Back to Journals » Clinical Ophthalmology » Volume 13
Brittle cornea syndrome: current perspectives
Authors Walkden A ![]() , Burkitt-Wright E, Au L
, Burkitt-Wright E, Au L
Received 21 May 2019
Accepted for publication 12 July 2019
Published 12 August 2019 Volume 2019:13 Pages 1511—1516
DOI https://doi.org/10.2147/OPTH.S185287
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Andrew Walkden,1,2 Emma Burkitt-Wright,3,4 Leon Au1,2
1Manchester Royal Eye Hospital, Manchester University Foundation Trust, Manchester, UK; 2Medical Academic Health Sciences Centre, University of Manchester, UK; 3Genetic Medicine, Institute of Human Development, Faculty of Medical and Human Sciences, University of Manchester, Manchester, UK; 4Genetic Medicine, St Mary’s Hospital, Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester, UK
Abstract: Brittle cornea syndrome (BCS) is a rare autosomal recessive connective tissue disorder characterised by severe corneal thinning, with the major ocular risk being spontaneous ocular perforation due to progressive stromal thinning and ectasia. It is a complex condition with limited treatment options. The purpose of this review is to highlight the difficulties associated with the condition and examine the available published evidence with regards to management.
Keywords: brittle Cornea, spontaneous corneal perforation, corneal ectasia
A Letter to the Editor has been published for this article
A Response to Letter has been published for this article
Method of literature search
A PubMed search was performed using the search terms “Brittle Cornea Syndrome” and “Ehlers-Danlos Syndrome”. A full systematic review of the literature using the PubMed database was conducted up until 10/03/19. The articles used were written in English, with all articles accessed in full. Both review articles and original articles were used for this review.
The prevalence of BCS is said to be less than 1 in 1,000,000,1 with only around 60 cases being reported within the literature. The initial cases were reported in families of Tunisian Jewish origin.2,3 More recently, cases have been reported worldwide, in patients of European and Asian origin.4–7 It is important to note that within the literature, BCS has sometimes historically been considered as a phenotypic variant of an autosomal recessive form of Ehlers-Danlos Syndrome (EDS) previously known as EDS VI, but with the identification of the molecular basis for many forms of EDS and clinical reclassification of these disorders, more recently it has been clarified that BCS is a separate entity.3–9 Whilst some patients with BCS have a purely ocular phenotype,10,11 others have additional phenotypes distinct from those described in EDS. It is likely that given the occult nature of the condition, the difficulty in making the diagnosis and similarity to other ectatic corneal conditions, BCS remains underdiagnosed.4 Abnormalities in the function of lysyl hydroxylase, an important agent of collagen post-translational modification, are found in the kyphoscoliotic form of EDS (formerly EDS VI), whereas activity of this enzyme is normal in BCS patients.2 The principal ocular fragility in the kyphoscoliotic form of EDS is reported to be in the sclera, whereas for BCS it is the cornea that is most affected.4,12
Biallelic variants in two genes, ZNF46913–15 and PRDM512,16 have been identified as being responsible for BCS. Subsequently, it has been confirmed that ZNF469 plays a role in normal anterior segment and corneal development as a determinant of the highly heritable quantitative trait of corneal thickness.4,5,15,17,18 It has also been implicated in the development of keratoconus18,19 and abnormal corneal curvature, with a high frequency of variants in this gene (23%) reported in one keratoconus population.18 However this is disputed by other authors,20 and larger population-based case-controlled studies have not replicated this finding.21,22 Other variations in this gene are likely to contribute towards population variation in CCT.23
PRDM5 is critical for extracellular matrix development and maintenance and acts as a determinant of corneal thickness.5,12,15,24 It has also been suggested to play a role in the development of normal retinal microvasculature25 and Bruch’s membrane.26 Fibroblasts from BCS patients with these mutations show disruption in the deposition of several collagens, fibronectin and integrins,12 all of which play a major role in collagen biosynthesis. It has been hypothesised that fibronectin and integrins act as the ‘organisers’ for collagen deposition within the extracellular matrix and determine the site of collagen fibril assembly, with a mutation in these components resulting in abnormal or reduced collagen deposition.27
Clinical features
The clinical features of BCS show considerable overlap with other collagen disorders4,28 known to have ocular associations, most notably the kyphoscoliotic form of EDS and Stickler syndrome. Both sensorineural and conductive deafness are seen in BCS, in addition to hypermobile tympanic membranes which can result in devastating polysensory loss when coupled with ophthalmic complications. Developmental hip abnormalities and joint hypermobility are often present, alongside dermatological features such as excessive skin elasticity abnormal scarring. A full list of non-ocular features can be seen in Table 1. Although differentiation between forms of EDS and BCS may be difficult clinically, it may be important in terms of prognosis, as kyphoscoliotic and other severe forms of EDS may be associated with premature death from arterial or visceral rupture. Whilst caution is necessary due to the small number of patients described to date, no such complications have yet been described in BCS.4
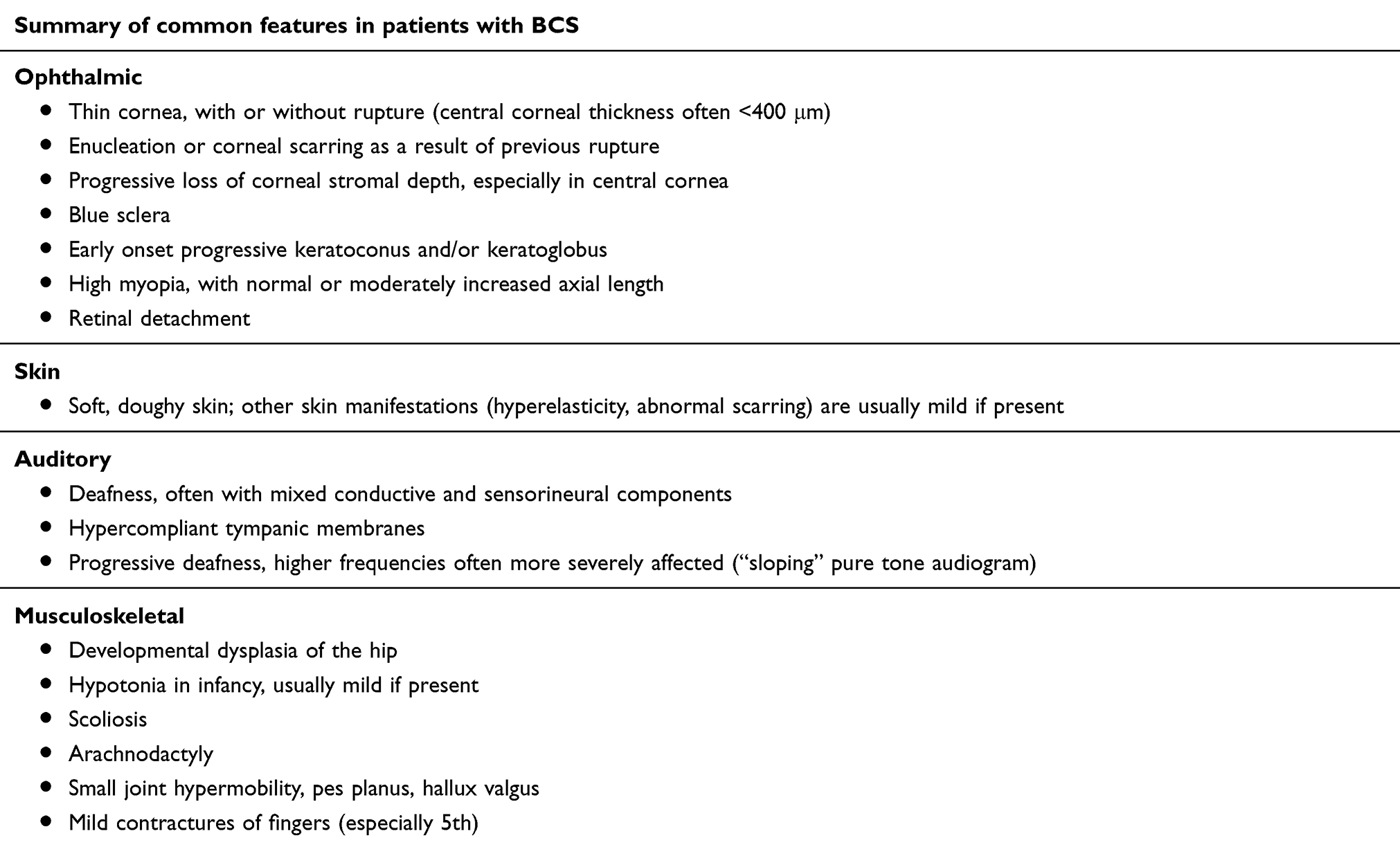 |
Table 1 Brittle cornea syndrome (BCS) manifestations |
Ocular features
The ophthalmic features of BCS are the most devastating. Extreme myopia, keratoglobus and keratoconus all have the potential to cause blindness, with corneal perforation reported from innocuous trauma. More worryingly, spontaneous rupture can occur due to BCS corneas being unable to withstand normal biomechanical stresses as result of abnormal corneal hysteresis.4 Any central corneal thickness (CCT) measurement of less than 400μm should raise suspicion of BCS. The largest case series of BCS patients in the literature showed that 100% of patients identified to have pathogenic variants in either PRDM5 or ZNF469 had a CCT of less than 400μm.4 Some patients have been found to have CCTs of less than 300μm,12 although the peripheral corneal thickness is said to be relatively well preserved in individuals with BCS.4 (Figure 1). Other connective tissue disorders within the EDS spectrum are known to have reduced CCT, but is said to be less pronounced than BCS patients. Interestingly, despite the similarities to isolated keratoconus, none of the cases within the literature report corneal striae being present. It is suggested that stromal striae are associated with the biomechanical properties of the cornea and they represent areas of structural integrity under mechanical stress maintaining corneal shape when affected by pathology.29 It is therefore interesting that they appear to be absent in BCS patients.
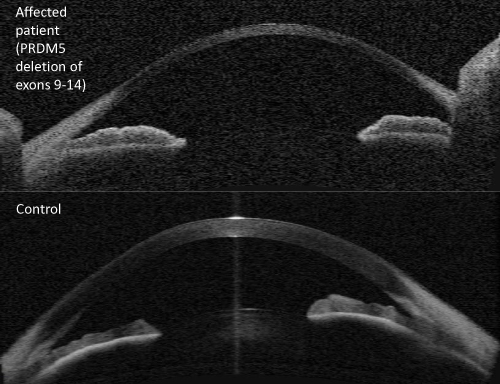 |
Figure 1 Ocular coherence tomography imaging (Zeiss Visante system) of (upper panel) patient with brittle cornea syndrome due to PRDM5 mutation, and (lower panel) normal control eye. Note extreme thinning throughout the central cornea of the affected eye, with relative sparing of the peripheral cornea. |
Although blue sclera is associated with this condition, it is by no means universal4 and can also disappear.30 Blue sclera is important to note clinically as it correlates with a CCT reduction of at least 33%.2 It can also occur in other conditions such as EDS, Marfan syndrome and osteogenesis imperfecta. Retinal detachments and secondary glaucoma have also been reported to be associated with levels of extreme myopia.14,31 Authors judiciously note that retinal detachment is a rare feature of BCS, although this is likely to be determined by the fact that patients often present at an early age with globe perforation.4 From the cases within the literature, reported perforations occur at a mean age of 4.3 years (range, 1.5–19 years), with more than half of the published cases permanently losing eyesight.32
Medical management
Due to the rarity of this condition and lack of robust evidence within the literature base, there is no agreed treatment protocol, although an excellent “management checklist” has been recommended by previous authors4 which can be used to guide clinicians when confronted with this rare condition. As discussed above, globe rupture tends to occur in early childhood from relatively innocuous trauma.32 Given the poor visual prognosis following globe rupture, the overarching recommendation from the literature is early diagnosis and recognition of risk factors to allow preventative measures. Relatives of affected individuals should be examined. It is important to recognise the high risk of rupture, with one case series reporting a risk of greater than 70%.4 This, when coupled with family and patient education, allows the opportunity to introduce preventative protective measures. Many authors advocate the use of protective polycarbonate glasses and lifestyle modifications such as avoiding contact sports and rough play.3,4,33,34 It is uncertain if these need to be worn at all times and this is probably an unrealistic expectation especially in younger children but at least it provides the family an opportunity to determine high risk activities and establish a routine of regular protective eyewear.
Regular follow up with serial refractions and topographical readings are required in order to ensure optimal visual correction and prevention of amblyopia. The irregular astigmatism will be difficult to correct with spectacles, and contact lenses can be used, although some authors advocate cautious contact lens use in extremely thin corneas.35,36 There are reports of combined use of larger diameter scleral contact lenses in conjunction with spectacles to provide better levels of visual acuity.4
Awareness needs to be raised amongst other subspecialties specifically anaesthetics and paediatrics. Recommendations from anaesthetic colleagues exist with regards to the perioperative management of such patients and the unique challenges these patients provide.37 Eyes are generally taped shut when general anaesthesia is administered and this must be done with extreme caution in individuals with BCS. Care must be made to avoid trauma to the globe whilst the patient is unconscious. It is also recommended that following emergence from anaesthesia, certain anaesthetic precautions are taken to reduce the risk of coughing during extubation, as violent coughing could lead to globe rupture due to an acute rise in intraocular pressure.37
Finally, it is important to consider BCS as a differential diagnosis for patients presenting with suspected non-accidental eye injuries (NAI). It is possible that the severity of the injury may not correlate well with the historical mechanism of the injury, which raises suspicion of NAI and there are cases of allegations being made when patients have presented with severe injuries following reports of relatively innocuous trauma.4 Although it is paramount to rule out NAI, careful examination for excessive corneal thinning in such injuries is necessary in such patients.
Surgical management
A few surgical techniques have been described for use in BCS eyes, and their applicability depends on the clinical setting. Following perforation, repair of the BCS cornea is notoriously difficult and sometimes impossible due to excessive tissue fragility. “Cheese wiring” of sutures and tissue breakdown32,38 has been reported by Izquierdo et al in addition to intraoperative perforation during suture rotation whilst attempting penetrating keratoplasty (PK).31 This required a scleral patch graft at the time of surgery. Corneal transplants in the setting of such a thin recipient bed are challenging and likely to be unable to completed in theatre39 or associated with excessive leaking post operatively. Authors have suggested using 11.0 nylon sutures with long suture bites in order to reduce the risk of post operative leak.31 Other cases exist describing intraoperative difficulty in performing PK, which required conversion to a 12mm sclero-corneal graft. This was successful and the patient was able to undergo Boston Type 1 KeratoProsthesis at a later date.40 PK can be used as an integrity restoring procedure with the hope of repeating the procedure later in order to visually rehabilitate the patient.38
Epikeratoplasty is historically a refractive procedure in which the host corneal epithelium is removed and a donor lenticule is placed onto the recipient cornea, secured with sutures until it heals into place resulting in this technique being referred to as a “living contact lens.” It is not used routinely as a refractive procedure nowadays given the advantages of modern laser refractive surgery, although it has been utilised as a temporary measure for tectonic restoration of perforated corneas following corneal melts.41
It has also been applied successfully in keratoglobus patients with associated connective tissue diseases, although BCS is not specifically mentioned.36 In one series, surgery was performed for tectonic support and/or visual improvement with successful outcomes in five of six patients over a follow-up period of 11 to 27 months (mean, 21 months).36 Only one lenticule was removed because the epithelium did not heal. No patient lost visual acuity as a result of the procedure and mean corneal flattening was 19D.36 In their discussion, the authors recommend the use of a 12.5mm length and 0.3mm thickness lenticule in order to protect the cornea and limbus.36 These outcomes suggest that this technique may be useful in stable patients.
In another trauma case, Macsai et al42 report using a similar technique of removing the donor Descemet’s membrane and endothelium and placing the graft on top of the host cornea once the epithelium had been removed with a surgical blade. It is worth noting that even careful epithelial debridement resulted in a small corneal rupture. The donor corneo-scleral button was secured using 9.0 nylon. A 360 degree conjunctival peritomy allowed the conjunctiva to be pulled up to the edge of the graft and tacked down. This procedure allows a scaffold to form of host and donor cornea, allowing a full-thickness penetrating keratoplasty to be performed four months later, with the patient reporting 20/80 visual acuity with follow up. It is worth noting some important differences however between the presentation of the patients in the two cases discussed above. Izquierdo et al case presented with a flat anterior chamber due to a full thickness perforation, whilst in contrast Macsai et al presented with a formed anterior chamber and corneal scarring.42 Despite the fantastic illustration of this technique to visually rehabilitate the patient, this may not be possible at the time of significant corneal rupture, if the anterior chamber cannot be restored. As described by Macsai et al, the initial corneal repair was performed with nylon sutures, with the epikeratoplasty being performed at a later date. Interestingly, this patient also required this procedure in their fellow eye after a subsequent perforating injury to this eye. In order to aid anterior chamber reformation, the use of intraocular C3F8 gas has been suggested to provide a tamponade effect. This required a three port vitrectomy in order to facilitate a maximal gas fill, with the aim to provide gas contact with the corneal wound to prevent aqueous egress, allowing wound integrity and healing time whilst keeping the globe formed.43
Onlay corneal grafts have also been described in order to improve extreme levels of myopia in non-perforated BCS eyes. An impressive reduction from −23 dioptres to −5 dioptres has been documented in one patient who was intolerant of spectacles or contact lenses. This patient’s other eye had suffered a penetrating injury in childhood rendering it not able to perceive light. Despite the initial improvement in refraction and navigational ability, the patient unfortunately developed glaucoma requiring cycloablation and later developed acute hydrops.33 Filtration or aqueous shunt surgery was deemed not possible due to the tissue status with the final visual acuity being 1.60 logMar.
Collagen crosslinking (CXL) has been reported to be effective in treating progressive keratoconus in children and adults.44–47 This procedure involves riboflavin (vitamin B2) and long wavelength ultraviolet A light (370 nm) application in order to induce chemical reactions in the corneal stroma. Covalent bonds between collagen molecules, fibres and microfibrils, form in the hope of strengthening the cornea, potentially stabilising ectatic disease.47 A few reports document the use of CXL in BCS patient with encouraging results. One child with BCS showed improved visual acuity post CXL,4 with another case report in a similar case showing a potentially promising outcome.32 The significance of these cases is that both patients had CCTs of less than 280μm which would normally contraindicate the use of CXL according to the Dresden protocol.48 CXL use in corneas under 400μm carries the significant risk of endothelial cell toxicity as a result of the UV light used. In an attempt to reduce endothelial injury, the authors opted for an epithelium on procedure in order to reduce the risk of corneal rupture during epithelial removal and to reduce UV penetrance into the cornea. The UV light exposure was also reduced in proportion to CCT. Fortunately in this case, stable endothelial cell counts were measured at post procedure visits.32
Conclusion
BCS is a complex disease that may have devastating consequences. It is likely to be underdiagnosed due to its rarity and minimal or nonspecific extraocular systemic features. The overwhelming message from the available body of research is the need for diagnosis and therefore prevention of ocular injuries. Genotypic differentiation from other similar connective tissue disorders allows tailored advice for the patient, particularly regarding other systemic health considerations. If ocular injury occurs, it is likely to result in poor visual outcomes. Different techniques have been described for use in emergency settings which can often present challenging surgical scenarios. There appears to be the potential for corneal strengthening procedures such as epikeratoplasty and corneal crosslinking to play a role in ocular protection and rehabilitation, but this opportunity can only be afforded to the patient once the diagnosis has been made.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Orphanet. Orphanet website. 2019; Orphanet.
2. Abu A, Frydman M, Marek D, et al. Mapping of a gene causing brittle cornea syndrome in Tunisian jews to 16q24. Invest Ophthalmol Vis Sci. 2006;47(12):5283–5287. doi:10.1167/iovs.06-0206
3. Zlotogora J, BenEzra D, Cohen T, Cohen E. Syndrome of brittle cornea, blue sclera, and joint hyperextensibility. Am J Med Genet. 1990;36(3):269–272. doi:10.1002/ajmg.1320360303
4. Burkitt Wright EM, Porter LF, Spencer HL, et al. Brittle cornea syndrome: recognition, molecular diagnosis and management. Orphanet J Rare Dis. 2013;8:68. doi:10.1186/1750-1172-8-68
5. Swierkowska J, Gajecka M. Genetic factors influencing the reduction of central corneal thickness in disorders affecting the eye. Ophthalmic Genet. 2017;38(6):501–510. doi:10.1080/13816810.2017.1313993
6. Royce PM, Steinmann B, Vogel A, Steinhorst U, Kohlschuetter A. Brittle cornea syndrome: an heritable connective tissue disorder distinct from Ehlers-Danlos syndrome type VI and fragilitas oculi, with spontaneous perforations of the eye, blue sclerae, red hair, and normal collagen lysyl hydroxylation. Eur J Pediatr. 1990;149(7):465–469.
7. Ticho U, Ivry M, Merin S. Brittle cornea, blue sclera, and red hair syndrome (the brittle cornea syndrome). Br J Ophthalmol. 1980;64(3):175–177.
8. Al-Hussain H, Zeisberger SM, Huber PR, Giunta C, Steinmann B. Brittle cornea syndrome and its delineation from the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI): report on 23 patients and review of the literature. Am J Med Genet A. 2004;124A(1):28–34.
9. Ramappa M, Wilson ME, Rogers RC, Trivedi RH. Brittle cornea syndrome: a case report and comparison with Ehlers Danlos syndrome. J Aapos. 2014;18(5):509–511.
10. Micheal S, Khan MI, Islam F, et al. Identification of Mutations in the PRDM5 Gene in Brittle Cornea Syndrome. Cornea. 2016;35(6):853–859. doi:10.1097/ICO.0000000000000824
11. Khan AO, Aldahmesh MA, Alkuraya FS. Brittle cornea without clinically-evident extraocular findings in an adult harboring a novel homozygous ZNF469 mutation. Ophthalmic Genet. 2012;33(4):257–259. doi:10.3109/13816810.2012.670362
12. Burkitt Wright EMM, Spencer HL, Daly SB, et al. Mutations in PRDM5 in brittle cornea syndrome identify a pathway regulating extracellular matrix development and maintenance. Am J Hum Genet. 2011;88(6):767–777. doi:10.1016/j.ajhg.2011.05.007
13. Abu A, Frydman M, Marek D, et al. Deleterious mutations in the Zinc-Finger 469 gene cause brittle cornea syndrome. Am J Hum Genet. 2008;82(5):1217–1222. doi:10.1016/j.ajhg.2008.04.001
14. Christensen AE, Knappskog PM, Midtbo M, et al. Brittle cornea syndrome associated with a missense mutation in the zinc-finger 469 gene. Invest Ophthalmol Vis Sci. 2010;51(1):47–52. doi:10.1167/iovs.09-4251
15. Hoehn R, Zeller T, Verhoeven VJ, et al. Population-based meta-analysis in Caucasians confirms association with COL5A1 and ZNF469 but not COL8A2 with central corneal thickness. Hum Genet. 2012;131(11):1783–1793. doi:10.1007/s00439-012-1201-3
16. Aldahmesh MA, Mohamed JY, Alkuraya FS. A novel mutation in PRDM5 in brittle cornea syndrome. Clin Genet. 2012;81(2):198–199. doi:10.1111/j.1399-0004.2011.01808.x
17. Micheal S, Siddiqui SN, Zafar SN, et al. Whole exome sequencing identifies a heterozygous missense variant in the PRDM5 gene in a family with Axenfeld-Rieger syndrome. Neurogenetics. 2016;17(1):17–23. doi:10.1007/s10048-015-0462-0
18. Vincent AL, Jordan CA, Cadzow MJ, Merriman TR, McGhee CN. Mutations in the zinc finger protein gene, ZNF469, contribute to the pathogenesis of keratoconus. Invest Ophthalmol Vis Sci. 2014;55(9):5629–5635.
19. Lechner J, Porter LF, Rice A, et al. Enrichment of pathogenic alleles in the brittle cornea gene, ZNF469, in keratoconus. Hum Mol Genet. 2014;23(20):5527–5535. doi:10.1093/hmg/ddu253
20. Davidson AE, Borasio E, Liskova P, et al. Brittle cornea syndrome ZNF469 mutation carrier phenotype and segregation analysis of rare ZNF469 variants in familial keratoconus. Invest Ophthalmol Vis Sci. 2015;56(1):578–586. doi:10.1167/iovs.14-15792
21. Lucas SEM, Zhou T, Blackburn NB, et al. Rare, potentially pathogenic variants in ZNF469 are not enriched in keratoconus in a large australian cohort of european descent. Invest Ophthalmol Vis Sci. 2017;58(14):6248–6256. doi:10.1167/iovs.17-22417
22. Karolak JA, Gambin T, Rydzanicz M, et al. Evidence against ZNF469 being causative for keratoconus in Polish patients. Acta Ophthalmol. 2016;94(3):289–294. doi:10.1111/aos.12968
23. Lu Y, Dimasi DP, Hysi PG, et al. Common genetic variants near the brittle cornea syndrome locus ZNF469 influence the blinding disease risk factor central corneal thickness. PLoS Genet. 2010;6(5):e1000947. doi:10.1371/journal.pgen.1000947
24. Vithana EN, Aung T, Khor CC, et al. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum Mol Genet. 2011;20(4):649–658. doi:10.1093/hmg/ddq511
25. Porter LF, Galli GG, Williamson S, et al. A role for repressive complexes and H3K9 di-methylation in PRDM5-associated brittle cornea syndrome. Hum Mol Genet. 2015;24(23):6565–6579. doi:10.1093/hmg/ddv345
26. Porter LF, Gallego-Pinazo R, Keeling CL, et al. Bruch’s membrane abnormalities in PRDM5-related brittle cornea syndrome. Orphanet J Rare Dis. 2015;10:145. doi:10.1186/s13023-015-0360-4
27. Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20(5):495–501. doi:10.1016/j.ceb.2008.06.008
28. Al-Owain M, Al-Dosari MS, Sunker A, Shuaib T, Alkuraya FS. Identification of a novel ZNF469 mutation in a large family with Ehlers-Danlos phenotype. Gene. 2012;511(2):447–450. doi:10.1016/j.gene.2012.09.022
29. Grieve K, Ghoubay D, Georgeon C, et al. Stromal striae: a new insight into corneal physiology and mechanics. Sci Rep. 2017;7(1):13584. doi:10.1038/s41598-017-13194-6
30. Cameron JA. Corneal abnormalities in Ehlers-Danlos syndrome type VI. Cornea. 1993;12(1):54–59.
31. Izquierdo L
32. Kaufmann C, Schubiger G, Thiel MA. Corneal cross-linking for brittle cornea syndrome. Cornea. 2015;34(10):1326–1328. doi:10.1097/ICO.0000000000000577
33. Muthusamy K, Tuft S. Use of an onlay corneal lamellar graft for brittle cornea syndrome. BMJ Case Rep. 2018;2018.
34. Wan Q, Tang J, Han Y, Xiao Q, Deng Y. Brittle cornea syndrome: a case report and review of the literature. BMC Ophthalmol. 2018;18(1):252. doi:10.1186/s12886-018-0903-2
35. Javadi MA, Kanavi MR, Ahmadi M, Yazdani S. Outcomes of epikeratoplasty for advanced keratoglobus. Cornea. 2007;26(2):154–157. doi:10.1097/01.ico.0000244878.38621.fc
36. Cameron JA, Cotter JB, Risco JM, Alvarez H. Epikeratoplasty for keratoglobus associated with blue sclera. Ophthalmology. 1991;98(4):446–452.
37. Ioscovich A, Grisaru-Granovsky S, Halpern S, Shapiro Y. Peripartum anesthetic management of a patient with brittle cornea syndrome. Arch Gynecol Obstet. 2011;283(Suppl 1):49–52. doi:10.1007/s00404-011-1838-5
38. Natarajan R, Shah GY, Rao SK, Padamanabhan P. Penetrating keratoplasty as a globe-saving procedure in fragile cornea. Cornea. 2003;22(2):164–165.
39. Avgitidou G, Siebelmann S, Bachmann B, Kohlhase J, Heindl LM, Cursiefen C. Brittle cornea syndrome: case report with novel mutation in the PRDM5 gene and review of the literature. Case Rep Ophthalmol Med. 2015;2015:637084.
40. Joshi SA, Uppapalli S, More P, Deshpande M. Unusual case of globe perforation: the brittle cornea without systemic manifestations. BMJ Case Rep. 2016;2016.
41. Lazaridis A, Brouzas D, Sekundo W, et al. Tectonic epikeratoplasty with ethanol-stored donor corneas. Cell Tissue Bank. 2018;19(4):637–644. doi:10.1007/s10561-018-9714-1
42. Macsai MS, Lemley HL, Schwartz T. Management of oculus fragilis in Ehlers-Danlos type VI. Cornea. 2000;19(1):104–107.
43. Hussin HM, Biswas S, Majid M, Haynes R, Tole D. A novel technique to treat traumatic corneal perforation in a case of presumed brittle cornea syndrome. Br J Ophthalmol. 2007;91(3):399. doi:10.1136/bjo.2006.102178
44. Kobashi H, Rong SS. Corneal collagen cross-linking for keratoconus: systematic review. Biomed Res Int. 2017;2017:8145651. doi:10.1155/2017/8145651
45. Caporossi A, Mazzotta C, Baiocchi S, Caporossi T. Long-term results of riboflavin ultraviolet a corneal collagen cross-linking for keratoconus in Italy: the Siena eye cross study. Am J Ophthalmol. 2010;149(4):585–593. doi:10.1016/j.ajo.2009.10.021
46. Caporossi A, Mazzotta C, Baiocchi S, Caporossi T, Denaro R, Balestrazzi A. Riboflavin-UVA-induced corneal collagen cross-linking in pediatric patients. Cornea. 2012;31(3):227–231.
47. Wollensak G. Crosslinking treatment of progressive keratoconus: new hope. Curr Opin Ophthalmol. 2006;17(4):356–360. doi:10.1097/01.icu.0000233954.86723.25
48. Wollensak G, Spoerl E, Seiler T. Riboflavin/ultraviolet-a-induced collagen crosslinking for the treatment of keratoconus. Am J Ophthalmol. 2003;135(5):620–627. doi:10.1016/s0002-9394(02)02220-1
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.