Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 12
Bronchodilator efficacy of extrafine glycopyrronium bromide: the Glyco 2 study
Authors Singh D, Scuri M, Collarini S, Vezzoli S, Mariotti F, Muraro A, Acerbi D
Received 22 March 2017
Accepted for publication 20 May 2017
Published 7 July 2017 Volume 2017:12 Pages 2001—2014
DOI https://doi.org/10.2147/COPD.S137659
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Richard Russell
Dave Singh,1 Mario Scuri,2 Sara Collarini,2 Stefano Vezzoli,2 Fabrizia Mariotti,2 Annamaria Muraro,2 Daniela Acerbi2
1Medicines Evaluation, University Of Manchester, University Hospital of South Manchester, Manchester, UK; 2Global Clinical Development, Chiesi Farmaceutici SpA, Parma, Italy
Abstract: An extrafine formulation of the long-acting muscarinic antagonist glycopyrronium bromide (GB) is in development for chronic obstructive pulmonary disease (COPD), in combination with beclometasone dipropionate and formoterol fumarate – a “fixed triple”. This two-part study was randomized, double blind, placebo controlled in patients with moderate-to-severe COPD: Part 1: single-dose escalation, GB 12.5, 25, 50, 100 or 200 µg versus placebo; Part 2: repeat-dose (7-day), four-period crossover, GB 12.5, 25 or 50 µg twice daily (BID) versus placebo, with an open-label extension in which all patients received tiotropium 18 µg once daily. On the morning of Day 8 in all five periods, patients also received formoterol 12 µg. In study Part 1, 27 patients were recruited. All GB doses significantly increased from baseline forced expiratory volume in 1 second (FEV1) area under the curve (AUC0–12h) and peak FEV1, with a trend toward greater efficacy with higher GB dose. All adverse events were mild–moderate in severity, with a lower incidence with GB than placebo and no evidence of a dose–response relationship. In study Part 2, of 38 patients recruited, 34 completed the study. Adjusted mean differences from placebo in 12 h trough FEV1 on Day 7 (primary) were 115, 142 and 136 mL for GB 12.5, 25 and 50 µg BID, respectively (all P<0.001). GB 25 and 50 µg BID were superior (P<0.05) to GB 12.5 µg BID for pre-dose morning FEV1 on Day 8. For this endpoint, GB 25 and 50 µg BID were also superior to tiotropium. Compared with Day 7, addition of formoterol significantly increased Day 8 FEV1 peak and AUC0–12h with all GB doses and placebo (all P<0.001). All adverse events were mild–moderate in severity and there was no indication of a dose-related relationship. This study provides initial evidence on bronchodilation, safety and pharmacokinetics of extrafine GB BID. Overall, the results suggest that GB 25 µg BID is the optimal dose in patients with COPD.
Keywords: glycopyrronium, COPD, bronchodilator, dose-ranging
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by persistent airflow limitation, with long-acting bronchodilators (muscarinic antagonists [LAMAs] or β2-agonists [LABAs]) central to maintenance treatment, either alone or in combination.1 LAMAs such as tiotropium have demonstrated efficacy in a range of measures, including lung function, exacerbations, dyspnea and health status.2–7 “Open” triple therapy, consisting of a LAMA in one inhaler and an inhaled corticosteroid (ICS) plus a LABA in a second, is often used in clinical practice, and is recommended as a treatment option for patients who have the highest level of symptoms and risk of future exacerbations.1 Clinical trials have shown a benefit for the addition of a LAMA to an ICS/LABA; in particular, the addition of the LAMA glycopyrronium to a LABA/ICS significantly improved lung function, health status and rescue medication use.8
An extrafine formulation of glycopyrronium bromide (GB) is in development for management of COPD, to be used in a pressurized metered dose inhaler (pMDI) in combination with the ICS beclometasone dipropionate (BDP) and the LABA formoterol fumarate (FF) – a “fixed triple”. This combination has demonstrated improvements in lung function and exacerbation rates compared with BDP/FF9 and compared with tiotropium.10 The Phase II study described here was conducted as part of the development program of the BDP/FF/GB fixed triple. The overall aim of the study was to assess the bronchodilator efficacy of extrafine GB, to identify the optimal dose to be subsequently combined with BDP/FF. In addition, the effects of GB were compared to those of LAMA, tiotropium, and the additive bronchodilator effect of using formoterol with GB was also evaluated.
Methods
Participants
The study recruited males and females, 40–75 years of age, with moderate to severe COPD, specifically with post-bronchodilator forced expiratory volume in 1 second (FEV1) 40%–80% predicted and FEV1/forced vital capacity ≤0.70. Eligible patients had reversibility ≥100 mL within 30–45 min after inhalation of ipratropium 80 μg and were current or ex-smokers with a smoking history of ≥10 pack-years. Exclusion criteria included: other significant lung diseases (such as asthma); a history of chronic or seasonal allergy; blood eosinophils >0.6×109/L; clinically relevant findings on physical examination or from laboratory or electrocardiogram (ECG) evaluations; or a respiratory tract infection 4 weeks prior to entry or hospitalization 6 weeks prior to entry. In addition, patients were excluded if they had significant 24 h Holter ECG abnormalities at screening (described in the Supplementary materials). All patients signed written informed consent for this study, and the study was approved by the local research ethics committee (North West – Greater Manchester Central, UK; Ref: 10/H1008/47). All the inclusion and exclusion criteria are given in the Supplementary materials. Patients who completed study Part 1 were eligible for study Part 2, provided they still met the inclusion and exclusion criteria.
Trial design
The two-part study was randomized, double blind and placebo controlled (ClinicalTrials.gov registration: NCT01176903). Part 1 was a two-cohort, single-dose escalation design, with the dose escalated only if the safety and tolerability profile of the lower dose was acceptable. Cohort A was randomized to receive GB 12.5, 50 or 200 μg, via pMDI, or matching placebo; there were three treatment periods separated by washouts of ≥7 days. Cohort B was randomized to receive GB 25 or 100 μg or placebo in two treatment periods. Treatment sequences were defined to ensure a 10:2 ratio between GB and placebo in each period.
Part 2, which started only after a positive evaluation of safety data from Part 1, was repeat-dose, four-period crossover design, followed by an open-label extension period. In the first four (core) periods, patients were randomized to receive GB 12.5, 25 or 50 μg, or matching placebo, twice daily (BID) for 7 days (total daily doses of 25, 50 and 100 μg), with a further dose on the morning of Day 8. In the fifth (extension) period, all patients received tiotropium 18 μg once daily (OD) for 8 days, open-label via the manufacturer’s single-dose dry powder inhaler. On the morning of Day 8 in all five treatment periods, patients also received a single dose of formoterol 12 μg via pMDI. Treatment periods were separated by a 7±2 day washout.
Patients were assigned to treatment sequences according to randomization lists prepared by the study sponsor. Patients, investigators and site staff, monitors, and the sponsor’s clinical team were all blinded to GB and placebo treatment.
Visit schedule
After completing the screening visit, eligible patients entered a 7-day run-in, when LAMAs and LABAs were discontinued; LAMAs were replaced with ipratropium four times a day (withheld from 24 h before each study visit until the end of the treatment period) and LABAs were replaced with salbutamol as needed (prn). All patients were permitted salbutamol prn during the treatment periods, with a 6 h washout prior to any pulmonary function test. ICS use was permitted throughout the study, provided the dose had been stable for ≥4 weeks prior to entry (patients taking ICS/LABA fixed combination were prescribed ICS at an equivalent dose plus salbutamol prn).
In study Part 1, lung function was assessed pre-dose, and 15, 30 and 45 min and 1, 2, 4, 6, 8, 10, 12, 12.5 and 24 h post-dose. In study Part 2, lung function was assessed pre-dose on Day 1, and pre-dose and 15, 30 and 45 min and 1, 2, 4, 6, 8, 10 and 12 h post-dose on days 7 and 8. Blood samples were collected for pharmacokinetic (PK) analysis on days 1 and 7 of study Part 2 at pre-dose and at 5, 10, 15 and 30 min and 1, 2, 4, 6, 8 and 12 h post-dose, and pre-dose on Day 6. Urine samples were collected pre-dose on Day 1, and at 0–4 h and 4–12 h on days 1 and 7.
Objectives and outcomes
Study Part 1
The primary objective was to assess the safety and tolerability of single-dose GB at five dose levels. Secondary objectives were to assess the bronchodilator effects of GB, compared with placebo, in terms of FEV1 area under the curve from 0 to 12 h (AUC0–12h) normalized by time, and peak FEV1.
Study Part 2
The primary objective was to assess the bronchodilator efficacy of GB compared to placebo, in terms of 12 h trough FEV1 on Day 7 (using data collected at 12 h after morning administration of medication on Day 7).
Secondary objectives included: evaluation of the bronchodilator effects of GB versus placebo on Day 7, in terms of FEV1 AUC0–12h normalized by time, and peak FEV1, and comparison of GB with placebo and tiotropium, with respect to pre-dose morning FEV1 on Day 8. Safety and tolerability was assessed by adverse events (AEs), ECGs (12-lead and 24 h Holter), vital signs and laboratory tests. The PK profile of GB was analyzed after single (on Day 1) and repeated administration (ie, at steady state, assessed on Day 7).
An exploratory objective was to evaluate the additive effect of single-dose formoterol over GB; the difference between 8 and 7 days (ie, with and without formoterol) in peak FEV1, FEV1 AUC0–12h normalized by time and 12 h trough FEV1 was determined.
Sample size
There was no formal power calculation for study Part 1, as this was primarily performed for safety evaluation. A sample size of 24 patients (12 patients per cohort: 10 on GB and 2 on placebo in each treatment period) was considered sufficient for dose escalation. Dropouts were replaced to have at least 11 evaluable patients per cohort.
For the primary endpoint of study Part 2 (12 h trough FEV1 on Day 7), 32 evaluable patients provided 80% power to detect a mean difference of 120 mL between at least one dose of GB and placebo at a two-sided significance level of 0.05, adjusting for multiplicity using Hommel’s method and assuming a within-subject standard deviation (SD) of 140 mL. Assuming a nonevaluable rate of 20%, a total of 40 patients would need to be randomized. Patients prematurely withdrawing were not replaced.
Statistical methods
In study Part 1, FEV1 AUC0–12h (normalized by time) and peak FEV1 were analyzed descriptively, and the mean change from baseline and 95% confidence intervals are presented. In study Part 2, trough FEV1 at 12 h post-dose on Day 7 was analyzed using an analysis of covariance (ANCOVA) model (core periods only), with treatment, period and patient as fixed effects and baseline FEV1 (pre-dose on Day 1) as the covariate. The P-values of the comparisons between each dose of GB and placebo were adjusted for multiplicity using Hommel’s method. FEV1 AUC0–12h (normalized by time) and peak FEV1 on Day 7 were analyzed using the same ANCOVA model as for the primary variable, but without adjustment for multiplicity. Pre-dose morning FEV1 on Day 8 was submitted to an ANCOVA model with treatment and patient as fixed effects and baseline FEV1 as the covariate. For the exploratory objective, data on Day 8 (ie, after administration of formoterol) were compared with the respective values on Day 7 using paired t-tests. PK parameters were calculated using standard noncompartmental methods.
AEs, serious AEs (SAEs) and AEs leading to study withdrawal were presented descriptively; absolute values and mean changes from baseline were calculated for other safety parameters, with abnormal findings summarized by treatment. In both parts of the study, all randomized patients were included in the efficacy, safety and PK analyses.
Results
Participants
In study Part 1, 66 patients were screened, and 27 were recruited (23 were initially randomized; 4 prematurely withdrew and were replaced), as shown in Figure 1A. In study Part 2, 145 patients were screened and 38 were randomized, with 34 completing it (Figure 1B). Baseline demographics and disease characteristics of the randomized patients are shown in Table 1.
 | Figure 1A Patient flow through Part 1 of the study. |
 | Figure 1B Patient flow through Part 2 of the study. |
 | Table 1 Patient baseline demographics and disease characteristics |
Efficacy
Study Part 1
All GB doses were associated with statistically significant increases from baseline in FEV1 AUC0–12h (Figure 2) and peak FEV1 (Figure 3). The largest differences compared to placebo were observed with the three highest GB doses (50, 100 and 200 μg); both the FEV1 AUC0–12h and peak FEV1 data suggest the plateau of the dose response curve had been reached at these doses.
 | Figure 2 Study Part 1: Change from baseline in FEV1 AUC0–12h after a single dose. |
 | Figure 3 Study Part 1: Change from baseline in peak FEV1 after a single dose. |
Study Part 2
Primary endpoint
All three GB doses resulted in statistically significant increases versus placebo in terms of 12 h trough FEV1 on Day 7, with no significant differences between GB doses (Figure 4). The adjusted mean (95% confidence interval) differences compared to placebo were 115 (76, 155), 142 (102, 182) and 136 (96, 176) mL for the 12.5, 25 and 50 μg BID doses, respectively.
 | Figure 4 Study Part 2: Adjusted mean 12 h trough FEV1 on Day 7. |
Secondary endpoints
All three GB doses were associated with statistically significant improvements versus placebo in terms of FEV1 AUC0–12h normalized by time on Day 7, with no significant difference between GB doses (Figure 5). Mean changes from baseline in peak FEV1 values on Day 7 were 292, 320 and 332 mL for GB 12.5, 25 and 50 μg BID, respectively, compared with 83 mL for placebo (P<0.001 for all GB doses versus placebo). The difference between GB 12.5 and 25 μg BID was significant (P=0.034).
 | Figure 5 Study Part 2: Adjusted mean FEV1 AUC0–12h on Day 7. |
Comparisons with tiotropium
For pre-dose morning FEV1 on Day 8, all three GB doses were associated with statistically significant increases versus placebo, with the two highest GB doses being statistically superior to both tiotropium and GB 12.5 μg BID (Figure 6). The adjusted mean differences versus tiotropium were 71 and 47 mL with GB 25 and 50 μg BID, respectively.
 | Figure 6 Study Part 2: Adjusted mean pre-dose morning FEV1 on Day 8. |
Add-on formoterol
Compared with the Day 7 data, the addition of formoterol resulted in statistically significant increases in Day 8 peak (Figure 7), FEV1 AUC0–12h (Figure 8) and 12 h trough FEV1 (Figure S1) with all treatments. The magnitude of increase caused by formoterol added to placebo was approximately double that of formoterol added to GB; for example, for FEV1 AUC0–12h, the increases were ~200 and 100 mL, respectively.
 | Figure 7 Study Part 2: Difference in peak FEV1 between Day 7 and Day 8, following administration of formoterol. |
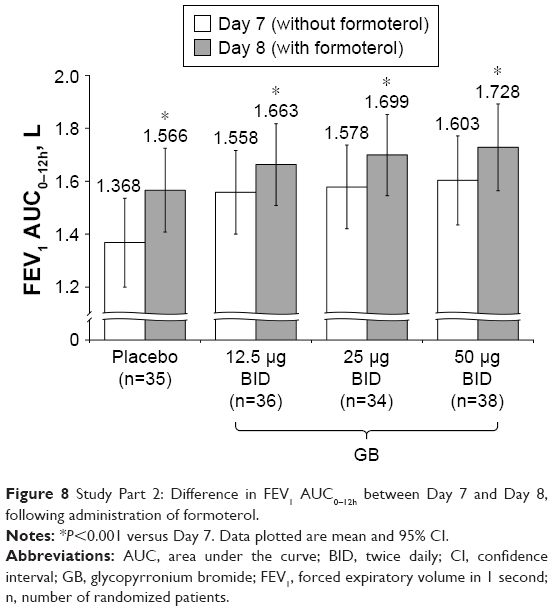 | Figure 8 Study Part 2: Difference in FEV1 AUC0–12h between Day 7 and Day 8, following administration of formoterol. |
Safety and tolerability
Study Part 1
All of the AEs during study Part 1 were mild or moderate in severity, with a lower incidence with GB than placebo and no evidence of a dose-related trend (Table 2). Mean changes over time in the ECG parameters were similar following all treatments (including placebo). No patient had a Fridericia’s corrected QTc (QTcF) interval value above 480 ms or a change from baseline of more than 60 ms. Two patients withdrew due to AEs – atrial fibrillation in a patient receiving placebo and ventricular extrasystoles in a patient receiving GB 50 μg. Two more patients withdrew due to ECG findings – one with a first-degree atrioventricular block following 12.5 μg GB and with a QTc value above normal at screening (the patient had completed the first treatment period by the time the decision was taken to withdraw the patient, and therefore was included in the analyses).
 | Table 2 Study Part 1: Overall experience of AEs and SAEs |
Study Part 2
There was no dose-related trend in AE incidence. All AEs were mild or moderate in severity, with no deaths or SAEs (Table 3). Few changes in laboratory parameters were observed, with no relevant differences in treatment-emergent laboratory abnormalities between any GB dose, tiotropium or placebo. Mean changes over time in all ECG parameters were similar over time for all treatments. No patient had a QTcF interval value above 480 ms; one patient had a change from baseline in QTcF interval >60 ms (during treatment with GB 50 μg BID).
 | Table 3 Study Part 2: Overall experience of AEs and SAEs |
PK (Study Part 2)
Pre-dose mean plasma GB concentrations were similar on days 6 and 7, indicating that plasma steady state was reached within 6 days. GB plasma and urine PK parameters on days 1 and 7 are shown in Table 4. At a steady state, the mean maximum plasma concentration (Cmax,ss) increased proportionally with dose, as did systemic exposure over the first 30 min post-dose (AUC0–30 min,ss). Systemic exposure assessed by both AUC0–12h,ss and AUC0–t,ss increased more than proportionally across the full dose range, but approximately proportionally between 25 and 50 μg BID. In contrast, although the plasma elimination half-life (t½,ss) increased from 12.5 to 25 μg BID, the values were similar with the two higher doses. The median accumulation ratio (Rac) was also similar across doses (note that due to high variability of Rac values with the 25 and 50 μg doses, the median is more reliable than the mean). Mean urinary GB excretion increased proportionally with dose; in contrast, renal clearance decreased slightly with increasing dose.
 | Table 4 Study Part 2: PK parameters of GB on Day 1 and Day 7 |
Discussion
The dose ranging study reported here provides information on bronchodilation, safety and PK of single and multiple doses of a novel extrafine GB formulation delivered by pMDI. This formulation was well tolerated in both parts of the study. Study Part 2 was a multiple-dose, BID-dosing, crossover study that provided important efficacy data; on Day 7, 25 μg BID was the lowest to meet the predefined threshold for clinical relevance for the primary endpoint (12 h trough FEV1) with a difference of ≥120 mL from placebo.
The dose response for GB in study Part 2 was also evaluated by considering the FEV1 AUC0–12h and peak FEV1 on Day 7. Consistent with the primary endpoint showing improvement of ≥120 mL compared to placebo for GB 25 and 50 μg BID, but not 12.5 μg BID, peak FEV1 showed a significantly greater improvement with GB 25 μg compared to 12.5 μg BID. The FEV1 AUC0–12h on Day 7 showed no significant difference between the GB doses, although numerically, the effect of GB 12.5 μg BID was the lowest. Overall, these data indicate that GB 25 and 50 μg BID caused greater lung function improvement than GB 12.5 μg BID, a finding supported by the Day 8 trough FEV1 analysis of GB compared to tiotropium. This suggests that GB 25 μg BID in this formulation may provide at least similar efficacy to tiotropium. This is consistent with data for the dry powder formulation of glycopyrronium, in which 50 μg OD was comparable to tiotropium in terms of lung function, dyspnea and health status.11 Overall, therefore, these results suggest that GB 25 μg BID is the optimal dose in patients with COPD.
The bronchodilator effects of inhaled glycopyrronium have been studied using both once and twice a day administration, with one study showing little difference between these regimens when the same daily dose was administered entirely in the morning or divided between morning and evening.12 The results reported here for twice a day administration are consistent with those for a porous particle pMDI formulation (in development for twice daily dosing), in which a single 28.8 μg dose resulted in differences from placebo of 158 mL in peak FEV1 and ~140 mL in FEV1 AUC0–12h.13
Given that we sought to evaluate the dose–response of GB, we enriched the population with individuals with a greater response to muscarinic blockade, identified by an increase in FEV1 ≥100 mL following inhalation of ipratropium at screening. Similar inclusion criteria have often been used in other early-phase clinical trials of novel LAMA formulations.14,15 This potentially limits the generalizability of the findings to a wider population, but this aspect can be addressed in later-phase clinical trials of longer duration with larger samples sizes. Furthermore, we used a tiotropium arm in order to help select GB doses that would be more likely to achieve at least comparable lung function improvements to those achieved with a commonly used LAMA.
The addition of a single dose of formoterol was associated with significant increases in peak FEV1, FEV1 AUC0–12h and 12 h trough FEV1 for all treatments. Although this study only investigated bronchodilation, other studies have demonstrated the broad clinical benefits of combining long-acting bronchodilators, either in separate devices16,17 or as a fixed combination in the same inhaler,18–28 in terms of dyspnea,18,21,24 exercise endurance time22 and health status.19,23,28,29
Triple therapy with LABA, LAMA and ICS has shown additional advantages to patients in terms of bronchodilation, dyspnea and exacerbations,8–10,29–32 and is one of the most commonly prescribed maintenance treatments for COPD.33 A combination of BDP and FF is currently available in the same device as used for GB in this study (Foster®; Chiesi Farmaceutici S.p.A., Parma, Italy) and has a BID dosing regimen. A logical development is, therefore, the co-formulation of BDP/FF and GB in the same inhaler.9,10 Furthermore, the extrafine formulation has potential advantages over conventional large particle standard formulations in that it allows higher total lung deposition as well as for deeper and more uniform distribution within the airways.34
An unusual characteristic of the population recruited into both parts of this study is the high proportion of female patients. Most interventional clinical trials in COPD have recruited predominantly male populations – and as a consequence, much of the data available on the efficacy of bronchodilators is generated from men. Although a number of epidemiology studies have identified gender influences on the risk or progression of COPD,35–37 post hoc analyses of clinical trials suggest that gender does not seem to influence the response to bronchodilators.38,39
In the PK analyses, plasma concentration, systemic exposure and urinary excretion all increased with increasing dose – in most cases, the increase being proportional to GB dose. This is also consistent with the PK analysis of the dry powder glycopyrronium formulation, dosed OD.40 The main purpose of study Part 1 was to provide safety data in order to allow continuation to study Part 2 for efficacy analysis. Study Part 1 showed that a single dose (200 μg), greater than doses used in study Part 2, had a good overall safety and tolerability profile. Few AEs were considered related to treatment, and no SAEs were reported during GB treatment in either part of the study. There was no evidence of a dose–response in terms of the AE occurrence in either part of the study – although it should be noted that the study was not designed to evaluate such a dose–response (this would probably require much higher doses to be administered for a longer period). In addition, the exclusion criteria, typical of Phase II studies, meant that patients with clinically significant comorbidities were excluded from participation, and so, confirmation of the observed good overall safety and tolerability profile would need further studies in wider populations. However, in longer-term studies, inhaled GB (in a different formulation) has demonstrated an overall good safety profile.11,41,42
Although this study was relatively small and a short-term one, two studies have been conducted that provide long-term evidence on the efficacy and safety of GB 25 μg BID as part of a fixed triple combination of BDP/FF/GB.9,10 The study reported here was focused on lung function in order to identify the optimum dose for later-phase studies with larger sample sizes and longer duration that could also evaluate symptoms and exacerbations.9,10,43
In conclusion, this study provides initial evidence of the bronchodilator efficacy and safety and tolerability profile of this extrafine formulation of GB.
Acknowledgments
This study was funded by Chiesi Farmaceutici SpA. Writing support was provided by David Young of Young Medical Communications and Consulting Ltd. This support was funded by Chiesi Farmaceutici SpA.
Disclosure
DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Merck, NAPP, Novartis, Pfizer, Skypharma, Takeda, Teva, Therevance and Verona. MS, SC, SV, FM, AM and DA are employees of Chiesi Farmaceutici SpA, the study sponsor. The authors report no other conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Available from: www.goldcopd.org. Accessed December 21, 2016. | ||
Niewoehner DE, Rice K, Cote C, et al. Prevention of exacerbations of chronic obstructive pulmonary disease with tiotropium, a once-daily inhaled anticholinergic bronchodilator: a randomized trial. Ann Intern Med. 2005;143(5):317–326. | ||
Dusser D, Bravo ML, Iacono P. The effect of tiotropium on exacerbations and airflow in patients with COPD. Eur Respir J. 2006;27(3):547–555. | ||
Halpin D, Menjoge S, Viel K. Patient-level pooled analysis of the effect of tiotropium on COPD exacerbations and related hospitalisations. Prim Care Respir J. 2009;18(2):106–113. | ||
Casaburi R, Mahler DA, Jones PW, et al. A long-term evaluation of once-daily inhaled tiotropium in chronic obstructive pulmonary disease. Eur Respir J. 2002;19(2):217–224. | ||
Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543–1554. | ||
Brusasco V, Hodder R, Miravitlles M, Korducki L, Towse L, Kesten S. Health outcomes following treatment for six months with once daily tiotropium compared with twice daily salmeterol in patients with COPD. Thorax. 2003;58(5):399–404. | ||
Frith PA, Thompson PJ, Ratnavadivel R, et al. Glycopyrronium once-daily significantly improves lung function and health status when combined with salmeterol/fluticasone in patients with COPD: the GLISTEN study, a randomized controlled trial. Thorax. 2015;70(6):519–527. | ||
Singh D, Papi A, Corradi M, et al. Single inhaler triple therapy vs inhaled corticosteroid plus long-acting β2-agonist therapy for chronic obstructive pulmonary disease (TRILOGY): a double-blind, parallel group, randomized controlled trial. Lancet. 2016;388(10048):963–973. | ||
Vestbo J, Papi A, Corradi M, et al. Single inhaler extrafine triple therapy vs long-acting muscarinic antagonist therapy for chronic obstructive pulmonary disease (TRINITY): a double-blind, parallel group, randomized controlled trial. Lancet. 2017;389(10082):1919–1929. | ||
Kerwin E, Hébert J, Gallagher N, et al. Efficacy and safety of NVA237 vs placebo and tiotropium in patients with COPD: the GLOW2 study. Eur Respir J. 2012;40(5):1106–1114. | ||
Arievich H, Overend T, Renard D, et al. A novel model-based approach for dose determination of glycopyrronium bromide in COPD. BMC Pulm Med. 2012;12(1):74. | ||
Rennard S, Fogarty C, Reisner C, et al. Randomized study of the safety, pharmacokinetics, and bronchodilatory efficacy of a proprietary glycopyrronium metered-dose inhaler in study patients with chronic obstructive pulmonary disease. BMC Pulm Med. 2014;14:118. | ||
Leaker BR, Barnes PJ, Jones CR, Tutuncu A, Singh D. Efficacy and safety of nebulized glycopyrrolate for administration using a high efficiency nebulizer in patients with chronic obstructive pulmonary disease. Br J Clin Pharmacol. 2015;79(3):492–500. | ||
Singh D, Ravi A, Reid F, Buck H, O’Connor G, Down G. Bronchodilator effects, pharmacokinetics and safety of PSX1002-GB, a novel glycopyrronium bromide formulation, in COPD patients; a randomized crossover study. Pulm Pharmacol Ther. 2016;37:9–14. | ||
Rabe KF, Timmer W, Sagkriotis A, Viel K. Comparison of a combination of tiotropium plus formoterol to salmeterol plus fluticasone in moderate COPD. Chest. 2008;134(2):255–262. | ||
Mahler DA, D’Urzo A, Bateman ED, et al; INTRUST-1 and INTRUST-2 study investigators. Concurrent use of indacaterol plus tiotropium in patients with COPD provides superior bronchodilation compared with tiotropium alone: a randomized, double-blind comparison. Thorax. 2012;67(9):781–788. | ||
Bateman ED, Ferguson GT, Barnes N, et al. Dual bronchodilation with QVA149 vs single bronchodilator therapy: the SHINE study. Eur Respir J. 2013;42(6):1484–1494. | ||
Buhl R, Maltais F, Abrahams R, et al. Tiotropium and olodaterol fixed-dose combination vs mono-components in COPD (GOLD 2–4). Eur Respir J. 2015;45(4):969–979. | ||
Decramer M, Anzueto A, Kerwin E, et al. Efficacy and safety of umeclidinium plus vilanterol vs tiotropium, vilanterol, or umeclidinium monotherapies over 24 weeks in patients with chronic obstructive pulmonary disease: results from two multicentre, blinded, randomized controlled trials. Lancet Respir Med. 2014;2(6):472–486. | ||
Mahler DA, Decramer M, D’Urzo A, et al. Dual bronchodilation with QVA149 reduces patient-reported dyspnoea in COPD: the BLAZE study. Eur Respir J. 2014;43(6):1599–1609. | ||
Maltais F, Singh S, Donald AC, et al. Effects of a combination of umeclidinium/vilanterol on exercise endurance in patients with chronic obstructive pulmonary disease: two randomized, double-blind clinical trials. Ther Adv Respir Dis. 2014;8(6):169–181. | ||
Siler TM, Kerwin E, Sousa AR, Donald A, Ali R, Church A. Efficacy and safety of umeclidinium added to fluticasone furoate/vilanterol in chronic obstructive pulmonary disease: results of two randomized studies. Respir Med. 2015;109(9):1155–1163. | ||
Singh D, Jones PW, Bateman ED, et al. Efficacy and safety of aclidinium bromide/formoterol fumarate fixed-dose combinations compared with individual components and placebo in patients with COPD (ACLIFORM-COPD): a multicentre, randomized study. BMC Pulm Med. 2014;14(1):178. | ||
Tashkin DP, Varghese ST. Combined treatment with formoterol and tiotropium is more efficacious than treatment with tiotropium alone in patients with chronic obstructive pulmonary disease, regardless of smoking status, inhaled corticosteroid use, baseline severity, or gender. Pulm Pharmacol Ther. 2011;24(1):147–152. | ||
van Noord JA, Aumann JL, Janssens E, et al. Effects of tiotropium with and without formoterol on airflow obstruction and resting hyperinflation in patients with COPD. Chest. 2006;129(3):509–517. | ||
van Noord JA, Buhl R, Laforce C, et al. QVA149 demonstrates superior bronchodilation compared with indacaterol or placebo in patients with chronic obstructive pulmonary disease. Thorax. 2010;65(12):1086–1091. | ||
ZuWallack R, Allen L, Hernandez G, Ting N, Abrahams R. Efficacy and safety of combining olodaterol Respimat(®) and tiotropium HandiHaler(®) in patients with COPD: results of two randomized, double-blind, active-controlled studies. Int J Chron Obstruct Pulmon Dis. 2014;9:1133–1144. | ||
Jung KS, Park HY, Park SY, et al. Comparison of tiotropium plus fluticasone propionate/salmeterol with tiotropium in COPD: a randomized controlled study. Respir Med. 2012;106(3):382–389. | ||
Singh D, Brooks J, Hagan G, Cahn A, O’Connor BJ. Superiority of “triple” therapy with salmeterol/fluticasone propionate and tiotropium bromide vs individual components in moderate to severe COPD. Thorax. 2008;63(7):592–598. | ||
Chatterjee A, Shah M, D’Souza AO, Bechtel B, Crater G, Dalal AA. Observational study on the impact of initiating tiotropium alone vs tiotropium with fluticasone propionate/salmeterol combination therapy on outcomes and costs in chronic obstructive pulmonary disease. Respir Res. 2012;13:15. | ||
Welte T, Miravitlles M, Hernandez P, et al. Efficacy and tolerability of budesonide/formoterol added to tiotropium in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2009;180(8):741–750. | ||
Price D, West D, Brusselle G, et al. Management of COPD in the UK primary-care setting: an analysis of real-life prescribing patterns. Int J Chron Obstruct Pulmon Dis. 2014;9:889–905. | ||
Scichilone N, Benfante A, Morandi L, Bellini F, Papi A. Impact of extrafine formulations of inhaled combinations on patient-related outcomes in asthma and COPD. Patient Relat Outcome Meas. 2014;5:153–162. | ||
Sørheim IC, Johannessen A, Gulsvik A, Bakke PS, Silverman EK, Demeo DL. Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax. 2010;65(6):480–485. | ||
Aryal S, Diaz-Guzman E, Mannino DM. Influence of sex on chronic obstructive pulmonary disease risk and treatment outcomes. Int J Chron Obstruct Pulmon Dis. 2014;9:1145–1154. | ||
Jain NK, Thakkar MS, Jain N, Rohan KA, Sharma M. Chronic obstructive pulmonary disease: Does gender really matter? Lung India. 2011;28(4):258–262. | ||
Vestbo J, Soriano JB, Anderson JA, Calverley P, Pauwels R, Jones P. Gender does not influence the response to the combination of salmeterol and fluticasone propionate in COPD. Respir Med. 2004;98(11):1045–1050. | ||
Tashkin D, Celli B, Kesten S, Lystig T, Decramer M. Effect of tiotropium in men and women with COPD: Results of the 4-year UPLIFT® trial. Respir Med. 2010;104(10):1495–1504. | ||
Dumitras S, Sechaud R, Drollmann A, et al. Effect of cimetidine, a model drug for inhibition of the organic cation transport (OCT2/MATE1) in the kidney, on the pharmacokinetics of glycopyrronium. Int J Clin Pharmacol Ther. 2013;51(10):771–779. | ||
D’Urzo A, Ferguson GT, van Noord JA, et al. Efficacy and safety of once-daily NVA237 in patients with moderate-to-severe COPD: the GLOW1 trial. Respir Res. 2011;12:156. | ||
Wang C, Sun T, Huang Y, et al. Efficacy and safety of once-daily glycopyrronium in predominantly Chinese patients with moderate-to-severe chronic obstructive pulmonary disease: the GLOW7 study. Int J Chron Obstruct Pulmon Dis. 2015;10:57–68. | ||
Singh D, Schröder-Babo W, Cohuet G, et al. The bronchodilator effects of extrafine glycopyrronium added to combination treatment with beclometasone dipropionate plus formoterol in COPD: a randomized crossover study (the TRIDENT study). Respir Med. 2016;114:84–90. |
Supplementary materials
Inclusion criteria
Subjects had to meet all of the following inclusion criteria (applicable for study Parts 1 and 2):
- Male and female subjects, aged 40–75 years.
- Written informed consent obtained before the first study-related activity.
- Diagnosis of moderate–severe COPD, according to the Global Initiative for Chronic Obstructive Lung Disease guidelines (2009).
- Able to understand the study procedures, the risks involved and the ability to be trained to use the devices correctly.
- Body mass index between 18 and 35 kg/m2.
- Current or ex-smokers with a smoking history of ≥10 pack-years (eg, ≥20 cigarettes per day for 10 years and 40 cigarettes per day for 5 years).
- Vital signs within the following ranges:
- 90 mmHg ≤ systolic blood pressure ≤160 mmHg,
- 50 mmHg ≤ diastolic blood pressure ≤90 mmHg,
- 50 beats per min ≤ heart rate ≤100 beats per min.
- Twelve-lead electrocardiogram (ECG) with computerized protocol interpretation considered as normal: 120 ms ≤ PR ≤200 ms; QRS ≤120 ms; QTcF ≤450 ms. Minor deviations were acceptable, provided that they were not judged clinically significant by the cardiologist.
- Post-bronchodilator forced expiratory volume in 1 second (FEV1) between 40% and 80% predicted values (40% ≤ FEV1 ≤80%), documented at the screening visit.
- Post-bronchodilator FEV1/forced vital capacity: ≤70 (absolute value) documented at the screening visit.
- Airway reversibility of at least 100 mL within 30–45 min after inhalation of ipratropium 80 μg (for study Part 2: historical reversibility was acceptable for subjects who performed study Part 1). If the reversibility criteria were not met and if the investigator deemed it appropriate, the testing could be repeated once. This requirement had to be met after retesting during the run-in period at least 24 h prior to randomization.
Exclusion criteria
Subjects meeting at least one of the following criteria could not be enrolled (applicable for study Parts 1 and 2):
- History of chronic or seasonal allergy.
- Blood eosinophil count above 0.6×109/L.
- Clinically relevant findings on physical examination, laboratory or ECG parameters at screening. (Note that one patient had a prolonged QTc at screening and was allowed to enter the study by the investigator, who considered this to be not clinically relevant. The subject was later withdrawn from the study, as the study was designed to exercise caution in subjects who may be predisposed to an increased risk of cardiovascular events.)
- Occurrence of one of the following 24 h Holter ECG abnormalities at screening:
- More than 200 ventricular ectopics in 24 h;
- Ventricular tachycardia;
- Second-degree heart block;
- Sustained cardiac arrhythmias (atrial fibrillation, supraventricular tachycardia, complete heart block);
- Any symptomatic arrhythmia (except isolated extrasystoles);
- Sinus pauses ≥2.5 s.
- Significant disease not related to COPD (eg, myocardial infarction, stroke within the preceding 6 months).
- Respiratory tract infection (including upper tract) 4 weeks prior to the screening visit, requiring change of treatment.
- Subjects requiring oxygen therapy on a daily basis for chronic hypoxemia.
- Subjects who had been hospitalized in the 6 weeks prior to the screening visit.
- Having received an investigational product within the last 8 weeks before the screening visit.
- Inability to comply with the study procedures or with the study treatment intake, including inability to be trained with the Vitalograph® Aerosol Inhalation Monitor.
- History of cystic fibrosis, bronchiectasis, alpha-1 antitrypsin deficiency or any other significant lung disease that was considered to be clinically significant by the investigator.
- Intolerance/hypersensitivity or any contraindication (eg, history of glaucoma) to treatment with an M3 antagonist or any of the excipients contained in the formulations used in the study.
- History of alcohol or substance abuse that, in the opinion of the investigator, could be of clinical significance.
- Subjects who had undergone major surgery in the 12 weeks before the screening visit.
- Subjects treated with oral (slow-release included) or parenteral steroids 8 weeks prior to the screening visit.
- Subjects treated with oral β2-adrenergics, antihistamines or theophyllines 1 month prior to the screening visit or enzyme-inducing or -inhibiting drugs 2 months before the first administration.
- Subjects treated with tiotropium in the 10 days prior to the screening visit.
- (a) Pregnant or lactating women (pregnancy defined as the state of a female after conception and until the termination of the gestation, confirmed by a positive serum human chorionic gonadotropin laboratory test [>5 mIU/mL]). Serum pregnancy test was to be done at screening for verification.(b) Women of childbearing potential (ie, all women physiologically capable of becoming pregnant), including women whose career, lifestyle or sexual orientation precluded intercourse with a male partner, unless they were postmenopausal (defined as 12 months of natural [spontaneous] amenorrhea) or were using an acceptable method of contraception.(c) Male subjects had to be sterile or they or their partner had to be willing to use an approved method of contraception from the time of dose administration until 30 days after the last dose of study medication. Subjects were not allowed to donate sperm for 30 days after the last dose of study drug. A reliable method of contraception for male and female subjects (or their partner) could be one or more of the following ones:
- Surgical sterilization (ie, bilateral tubal ligation or hysterectomy for females, vasectomy for males);
- Hormonal contraception (implantable, patch, or oral);
- Double-barrier methods (any double combination of intrauterine device, male or female condom, diaphragm, sponge, cervical cap, condom or spermicide).
 | Figure S1 Study Part 2: Difference in 12 h trough FEV1 between Day 7 and Day 8, following administration of formoterol. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.