclerosis.2009.07.030
44. Martínez-López D, Roldan-Montero R, García-Marqués F. Complement C5 Protein as a Marker of Subclinical Atherosclerosis. J Am Coll Cardiol. 2020;75(16):1926–1941. doi:10.1016/j.jacc.2020.02.058
45. Lim J, Iyer A, Suen JY, et al. C5aR and C3aR antagonists each inhibit diet‐induced obesity, metabolic dysfunction, and adipocyte and macrophage signaling. FASEB J. 2013;27(2):822–831. doi:10.1096/fj.12-220582
46. Ruan CC, Gao PJ. Role of complement-related inflammation and vascular dysfunction in hypertension. Hypertension. 2019;73(5):965–971. doi:10.1161/HYPERTENSIONAHA.118.11210
47. Palikhe A, Sinisalo J, Seppänen M, et al. Serum complement C3/C4 ratio, a novel marker for recurrent cardiovascular events. Am J Cardiol. 2007;99(7):890–895. doi:10.1016/j.amjcard.2006.11.034
48. Casas JP, Cooper J, Miller GJ, Hingorani AD, Humphries SE. Investigating the genetic determinants of cardiovascular disease using candidate genes and meta-analysis of association studies. Ann Hum Genet. 2006;70(2):145–169. doi:10.1111/j.1469-1809.2005.00241.x
49. Bare LA, Morrison AC, Rowland CM. Five common gene variants identify elevated genetic risk for coronary heart disease. Gene Med. 2007;9(10):682–689. doi:10.1097/GIM.0b013e318156fb62
50. Lluis-Ganella C, Subirana I, Lucas G. Assessment of the value of a genetic risk score in improving the estimation of coronary risk. Atherosclerosis. 2012;222(2):456–463. doi:10.1016/j.atherosclerosis.2012.03.024
51. Zareba KM. Eculizumab: a novel therapy for paroxysmal nocturnal hemoglobinuria. Drugs Today (Barc). 2007;43(8):539–546. doi:10.1358/dot.2007.43.8.1130446
52. Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152(4):429–448. doi:10.1038/sj.bjp.0707332
53. Kumar V, Lee JD, Clark RJ, Noakes PG, Taylor SM, Woodruff TM. Preclinical Pharmacokinetics of Complement C5a Receptor Antagonists PMX53 and PMX205 in Mice. ACS Omega. 2020;5(5):2345–2354. doi:10.1021/acsomega.9b03735
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.