Back to Journals » Open Access Journal of Clinical Trials » Volume 18
Bridging the Generalizability Gap: A Methodologic Framework for Enhancing Generalizability in Clinical Trials Across Underrepresented Populations
Authors Williams A ![]() , Horne C
, Horne C ![]() , Freeman K
, Freeman K
Received 11 March 2026
Accepted for publication 23 May 2026
Published 18 June 2026 Volume 2026:18 608458
DOI https://doi.org/10.2147/OAJCT.S608458
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Arthur E. Frankel
Andre Williams,1 Cassandre Horne,1 Katherine Freeman2
1Christine E. Lynn College of Nursing, Florida Atlantic University, Boca Raton, FL, USA; 2Charles E. Schmidt College of Medicine, Florida Atlantic University, Boca Raton, FL, USA
Correspondence: Andre Williams, Email [email protected]
Abstract: Clinical trials constitute a cornerstone of contemporary medicine and healthcare, as they generate the highest level of empirical evidence regarding the safety and efficacy of pharmacological agents and other therapeutic or preventive interventions. Although strategies to address underrepresentation exist, a critical gap in the literature is the lack of comprehensive, operationalized approaches that integrate study design, social determinants of health (SDOH) data, statistical methodologies, reporting practices, and applications of artificial intelligence and machine learning (AI/ML). To address this gap, the Perspective outlines populations that are typically underrepresented, discusses the ramifications of this underrepresentation, and presents a comprehensive set of strategies to enhance representation and promote generalizability in clinical trials. The five components detailed in the following sections are: (1) defining inclusion and exclusion criteria that balance internal and external validity and reflect the broader affected community; (2) collecting and operationalizing SDOH and demographic data to understand factors associated with treatment outcomes; (3) using statistical adjustment techniques to account for diverse representation in results; (4) ensuring comprehensive clinical trial reporting to support transparency and reproducibility; and (5) leveraging AI/ML to optimize study planning and data analysis.
Plain Language Summary: Making Medical Research Work For Everyone: Clinical trials test whether new medicines and treatments are safe and effective. However, many trials do not include enough participants from key groups, such as older adults, racial and ethnic minorities, people with lower incomes, rural residents, and women. When these groups are underrepresented, we cannot be sure if the treatment will work the same way for everyone in real-world settings.
This Perspective explains why this underrepresentation happens and why it matters. More importantly, it provides a practical guide to fix the problem. The authors propose five strategies: (1) using real-world health data to design fairer eligibility rules; (2) collecting and analyzing information about social and demographic factors that affect health; (3) using statistical methods to adjust for trial population imbalances; (4) following updated guidelines for transparent reporting of the clinical trial plan and results; and (5) using artificial intelligence carefully to reduce bias, not amplify it.
These strategies aim to ensure that future clinical trials include the people who need the treatments most. The goal is better, fairer healthcare for all.
Keywords: clinical trial methodology, external validity, underrepresented populations, generalizability, artificial intelligence in clinical research
Introduction
Clinical trials constitute a cornerstone of contemporary medicine and healthcare, as they generate the highest level of empirical evidence regarding the safety and efficacy of pharmacological agents and other therapeutic or preventive interventions.1 The design of a clinical trial is an inherently complex process, primarily aimed at formulating and addressing research questions that estimate the causal effect of a specific intervention on predefined outcomes in well-characterized populations, typically in comparison with established standards of care or placebo.1
Internal and external validity represent two fundamental methodological dimensions that underpin the design, implementation, and appraisal of clinical trials.2 Internal validity is the extent to which a study can establish a definitive cause-and-effect relationship between an intervention and the observed outcomes, ensuring that any changes are attributed solely to the treatment rather than confounding factors.3 To safeguard internal validity, clinical trials routinely incorporate key methodological features, such as randomization, blinding, and control groups, which help minimize confounding and reduce various forms of bias. External validity, by contrast, refers to the extent to which a trial’s results can be generalized to broader real-world patient populations and clinical settings. Internal validity is a necessary, but not sufficient, prerequisite for external validity.2
The relationship between internal and external validity is frequently conceptualized as a potential trade-off. In particular, the use of stringent inclusion and exclusion criteria often enhances internal validity by creating a more homogeneous study sample and reducing sources of variability. However, this increased methodological rigor can limit external validity by constraining the representativeness of the study population, thereby diminishing the applicability and relevance of the findings to routine clinical practice.4 Considerations of cardiovascular drug trials illustrate this concept. Decades of large cardiovascular drug trials have enrolled disproportionately few women relative to their burden of ischemic heart disease, resulting in efficacy and safety data from predominantly male cohorts being generalized to women in routine practice.5 This imbalance has contributed to sex-specific differences in drug response and adverse events, highlighting how non-representative trial populations can obscure true benefit-risk profiles for approximately half of the patient population.6 Additionally, in the field of oncology, many registration trials have excluded or under-enrolled older and frail adults, even though they account for most cancer diagnoses, leading to “standard” chemotherapy regimens that may be poorly tolerated in real-world geriatric populations.7
While internal validity is essential for establishing the efficacy of an intervention within the controlled environment of the trial, it does not guarantee that these findings will apply to the broader population. The external validity of a clinical trial is critically dependent on how well the study’s participant demographics reflect the disease’s prevalence, making underrepresentation a direct threat to the research’s applicability and utility.8 Despite the essential role of clinical trials, a pervasive problem is the underrepresentation of diverse groups, which severely compromises external validity.9 Although strategies to address underrepresentation exist, a critical gap in the literature is the lack of comprehensive, operationalized approaches that integrate study design, social determinants of health (SDOH) data, statistical methodologies, reporting practices, and applications of artificial intelligence and machine learning (AI/ML).
To address this gap, this Perspective outlines populations that are typically underrepresented, discusses the ramifications of this underrepresentation, and presents a comprehensive set of strategies to enhance representation and promote generalizability in clinical trials.
Dimensions of Exclusion: Assessing Underrepresentation
Underrepresentation of groups bearing a significant disease burden in clinical trials undermines a trial’s external validity, and the results cannot be used to accurately estimate its performance in real-world settings.10 As shown in Table 1, groups underrepresented in clinical trials include older adults (age 65 or greater), racial and ethnic minorities, people residing in rural locations, people with lower socioeconomic status (SES), and women.
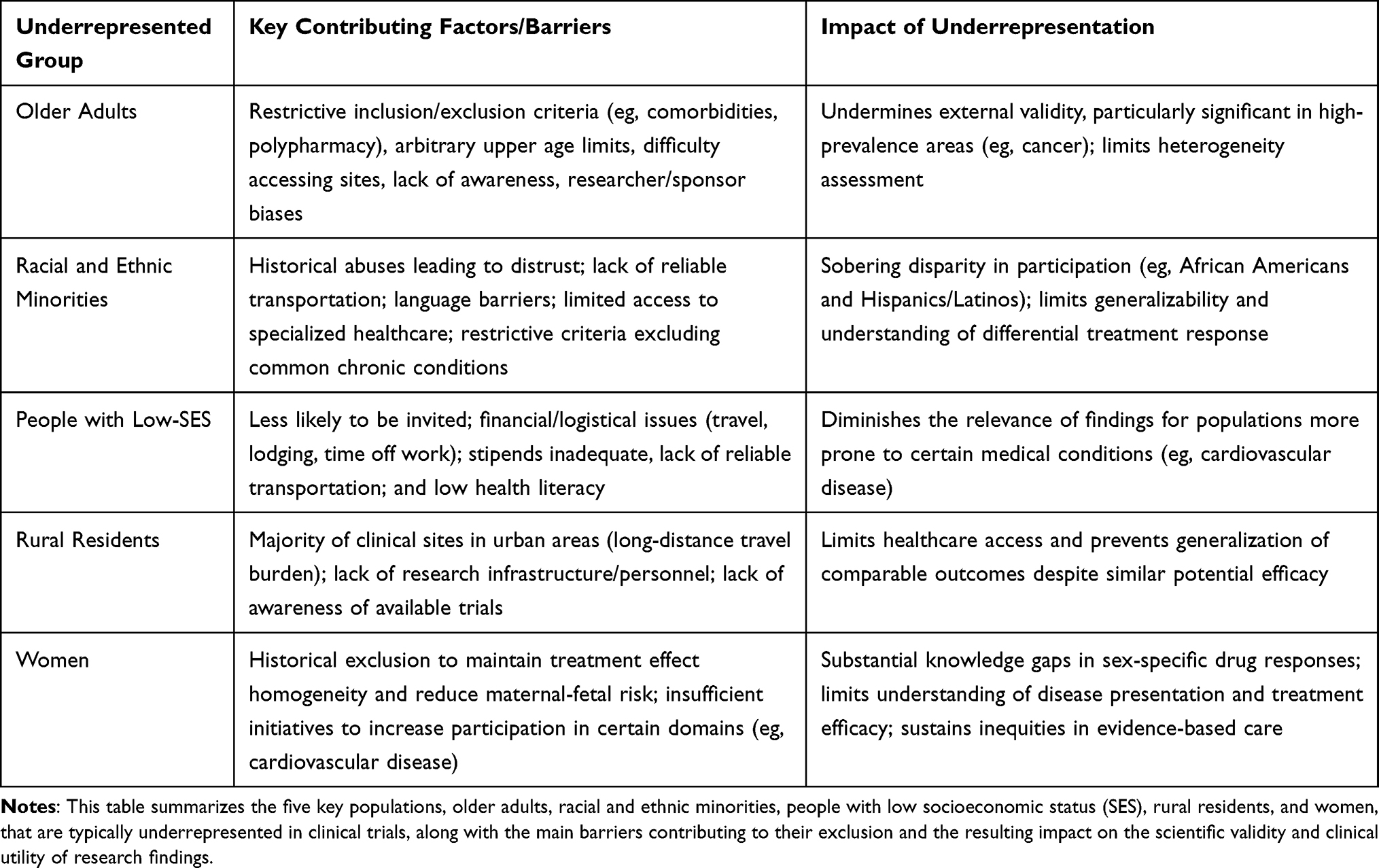 |
Table 1 Underrepresented Populations in Clinical Trials and Contributing Barriers |
Older Adults
Older adults are an essential, and often underrepresented, patient population in clinical trials, given the increased prevalence of chronic diseases and multimorbidity in this population.11 With increasing life expectancy, older adults comprise an increasingly large share of the population; consequently, the implications of their underrepresentation are likely to grow in significance.12 A 2017 systematic review of randomized controlled trials found that only 23% of trials reporting an average age had a mean age over 60 years.13 This underrepresentation is particularly acute in areas such as cancer, where an estimated 42% of cancer patients are 70 years of age or older; furthermore, less than 10% of National Cancer Institute (NCI)-sponsored clinical trials participants are in this age group.14 Factors that may contribute to this underrepresentation include restrictive inclusion and exclusion criteria (eg, prohibitions on comorbidities, polypharmacy, and functional or cognitive limitations)15 and arbitrary upper age limits.15 Other significant barriers include difficulty accessing trial sites and a lack of awareness of trial opportunities.16 Biases and misconceptions of researchers and sponsors (eg, older adults are a difficult, disease-prone population, and fear of an increase in adverse events) contribute to underrepresentation as well.16
Racial and Ethnic Minorities
The underrepresentation of ethnic and racial minorities in clinical research is a profound and longstanding issue.17 Statistics reveal a sobering disparity between the proportion of these groups in the population and their representation in trials. For example, African American and Hispanic/Latino minorities represent 32% of the US population, yet clinical trial participation is estimated at 19%.18 More specifically, Hispanics/Latinos, who make up approximately 18% of the US population, represent 11% of clinical trial participants, and African Americans, at 14% of the population, account for less than 6% of clinical trial participants.18 Many factors contribute to this disparity. Historical abuses like the Tuskegee Syphilis Study and the Guatemala Syphilis Experiments have led to a deeply rooted distrust of the medical establishment, particularly the conduct of clinical trials in communities of color.19–21 Beyond this historical legacy, barriers such as unreliable transportation, language barriers, and limited access to specialized healthcare centers also hinder participation.22 As with many other populations, restrictive eligibility criteria can further exacerbate this issue by frequently excluding those with chronic conditions like diabetes, high blood pressure, and kidney disease, which disproportionately affect racial and ethnic minority populations.9,23,24
People with Low SES
Individuals from low-SES backgrounds, defined by factors such as education, income, and occupation, often participate in clinical trials less often than their counterparts with higher-SES.25 This issue of underrepresentation exists even though there is evidence to indicate that these individuals are more prone to medical conditions, such as cardiovascular disease, than their more affluent and educated counterparts.25 Evidence suggests that those with low SES are less likely to be invited to participate in clinical trials.25 Additional barriers include financial and logistical issues impacting travel and lodging, as well as the inability to take time off work to attend study visits and receive treatment.26 Also, low-SES individuals may lack reliable transportation to study sites or have inflexible work schedules that conflict with clinic visits.23,27–29 Although trials may provide compensation for participants’ time and expense, this does not offset all expenses incurred. Low health literacy is also a common barrier for this demographic group.29 Without a complete understanding of the research process, many potential participants may view the lengthy consent forms and complex trial protocols with alarm and confusion, thereby reducing their likelihood of participating or complying with the study requirements.30 These factors contribute to underrepresentation and diminish the relevance of clinical research findings to people with low SES.
Rural Residents
Approximately 20% of the US population lives in rural areas and is consistently underrepresented in clinical trials.31,32 The main barrier is healthcare access because most clinical sites are in urban areas, and long-distance travel can be financially and logistically overwhelming for those who are ill.33 In addition to geographic barriers, the rural healthcare system may lack research infrastructure and personnel, which can limit the initiation and implementation of clinical trials in rural areas.34 A lack of research infrastructure often makes it difficult for rural residents to have access to clinical trials, even though rural and urban participants achieve comparable outcomes.35,36 This underrepresentation is also compounded by a lack of awareness of available trials in rural populations.37
Women
Historically, medical research has primarily focused on including male participants, frequently excluding women from clinical trials to maintain treatment effect homogeneity and reduce potential maternal-fetal risks.38 This exclusion has resulted in substantial gaps in knowledge regarding sex-specific drug responses and disease manifestations, often requiring the extrapolation of medical recommendations from male-dominated data sets.39 Such bias has led to a limited understanding of disease presentation and treatment efficacy in women, as demonstrated by the delayed recognition of sex-specific symptoms of myocardial infarction.40 This sex bias has contributed to a skewed characterization of the underlying biological mechanisms and may have resulted in suboptimal or less effective therapeutic strategies for female patients.41
The underrepresentation of women in research affects treatment optimization, since pharmacological agents may have different efficacy and safety profiles in women due to variations in pharmacokinetics and pharmacodynamics.42 Although initiatives such as the US National Institutes of Health Revitalization Act of 1993 have sought to increase female participation, women remain underrepresented in many clinical research domains, particularly in cardiovascular disease trials.43 This ongoing disparity restricts the generalizability of clinical trial outcomes and sustains inequities in evidence-based care for women, especially in cardiovascular diseases, where sex-specific differences in disease progression and treatment response are well established.44
Compounding Vulnerabilities: An Intersectional Lens on Clinical Underrepresentation
The population groups previously described as typically underrepresented in clinical trials are not mutually exclusive, as people often share multiple attributes. Examples include an older African American male having low SES with multiple myeloma or a Hispanic/Latino female residing in a rural area with heart failure. Inclusion in multiple groups can complicate matters, as individuals belonging to multiple groups may face a higher burden of disease and an increased risk of exclusion from clinical research. As a result, those most in need of the study’s findings are often underrepresented. Designing studies to be “clean” and internally valid by excluding complex patients, such as older individuals or those with low SES, can skew estimates of treatment effects. This compromises external validity, hindering the generalization of findings to the broader population, a critical issue for effective public health interventions.
Extending Beyond a US-Centric Perspective
While the groups previously described are drawn primarily from the US context, where the preponderance of published research on underrepresentation originates, the patterns of underrepresentation are a global phenomena with region-specific manifestations. In Europe, Romani and socioeconomically disadvantaged populations face similar barriers to participation.45,46 Worldwide, participation in clinical trials is similarly uneven, with North American and European countries routinely accounting for a disproportionately large share of global clinical trial participants; differences between countries account for more than 90% of the variation in participation rates.47
Systemic Erosion: Impact on Scientific Integrity, Policy, and Precision Medicine
Table 2 illustrates four consequences of underrepresentation in clinical trials.
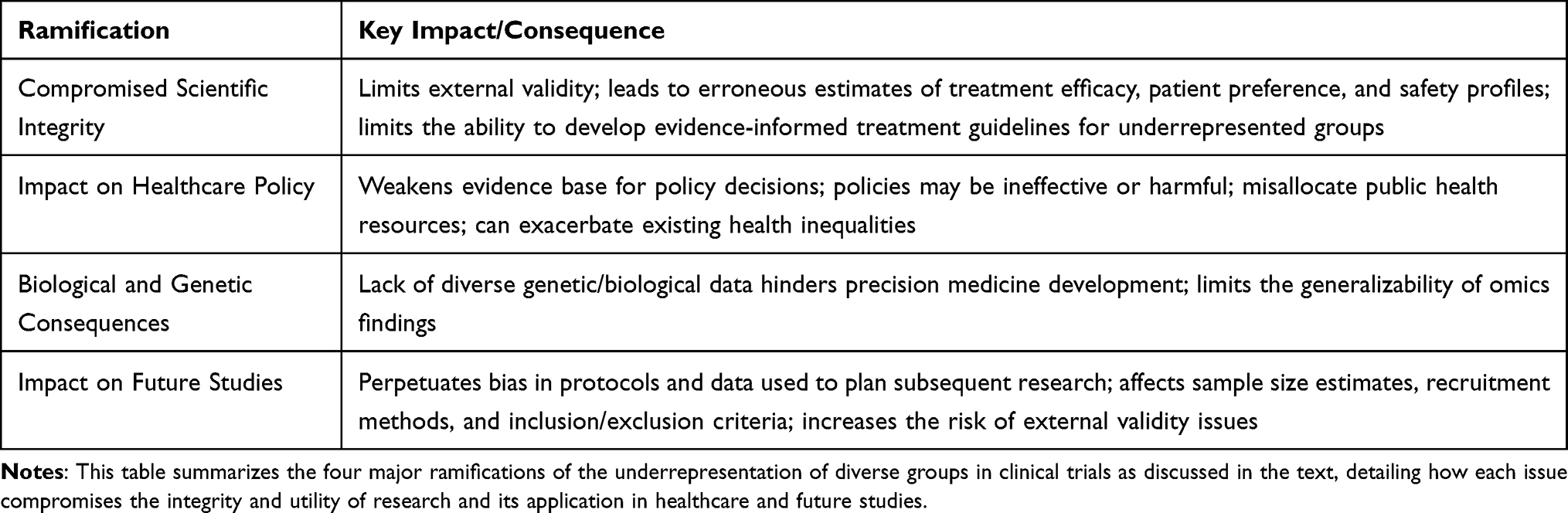 |
Table 2 Ramifications of Underrepresentation in Clinical Trials |
Compromised Scientific Integrity
Underrepresentation of different groups in clinical trials is not only a social or ethical issue; it also has a profound scientific effect on study results by limiting external validity.48 When trials are based on a homogeneous population or a population whose characteristics do not accurately reflect those of the population bearing the burden of the disease, the results may not be generalizable to the relevant patient population.10 This can lead to erroneous estimates of treatment efficacy, patient preferences, and the treatment’s safety profile. Without sufficient data from a particular population group, there is limited ability to assess how that group will respond to a treatment, and it is almost impossible to develop evidence-informed treatment guidelines for this group.
Impact on Healthcare Policy
Healthcare policy should be guided by research studies that clearly demonstrate the cause-and-effect relationship of a treatment and its applicability in real-world settings. If trials are conducted on populations whose traits do not closely match those of real-world groups, any policy based on that research may be ineffective, as the evidence supporting its value is weakened. Similarly, if trials focus on groups in which older patients are severely underrepresented but account for more than half of new disease cases, the treatment effect derived from the trial may not accurately reflect the response of this group. Consider a stark hypothetical. A government pours $100 million into treating an underserved community for a specific disease, confident, based on the overall clinical trial data, that 85% of patients will see a meaningful benefit. But buried in fine print is a crucial detail: only a small number of people from this underserved group were actually in the trial, and among them, the true success rate was just 45%. In reality, this policy fails. The money produces little to no societal benefit for a community that carries a heavy share of the disease burden. Worse, policies based on such unrepresentative data do not just “miss the mark”, so to speak; they can actively harm populations that were never properly included in the evidence to begin with. This can lead to the misallocation of public health resources, directing money toward treatments that are effective in the artificial setting of a clinical trial but are not effective in practice. It can also threaten to exacerbate existing health inequalities if policies based on unrepresentative data unintentionally disadvantage or harm underserved communities in the effort to achieve equitable healthcare for all.
Biological and Genetic Consequences
This issue of underrepresentation also has biological implications. Genetic and biological differences among populations can significantly impact how an individual responds to treatment.49 The more diverse the trial participants, the stronger the clinical trial data become for understanding a drug’s benefits and risks. Biological variations are becoming increasingly critical as medicine moves toward personalized and precision therapies tailored to an individual’s genotype; however, for this to happen, sufficient data must be collected on each genotype to fully understand its interaction with the treatment.50 Indeed, the disproportionate dominance of European ancestry individuals in large biorepositories and genome-wide association studies, particularly in cancer research, severely limits the generalizability of precision omics findings to diverse patient populations.51
Impact on Future Studies
Planning clinical trials requires a large investment of time, money, and staffing resources to improve the likelihood of internal and external validity. Inadequate planning, particularly regarding primary outcomes, decreases the likelihood of clinical trials’ success. Additionally, the trial protocol and other relevant documents should be carefully prepared, submitted, and approved by an institutional review board (IRB) and other relevant authorities before the first patient is enrolled. Regarding the statistical analysis plan (SAP), it should be drafted before the database lock and ideally pre-written, pre-registered, and submitted for publication before enrollment begins. This will enhance the study’s integrity, as observing data distributions after the database lock will affect SAP if it was not finalized beforehand. Although it is possible to make changes to the IRB post hoc, such changes should be kept to a minimum. To ensure consistent treatment effects, it is essential to maintain uniformity in experimental conditions from the first participant’s entry to the last patient’s exit, with a Data Monitoring Committee (DMC) in place.
When planning a study, it is common to use protocols and data from previous research.52 However, if bias existed in the referenced study’s protocols and data (defined as underrepresentation and the lack of consideration for key underrepresented groups bearing a significant disease burden), it would perpetuate bias in current and future study planning that relies on the reference for sample size estimates, recruitment and retention methods, and inclusion and exclusion criteria.
It is worth noting that obtaining a sufficient sample size for subgroup analyses is challenging, limiting the ability to accurately examine differences in treatment effects across subgroups of interest. Moreover, effective recruitment strategies are needed to improve the inclusion of underrepresented groups. The absence of these key study design concepts, or the presence of substantial in-group-related bias in past study protocols and SAPs, can unintentionally transfer these biases to new trial protocols and SAPs. Unless curbed, this unintentional, seamless transfer will continue, reinforcing biases in subsequent clinical trials and perpetuating underrepresentation and biased results.
Sample size calculations and preliminary analyses of historical data guide the planning of future trials by drawing on information from previous studies.53–55 If certain groups are underrepresented in the data from previous studies, statistical considerations could be affected – including variance, effect sizes, and other relevant factors used to determine the sample size. Sample size calculations and power analysis are also critical to ensure the study is adequately powered to detect a clinically and statistically meaningful treatment effect. Bias in historical data will influence both sample size calculation and any preliminary statistical analysis during the planning stages, and will also affect trial execution. Such bias can be perpetuated, and relying on these studies for future research increases the risk of external validity issues. Unless the methodology for protocol development and historical data analysis is corrected, these issues will persist and propagate into future research. A key step in addressing and removing these biases involves updating protocols and related documents to reduce or eliminate exclusionary practices for underrepresented groups. This ensures the research plan focuses on recruiting and retaining a study population that accurately reflects the characteristics of the real-world population affected by the disease under consideration.
A Methodological Blueprint for Inclusive Clinical Research
Conceptual Framework
This Perspective does not present primary empirical data; instead, we propose a methodological framework to enhance the representativeness and generalizability of clinical trials. We synthesized conceptual and practical guidance into a five-component methodological framework. As shown in Table 3, this framework addresses key areas of clinical trial design and execution that are necessary to enhance external validity while rigorously maintaining internal validity, thereby ensuring that research findings are generalizable to real-world populations bearing the burden of disease. The five components detailed in the following sections are: Rationalizing Inclusion and Exclusion via Real-World Data (RWD), Operationalizing SDOH as Primary Research Drivers, applying Statistical Guardrails (Weighting, Imputation, and Sensitivity Analysis), adhering to Transparency Standards (SPIRIT and CONSORT Guidelines), and leveraging Algorithmic Equity (AI/ML for Bias Mitigation and Synthetic Data Generation).
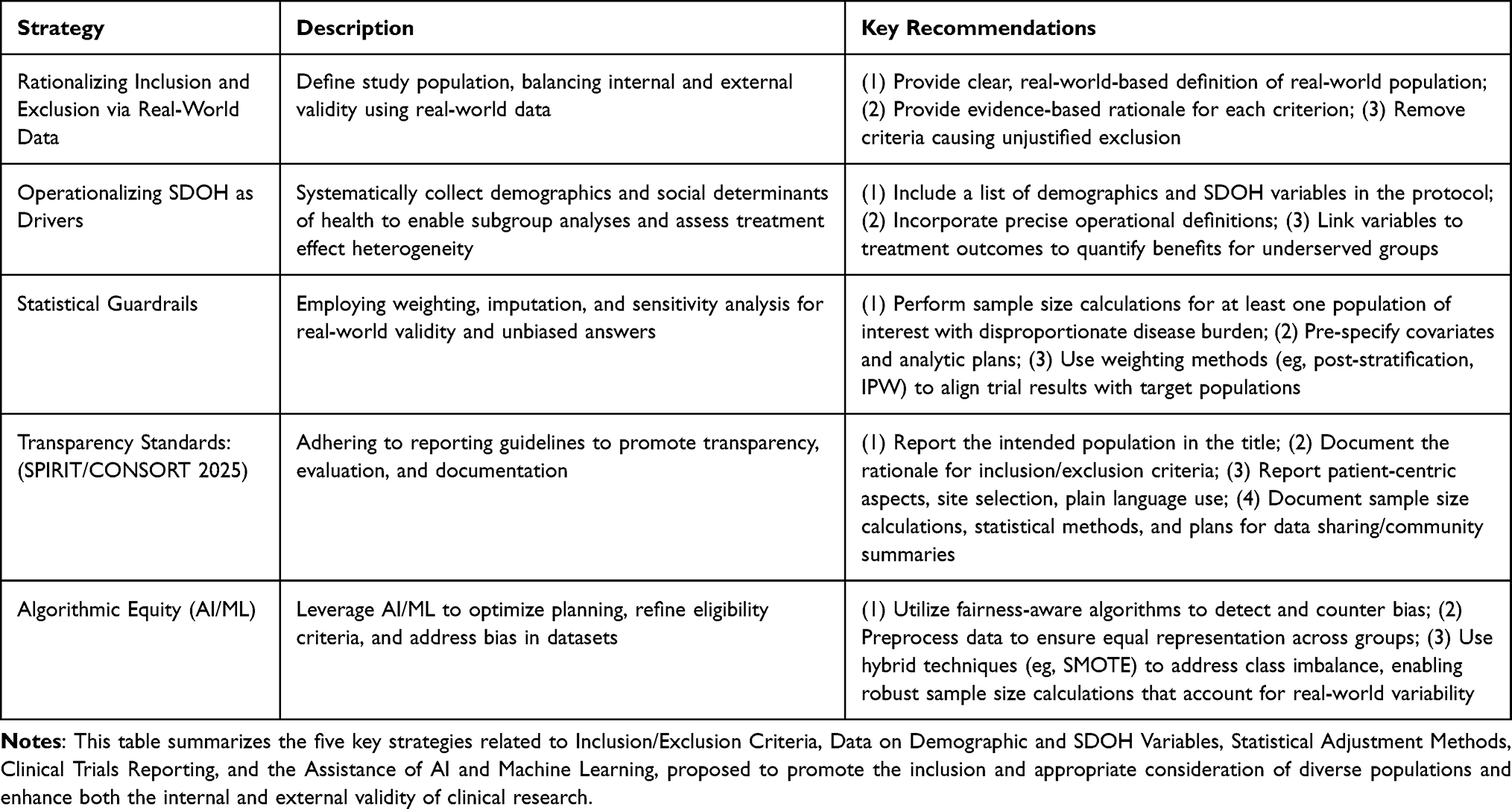 |
Table 3 Summary of Key Strategies for Enhancing Representation in Clinical Trials |
Rationalizing Inclusion and Exclusion via RWD
Establishing inclusion and exclusion criteria is a crucial part of trial planning.56 In addition to determining who is eligible to enter the study, the practice also helps define the ideal study population to support internal validity. When creating the inclusion and exclusion criteria, it is essential to ensure that both internal and external validity are preserved. It is recommended not to copy information from a previous trial protocol but to intentionally create these criteria. Some tips to accomplish this task include:
- Provide a clear and robust definition of the real-world population that bears the disease burden. RWD can be employed to accomplish this task. Typical sources include electronic health records, insurance claims, patient registries, and national health surveys. These sources help identify common comorbidities, concomitant medications, age ranges, and other pertinent attributes observed in the population bearing the disease burden. The study population, defined by the inclusion and exclusion criteria, should always be compared against its real-world counterpart.52
- Provide a clear, evidence-based rationale for each criterion.
- For each inclusion and exclusion rule, document its impact on the scientific, ethical, and safety elements of the study. How will the criteria affect the assessment of the treatment effect, generalizability, safety outcomes, measurement issues, and other related issues?
- If any inclusion and exclusion criteria lead to the unjustified exclusion of any group (eg, older adults, mild comorbid conditions, stable psychiatric conditions) without any clear mechanistic or safety justification, consider removing the criteria.
- Using real‑world data (RWD), prior clinical trial data, and relevant published evidence, systematically assess whether a proposed inclusion or exclusion criterion influences the estimation or interpretation of the treatment effect and/or modifies the composition of the study population in a way that meaningfully diverges from the characteristics of the populations that bear the primary burden of the disease.
Striking the right balance is crucial in any effort to create inclusion and exclusion criteria while maintaining both internal and external validity.
Operationalizing SDOH as Primary Research Drivers
SDOH are “the non-medical factors that influence health outcomes”.57,58 They are the “conditions in which people are born, grow, live, work, and age”.59 Relevant demographic and SDOH variables include age, race and ethnicity, gender (sex assigned at birth), income and socioeconomic status, food security, insurance status, perceived discrimination, stable employment, education level, health literacy, social isolation, housing stability, geographic location, and marital status. The Centers for Disease Control and Prevention (CDC)57 and Healthy People 203060 have categorized SDOH into five key domains. These domains are:
1. Economic Stability
2. Education Access and Quality
3. Social and Community Context
4. Neighborhood and the Built Environment
5. Healthcare Access and Quality
In clinical trials, an essential component is the evaluation of treatment effect heterogeneity. To assess the impact of the treatment on a group of interest, that group must be included in the data and identified via a relevant demographic or SDOH variable. By explicitly specifying SDOH and demographic variables for subgroup analysis, investigators can evaluate the treatment-outcome relationship across groups defined by economic stability, healthcare access, or any other variable of interest. This approach transforms SDOH and demographic fields from a necessary operational task to a primary driver of treatment effects in underserved groups. Such evidence can inform researchers, patients, and policymakers about the benefits of treatments for those groups who might need them most. Considerations for implementation are provided.
- Include a list of demographics and SDOH variables, as well as any additional relevant information, to be collected in the protocol.
- For the SDOH that will be assessed via survey, place a copy in the protocol appendix.
- Incorporate demographic and SDOH variables, accompanied by precise operational definitions, in the plan for data collection within the data management strategy. Also include them in a detailed plan for screening and baseline evaluation, a data dictionary, case report forms, and any additional pertinent data documentation.
- SDOH and demographic data collection should be conducted in a manner that is respectful, culturally sensitive, and patient-centered, with appropriate training for study staff to promote trust, minimize discomfort, and improve the completeness and accuracy of responses.
By rigorously collecting and linking demographic and SDOH variables to treatment outcomes, researchers can move beyond general population effects to explicitly quantifying the benefits of the intervention for the specific underserved groups who need them most, driving evidence-based health for all.
Statistical Guardrails: Weighting, Imputation, and Sensitivity Analysis
Statistical analysis aims to produce unbiased answers to research questions. The methodology should be chosen to focus on answering the research questions, while keeping the study design and data-related factors in mind (such as data quality and missingness). When writing the SAP, it is critical to describe how the analysis supports and maintains the study’s internal and external validity and how it provides concrete answers to research questions. The analytic plan should maintain internal validity by controlling for confounding and ensuring that the treatment’s true causal effect on the outcomes of interest is computed. When applied judiciously, analytical methodology should augment, rather than replace, essential study design safeguards in the protocol. The primary focus is on demographic and SDOH covariates; however, these considerations also apply to all other covariates considered.
- Sample size calculation considerations
- If pilot data are used, compare its demographic distribution with that of the target population (via RWD) for the trial to assess the relevance of the data for the calculations, with documentation included in the SAP.
- If estimates for effect sizes, variance, and the like are provided from scientific publications, examine the demographic descriptions and compare them with similar information for the target population to assess the relevance of the information for performing the calculations, with documentation included in SAP; discuss these limitations in manuscript limitations sections.
- For at least one population of interest that bears a disproportionate burden of the disease under consideration (eg, older adults, African Americans, individuals with low SES), perform sample size calculations to evaluate the minimum sample size needed to causally assess the treatment effect in that group.
- Pre-specify covariates and analytic plans
- Identify prognostic, stratification, and effect-modifying variables based on prior evidence or clinical logic. This information should be included in the SAP.
- The involvement of demographic and SDOH variables in the analysis to evaluate primary, secondary, tertiary, and exploratory (including subgroup analysis) aims should be clearly specified, and if demographic and SDOH variables are included in any sensitivity, interim, or other analyses, this should be clearly defined in the SAP.
- Finalize, pre-register, and even submit for publication, the SAP that defines all modeling, adjustment techniques, subgroup analyses, and sensitivity analyses to avoid data-driven bias. This should be completed before the database lock, with additional advice that it be done before the first patient enters the study.
- Modeling Considerations
- Ensure the sample size is large enough to estimate all main effects and interaction terms. A common rule of thumb is n = 10 (subjects or visits, depending on the data structure) per continuous covariate and per level of each categorical variable. Choose the number of main effects and interaction terms so that this requirement is met, given the projected sample size.
- Consider using shrinkage or regularization-based models, such as elastic-net regression, when including a relatively large set of covariates to prevent overfitting.
- Specify how the results of the modeling procedure should be interpreted. If the corresponding analysis addresses primary, secondary, exploratory, or sensitivity aims (including subgroup analyses), provide explicit instructions for interpreting the results in clinical trial and real-world contexts.
- Report the diagnostics and assessment of model fit for each model presented.
- Address nonadherence and protocol deviations carefully. As an exploratory analysis, perform a sensitivity analysis on the Per Protocol (PP) population. Evaluate if the characteristics of the PP group deviate from those of the intended population; are there particular groups that are not meeting adherence standards and becoming noncompliant? This will provide great insight into the ability of certain people (or groups) to adhere to the treatment regimen.
- Handling missing data. If missingness persists in the collected data, even after mitigation strategies have been implemented, perform multiple imputations under appropriate assumptions about the missingness pattern (missing at random). This will ensure that sufficient data are available to conduct the analysis and assess statistical significance.
- Use weighting methods to align trial results with trial populations
- Standardization and post-stratification involve adjusting trial estimates by reweighting them to align with the clinical and demographic characteristics of a specific target population, such as a national registry or health system. This process enhances real-world applicability by correcting discrepancies between the target population and study participants. Operationally, this is achieved by determining the joint distribution of SDOH and demographic variables within a population source, such as a national survey or an EHR network, and then applying weights to ensure the trial sample reflects that distribution.
- Inverse Probability Weighting (IPW): This method involves modeling participant inclusion probabilities based on essential demographic and SDOH covariates. By weighting each individual by the inverse of their calculated inclusion probability, researchers can apply statistical models that produce estimates that account for groups previously underrepresented in a trial. Ultimately, IPW aligns trial findings with broader populations by adjusting for these conditional inclusion factors.
These techniques lead to sample size calculations, analytic plans, and executed data analyses that promote external validity while maintaining internal validity. The aim is to ensure that the results are representative of the real-world population bearing the burden of the disease under consideration, while preserving internal validity, thus supporting evidence-based practice for all.
Transparency Standards: Adhering to SPIRIT and CONSORT Guidelines
When reporting a protocol designed to ensure internal and external validity, it is essential to adhere to the reporting guidelines to ensure consistency and facilitate comparison and evaluation across studies. This approach creates an ideal setting for a thorough evaluation of the value of studies in promoting both external and internal validity. An example of such a guideline is the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) reporting guideline.61 Evidence-based guidelines are provided.
- Describe the intended clinical trial population in the title. This is the first place to report on efforts to increase representativeness.
- Report on inclusion and exclusion criteria. Using previously recommended strategies, report on eligibility criteria (along with a rationale that explains how each will contribute to external and internal validity).
- Report on the patent-centric aspects of the study design. How are community partners and patient stakeholders involved in the design and execution of the study?
- Report on site selection. Report on using sites that represent actual care settings, such as community clinics and rural hospitals, and outline the representativeness strategy; how these sites ensure the retention and recruitment of a study population that will match its real-world counterpart. Additionally, summarize the plan to retain this population throughout the study.
- Report on the use of plain language in patient-focused materials. If the target population is multilingual, report on the translation of relevant materials into the appropriate languages and the use of interpreter services.
- Report on the development of culturally tailored outreach materials and consent scripts to address issues of trust and health literacy.
- Document the reductions of participant burden through flexible schedules, remote options, and simple data collection methods, ensuring uniform assessments across locations.
- Document the selection of patient-relevant outcomes (symptoms, function, quality of life, treatment burden) and ensure measures are validated, culturally adapted, and centrally adjudicated.
- Report on the use of teach-back checklists to be employed when communicating crucial information to study participants. Teach-back checklists are standardized tools used to verify a research subject’s understanding of key information by asking them to restate or demonstrate what they have learned.
- Document standardized data collection. The collection of demographics and SDOH data should be clearly explained.
- Document sample size calculations and study power on real-world variability and calculations that were performed for any subgroups of interest.
- Document the statistical methods in the study. For each primary and secondary aim, clearly and explicitly explain the analytic approach. Clearly specify who will be included in the analysis. Clearly explain how missing data will be dealt with. Also, clearly describe any preplanned exploratory, subgroup, and sensitivity analysis.
- Describe oversight operations employing dynamic risk-based strategies to meticulously identify site-level challenges that threaten representativeness, while simultaneously implementing retention measures to decisively thwart any potential bias in the study population.
- Commit to data sharing and community-accessible summaries in the dissemination plan. Include an assessment of representativeness and subgroup findings, with findings reviewed by patient and community partners. This ensures that community partners and patients can directly benefit from clinical trial research.
It is recommended to develop the protocol by using a checklist based on SPIRIT guidelines. Additionally, all relevant Standard Operating Procedures (SOPs) related to the development of this study’s protocol should be included, with copies placed in the protocol’s appendix. The Consolidated Standards of Reporting Trials (CONSORT) checklist should also be used to report clinical trial results and consulted during the planning stage for additional guidance.62 These recommendations comply with SPIRIT and CONSORT checklists and encompass design, operational, and analytical aspects that enhance external validity without compromising internal validity.
Algorithmic Equity: Leveraging AI/ML for Bias Mitigation and Synthetic Data Generation
AI/ML are being used to address many of the challenges in clinical research,63 such as high failure rates and high costs. By enhancing design and conduct, AI/ML technologies can improve the quality of clinical study planning and execution.64 Employing AI/ML to analyze RWD could yield insights to optimize clinical trial design and thereby increase the success rate of clinical trials.65 Optimizing design involves optimizing patient selection and recruitment, which are the main factors causing trial failure and extended timelines.64,65 For example, AI systems could analyze large datasets from past clinical trials to refine eligibility criteria, thereby informing ideal patient selection.66 AI/ML can help clinical trial designers pinpoint and apply the key drivers of success to create improved study designs, potentially shorten trial timelines, and lower overall study costs.64,67,68
While AI/ML offers remarkable capabilities, it is not without imperfections. The strength of AI/ML lies in its ability to analyze large volumes of data. However, complications arise when historical data are biased, and algorithms then absorb and amplify them. This results in what is termed algorithmic bias, in which algorithms yield biased or consistently unjust outcomes.69 If AI/ML systems are trained on data from biased or incomplete protocols and clinical trials, they will likewise recognize and perpetuate this bias in subsequent analyses, exacerbating the issue rather than effectively addressing it.70 Additionally, if the AI fails to adequately encounter concepts related to generalizability in the training data but is asked to generate information about them for future studies, this can potentially lead to AI hallucinations.71,72 This “hallucinations”-based output will be plausibly presented but will be grossly incorrect. As such, surreal or nonsensical content will be introduced into the study planning process, leading to additional errors in the study’s execution.
To combat these biases, several techniques can be utilized. For instance, applying fairness-aware algorithms explicitly designed to detect and counter bias can address the issue.73 Further, preprocessing data to ensure equal representation across various demographic and SDOH groups can also reduce bias.74,75 Regularly auditing AI/ML systems to evaluate their outputs for signs of bias is also key. Techniques such as reweighting, which adjusts data points to ensure fair representation, and adversarial debiasing, which uses a second AI to detect and mitigate bias in the initial model, can also help reduce algorithmic bias.76,77 These steps can help foster responsible AI/ML implementation, ensuring that clinical trials and other research efforts benefit from unbiased and fair insights.
One particularly relevant approach is to use data preprocessing techniques to reduce or eliminate bias in clinical study datasets before applying statistical or computational analyses. This is particularly useful for determining an appropriate sample size via sample size calculations and power analysis. These preprocessing techniques would be applied to historical data during the trial planning phase, rather than to data generated by the clinical trial itself. Sample size calculations in clinical studies typically utilize data from studies in which certain populations of interest are underrepresented. There is an imbalance in the data, with some groups having larger sample sizes while others (underrepresented populations) have very small sample sizes. Therefore, sample size calculations may not adequately account for the influence of such populations, and studies may be underpowered to detect a statistically significant difference between two treatments, even when a clinically meaningful difference exists.78,79 Data preprocessing techniques can powerfully elevate underrepresented groups by generating synthetic data points that mirror real-world patterns while staying safely distinct from the original entries. This approach facilitates more robust sample size calculations for studies aiming for representativeness.80 Simulations using real-world data, characterized by larger sample sizes and greater variability across underrepresented groups, yield more precise sample-size estimates for studies that include these populations.
There are various AI/ML-based approaches that can address these imbalance problems in research. These algorithms can be classified into three groups:
- Undersampling (Reducing the Majority). These methods aim to reduce the number of samples in the oversampled group.81
- Oversampling (Increasing the Minority). These methods aim to increase the number of samples in the underrepresented group.82
- Hybrid Techniques (A combination of the two previous groups).83
Hybrid Techniques are data-driven methods that address class imbalance by combining under- and oversampling. The goal is to provide the benefits of both techniques: increasing minority-class representation while reducing noise and overlap across population groups. This results in data that is more representative and has better-defined class boundaries. This modified data, with reduced bias, can then be used in AI/ML algorithms and statistical models to generate outputs that truly inform the planning of studies aimed at promoting external validity while preserving internal validity. A widely used method is the Synthetic Minority Oversampling Technique (SMOTE)84 for oversampling the minority class, combined with under-sampling techniques such as Tomek links and Edited Nearest Neighbors.81 Additional research is needed to evaluate the use of these hybrid techniques in generating clinical trial data with reduced bias. The use of hybrid sampling techniques to mitigate the underrepresentation of various patient populations in clinical trial data is beneficial for advancing clinical trials toward more robust external validity and ensuring that medical breakthroughs benefit all patients.65
Conclusion: Moving Towards Evidence-Based Health for All
This Perspective has provided a methodological framework to enhance the generalizability and representativeness of clinical trial results. We have documented the pervasive underrepresentation of various groups, namely older adults, racial and ethnic minorities, individuals with low SES status, rural residents, and women. We have demonstrated how this underrepresentation undermines external validity, compromises scientific integrity, distorts healthcare policy, perpetuates bias in future studies, and hinders the advancement of precision medicine.
Addressing underrepresentation in clinical research requires multiple strategies, as shown in Figure 1. Five were highlighted: (1) defining inclusion and exclusion criteria that balance internal and external validity and reflect the broader affected community; (2) collecting and operationalizing SDOH and demographic data to understand factors influencing outcomes; (3) using statistical adjustment techniques to account for diverse representation in results; (4) ensuring comprehensive clinical trial reporting to support transparency and reproducibility; and (5) leveraging AI/ML to optimize study planning and data analysis. These strategies, alone or combined, should identify and reduce exclusionary practices during planning and implementation. Any remaining bias should be documented in the limitations of the clinical study report and subsequent publications.
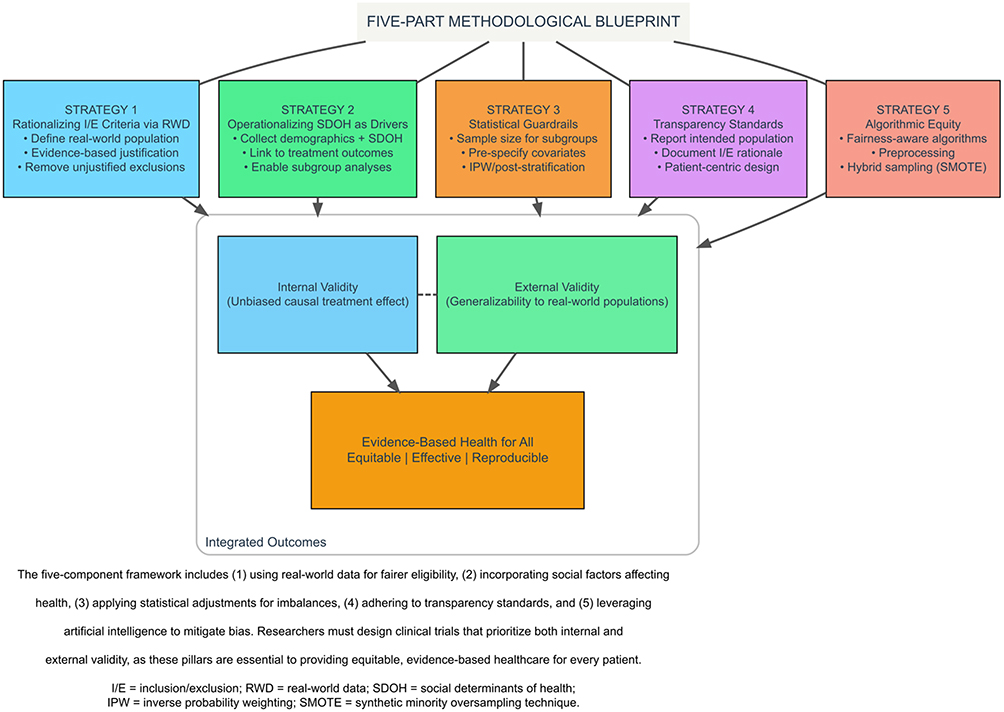 |
Figure 1 Balancing Internal and External Validity in Clinical Trials: A Comprehensive Methodological Framework. |
Implementing these strategies faces several key challenges. First, collecting SDOH data requires additional resources, training, and patient time, further straining limited research budgets and participant availability. Second, statistical adjustment methods like IPW and post-stratification require large sample sizes and correctly specified models; misspecification can introduce new biases. Third, AI/ML methods risk amplifying existing biases if the training data are unrepresentative or if bias-correction approaches are not used intentionally. Fourth, applying these strategies beyond the US requires adapting them to diverse healthcare systems, cultures, and regulatory frameworks, an important direction for future work.
Several valid criticisms of this framework merit acknowledgment. First, some methodologists argue that efforts to enhance generalizability inevitably weaken internal validity beyond what statistical adjustment can repair; we maintain that excluding whole populations is usually more costly than modest losses in internal validity, though this trade-off is context-dependent. Second, the operational demands of SDOH data collection, powered subgroup analyses, and decentralized infrastructure may be prohibitive in resource-limited settings; we therefore recommend incremental, context-sensitive adoption rather than universal implementation. Third, weighting, imputation, and AI/ML bias-mitigation methods can themselves introduce bias when assumptions are violated; rigorous sensitivity analyses and transparent reporting are essential. Fourth, the framework has not been tested outside high-income countries with strong data systems, underscoring the need for implementation research in low- and middle-income settings. Fifth, subgroup analyses are often underpowered and should generally be seen as hypothesis-generating. Finally, the ethical tension between inclusion and protection of vulnerable groups requires case-by-case judgment rather than automatic inclusion. These concerns do not invalidate the framework but highlight the need for empirical validation, ongoing methodological debate, and context-sensitive use.
Putting this framework into practice requires coordinated action. Researchers should pre-register SAPs with subgroup analyses for underrepresented groups, use RWD to justify eligibility criteria, report representativeness using SPIRIT and CONSORT, and co-design studies with patients. Sponsors should mandate SDOH data collection, require diversity plans with RWD-based enrollment targets, fund validation of AI/ML bias-mitigation methods, and support decentralized trials to reach rural and low-SES populations. Regulators should enforce diversity guidance with measurable milestones, require external validity assessments for marketing applications, and create approval pathways for AI/ML-based eligibility-optimization tools. Journal editors and reviewers should consistently enforce adherence to SPIRIT/CONSORT reporting guidelines, require plain-language summaries, and prioritize the publication of negative findings regarding generalizability. Patient and community partners should seek roles on trial steering committees, co-design recruitment and retention strategies, and demand accessible summaries of results. Only through these coordinated efforts can we bridge the gap between trial efficacy and real-world effectiveness, ensuring trials are both rigorous and representative of the diverse populations who will use the treatments.
The ultimate goal of this framework is to move beyond the current reality of underrepresentation and fulfill the promise of truly equitable evidence-based medicine. By proactively integrating external validity into trial design, the scientific community can secure the trust of diverse populations and accelerate the development of inclusive treatments. This approach also ensures that medical progress translates into tangible, better health outcomes for every individual, irrespective of their background or circumstance.
Declaration of Generative AI and AI-Assisted Technologies in the Manuscript Preparation Process
During the preparation of this work, the authors used Grammarly (Version v1.2.234.1829) and Writefull (Version 2025.50.0 (#1239)) to improve the manuscript’s language and readability. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the publication’s final content.
Abbreviations
AI/ML, Artificial Intelligence and Machine Learning; CDC, Centers for Disease Control and Prevention; CONSORT, Consolidated Standards of Reporting Trials; DMC, Data Monitoring Committee; IPW, Inverse Probability Weighting; IRB, Institutional Review Board; NCI, National Cancer Institute; PP, Per Protocol; RWD, Real World Data; SAP, Statistical Analysis Plan; SDOH, Social Determinants of Health; SES, Socioeconomic Status; SMOTE, Synthetic Minority Oversampling Technique; SOPs, Standard Operating Procedures; SPIRIT, Standard Protocol Items, Recommendations for Interventional Trials.
Acknowledgments
This paper is available as a preprint on SSRN at: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=6210032.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors did not receive financial support for this article’s research, writing, or publication.
Disclosure
All authors are employees of Florida Atlantic University. All authors report no financial interests or potential conflicts of interest for this work.
References
1. Esmail L, Barasky RJ, Mittman BS, Hickam DH. Improving comparative effectiveness research of complex health interventions: standards from the Patient-Centered Outcomes Research Institute (PCORI). J Gen Intern Med. 2020;35:875–17. doi:10.1007/s11606-020-06093-6
2. Kennedy-Martin T, Hulin-Curtis SL, Faries DE, Robinson S, Johnston JA. A literature review on the representativeness of randomized controlled trial samples and implications for the external validity of trial results. Trials. 2015;16(1). doi:10.1186/s13063-015-1023-4
3. Kite ME, Whitley BE. The internal validity of research. In. 2025;261–291. doi:10.4324/9781032720609-7
4. Steckler A, McLeroy KR. The importance of external validity. Am J Public Health. 2007;98(1):9–10. doi:10.2105/ajph.2007.126847
5. Tobb K, Kocher M, Bullock-Palmer RP. Underrepresentation of women in cardiovascular trials- it is time to shatter this glass ceiling. Am Heart J Plus Cardiol Res Pract. 2022;13:100109. doi:10.1016/j.ahjo.2022.100109
6. Tamargo J, Rosano G, Walther T, et al. Gender differences in the effects of cardiovascular drugs. Eur Heart J. 2017;3(3):163–182. doi:10.1093/ehjcvp/pvw042
7. Williams GR, Outlaw D, Harvey RD, Lichtman SM, Zamboni WC, Giri S. Chemotherapy dosing in older adults with cancer: one size does NOT fit all. J Geriatr Oncol. 2023;14(1):101363. doi:10.1016/j.jgo.2022.08.012
8. Routen A, Bodicoat DH, Willis A, Treweek S, Paget S, Khunti K. Tackling the lack of diversity in health research. Br J Gen Pract. 2022;72(722):444–447. doi:10.3399/bjgp22x720665
9. Peters U, Turner B, Álvarez D, et al. Considerations for embedding inclusive research principles in the design and execution of clinical trials. Ther Innov Regul Sci. 2022;57(2):186–195. doi:10.1007/s43441-022-00464-3
10. Stuart EA, Ackerman B, Westreich D. Generalizability of randomized trial results to target populations. Res Soc Work Pract. 2017;28(5):532–537. doi:10.1177/1049731517720730
11. Herrera AP, Snipes SA, King DW, Torres-Vigil I, Goldberg DS, Weinberg AD. Disparate inclusion of older adults in clinical trials: priorities and opportunities for policy and practice change. Am J Public Health. 2010;100:100. doi:10.2105/ajph.2009.162982
12. Lockett J, Sauma S, Radziszewska B, Bernard M. Adequacy of inclusion of older adults in NIH-funded phase III clinical trials. J Am Geriatr Soc. 2019;67(2):218–222. doi:10.1111/jgs.15786
13. van Deudekom FJ, Postmus I, van der Ham DJ, et al. External validity of randomized controlled trials in older adults, a systematic review. PLoS One. 2017;12(3):e0174053. doi:10.1371/journal.pone.0174053
14. Huang LW, Wang S. Cancer clinical trial enrollment in older vs younger adults. JAMA Network Open. 2022;5(10):e2235718. doi:10.1001/jamanetworkopen.2022.35718
15. Pitkälä K, Strandberg T. Clinical trials in older people. Age Ageing. 2022;51(5). doi:10.1093/ageing/afab282
16. LeCroy MN, Potter LN, Bandeen-Roche K, et al. Barriers to and solutions for representative inclusion across the lifespan and in life course research: the need for structural competency highlighted by the COVID-19 pandemic. J Clin Transl Sci. 2022;7(1). doi:10.1017/cts.2022.510
17. Oh SS, Galanter J, Thakur N, et al. Diversity in clinical and biomedical research: a promise yet to be fulfilled. PLoS Med. 2015;12(12):e1001918. doi:10.1371/journal.pmed.1001918
18. Kelsey MD, Patrick-Lake B, Abdulai RM, et al. Inclusion and diversity in clinical trials: actionable steps to drive lasting change. Contemp Clin Trials. 2022;116:106740. doi:10.1016/j.cct.2022.106740
19. Allison K, Patel D, Kaur R. Assessing multiple factors affecting minority participation in clinical trials: development of the clinical trials participation barriers survey. Cureus. 2022. doi:10.7759/cureus.24424
20. Bohler F, Aggarwal ND, Peters GW. Re: the FDA initiative to assure racial and ethnic diversity in clinical trials. J Am Board Fam Med. 2023;36(6):1095–1096. doi:10.3122/jabfm.2023.230210r0
21. Rodriguez MA, García R. First, do no harm: the US sexually transmitted disease experiments in Guatemala. Am J Public Health. 2013;103(12):2122–2126. doi:10.2105/AJPH.2013.301520
22. Adashi EY, Cohen IG. The FDA initiative to assure racial and ethnic diversity in clinical trials. J Am Board Fam Med. 2023;36(2):366–368. doi:10.3122/jabfm.2022.220290r1
23. Bodicoat DH, Routen A, Willis A, et al. Promoting inclusion in clinical trials—a rapid review of the literature and recommendations for action. Trials. 2021;22(1). doi:10.1186/s13063-021-05849-7
24. Fisher-Hoch SP, Below JE, North KE, McCormick JB. Challenges and strategies for recruitment of minorities to clinical research and trials. J Clin Transl Sci. 2023;7(1). doi:10.1017/cts.2023.559
25. Florez M, Botto E, Kim JY. Mapping strategies for reaching socioeconomically disadvantaged populations in clinical trials. JAMA Network Open. 2024;7(6):e2413962. doi:10.1001/jamanetworkopen.2024.13962
26. Donzo MW, Nguyen G, Nemeth J, et al. Effects of socioeconomic status on enrollment in clinical trials for cancer: a systematic review. Cancer Med. 2024;13(1). doi:10.1002/cam4.6905
27. Bierer BE, White S, Gelinas L, Strauss DH. Fair payment and just benefits to enhance diversity in clinical research. J Clin Transl Sci. 2021;5(1). doi:10.1017/cts.2021.816
28. Borno HT, Zhang S, Nieves E, et al. Broad inclusion leads to maximal value – examining travel costs associated with clinical trial participation. Int J Cancer Care Deliv. 2021;1(1). doi:10.53876/001c.27061
29. Williams CP, Geiger AM, Norton WE, de Moor JS, Senft N. Influence of cost-related considerations on clinical trial participation: results from the 2020 Health Information National Trends Survey (HINTS). J Gen Intern Med. 2022;38(5):1200–1206. doi:10.1007/s11606-022-07801-0
30. Ittenbach RF, Senft EC, Huang G, Corsmo JJ, Sieber JE. Readability and understanding of informed consent among participants with low incomes. J Empir Res Hum Res Ethics. 2015;10(5):444–448. doi:10.1177/1556264615615006
31. Bureau U. Race and ethnicity in the United Stats: 2010 census and 2020 census. Census. gov. 2022.
32. Williams C, Senft N, Shelburne N, Norton WE. Demographic and health behavior factors associated with clinical trial invitation and participation in the United States. JAMA Network Open. 2021;4(9):e2127792. doi:10.1001/jamanetworkopen.2021.27792
33. Bharucha AE, Wi CI, Srinivasan SG, et al. Participation of rural patients in clinical trials at a multisite academic medical center. J Clin Transl Sci. 2021;5(1). doi:10.1017/cts.2021.813
34. Noonan D, Lam WKK, Goodrich J, et al. A case study of inclusion of rural populations in research: implications for science and health equity. Clin Transl Sci. 2024;17(8). doi:10.1111/cts.13885
35. Brockman TA, Shaw O, Wiepert L, et al. Community engagement strategies to promote recruitment and participation in clinical research among rural communities: a narrative review. J Clin Transl Sci. 2023;7(1). doi:10.1017/cts.2023.16
36. Farris PE, Crist R, Miller S, Shannon J. Rural research capacity: a co-created model for research success. Health Res Policy Syst. 2023;21(1). doi:10.1186/s12961-023-01030-5
37. Croff R, Gowen LK, Lindauer A, Shofner S, Brown K, Eckstrom E. Including older rural adults in research: practical guidance for addressing the NIH inclusion across the lifespan policy. J Clin Transl Sci. 2020;4(5):431–436. doi:10.1017/cts.2020.12
38. Steinberg JR, Turner BE, Weeks BT, et al. Analysis of female enrollment and participant sex by burden of disease in US clinical trials between 2000 and 2020. JAMA Network Open. 2021;4(6):e2113749. doi:10.1001/jamanetworkopen.2021.13749
39. Feuerstein IM, Jenkins M, Kornstein SG, et al. Working together to address women’s health in research and drug development: summary of the 2017 women’s health congress preconference symposium. J Women Health. 2018;27(10):1195–1203. doi:10.1089/jwh.2018.29019.pcss
40. Golder S, O’Connor K, Wang Y, Stevens R, Gonzalez-Hernandez G. Best practices on big data analytics to address sex-specific biases in our understanding of the etiology, diagnosis, and prognosis of diseases. Annu Rev Biomed Data Sci. 2022;5(1):251–267. doi:10.1146/annurev-biodatasci-122120-025806
41. Msomphora MR, Larsen AIL. Disease prevention and health promotion strategies: the possible side effects of their good intentions. Glob J Health Sci. 2021;13(12):1. doi:10.5539/gjhs.v13n12p1
42. Broadwin C, Azizi Z, Rodríguez F. Clinical trial technologies for improving equity and inclusion in cardiovascular clinical research. Cardiol Ther. 2023;12(2):215–225. doi:10.1007/s40119-023-00311-y
43. Peters SAE, Woodward M. A roadmap for sex- and gender-disaggregated health research. BMC Med. 2023;21(1). doi:10.1186/s12916-023-03060-w
44. Rivera FB, Magalong JV, Bantayan NRB, et al. Participation of women in cardiovascular trials from 2017 to 2023. JAMA Network Open. 2025;8(8):e2529104. doi:10.1001/jamanetworkopen.2025.29104
45. Witham MD, Anderson E, Carroll C, et al. Developing a roadmap to improve trial delivery for underserved groups: results from a UK multi-stakeholder process. Res Sq Res Sq. 2020. doi:10.21203/rs.2.21325/v1
46. Schönthaler S, Spirovska I. The (Not So) hidden barriers behind research with romani minorities. Eurac Res Blogs. 2025. doi:10.57708/bzftd3uqfrzc5sdqb6de1-w
47. Lou W, Díaz-Faes AA, He J, Liu Z, Larivière V. Global inequalities in clinical trials participation. arXiv. 2026. doi:10.48550/ARXIV.2601.04660
48. Stronks K, Wieringa NF, Hardon A. Confronting diversity in the production of clinical evidence goes beyond merely including under-represented groups in clinical trials. Trials. 2013;14(1):177. doi:10.1186/1745-6215-14-177
49. Goldstein DB, Need AC, Singh R, Sisodiya SM. Potential genetic causes of heterogeneity of treatment effects. Am J Med. 2007;120(4):S21–S25. doi:10.1016/j.amjmed.2007.02.004
50. Shaaban S, Ji Y. Pharmacogenomics and health disparities, are we helping? Front Genet. 2023;14. doi:10.3389/fgene.2023.1099541
51. Aldrighetti CM, Niemierko A, Allen EMV, Willers H, Kamran SC. Racial and ethnic disparities among participants in precision oncology clinical studies. JAMA Network Open. 2021;4(11):e2133205. doi:10.1001/jamanetworkopen.2021.33205
52. Li Q, Guo Y, He Z, Zhang H, George TJ, Bian J. Using real-world data to rationalize clinical trials eligibility criteria design: a case study of alzheimer’s disease trials. MedRxiv Cold Spring Harb Lab. 2020. doi:10.1101/2020.08.02.20166629
53. Flight L, Julious SA. Practical guide to sample size calculations: an introduction. Pharm Stat. 2015;15(1):68–74. doi:10.1002/pst.1709
54. Pourhoseingholi MA, Vahedi M, Rahimzadeh M. Sample size calculation in medical studies. Gastroenterol Hepatol Bed Bench. 2013;6:14.
55. Röhrig B, du Prel JB, Wachtlin D, Kwiecien R, Blettner M. Sample size calculation in clinical trials. Dtsch Ärztebl Int. 2010. doi:10.3238/arztebl.2010.0552
56. Averitt A, Weng C, Ryan P, Perotte A. Translating evidence into practice: eligibility criteria fail to eliminate clinically significant differences between real-world and study populations. Npj Digit Med. 2020;3(1). doi:10.1038/s41746-020-0277-8
57. Hacker K, Thomas CW, Zhao G, Claxton JS, Eke P, Town M. Social determinants of health and health-related social needs among adults with chronic diseases in the united states, behavioral risk factor surveillance system, 2022. Prev Chronic Dis. 2024;21:240362. doi:10.5888/pcd21.240362
58. Marmot M, Wilkinson R. Social Determinants of Health.
59. Catalyst N. Social Determinants of Health (SDOH). NEJM Catal. 2017. doi:10.1056/CAT.17.0312
60. Pronk N, Kleinman DV, Goekler S, Ochiai E, Blakey C, Brewer KH. Promoting health and well-being in healthy people 2030. J Public Health Manag Pract. 2020;27. doi:10.1097/phh.0000000000001254
61. Hróbjartsson A, Boutron I, Hopewell S, et al. SPIRIT 2025 explanation and elaboration: updated guideline for protocols of randomised trials. BMJ. 2025:389. doi:10.1136/bmj-2024-081660
62. Hopewell S, Chan AW, Collins GS, et al. CONSORT 2025 explanation and elaboration: updated guideline for reporting randomised trials. PubMed. 2025:389. doi:10.1136/bmj-2024-081124
63. Askin S, Burkhalter D, Calado G, Dakrouni SE. Artificial intelligence applied to clinical trials: opportunities and challenges. Health Technol. 2023;13(2):203–213. doi:10.1007/s12553-023-00738-2
64. Harrer S, Shah P, Antony B, Hu J. Artificial intelligence for clinical trial design. Trends Pharmacol Sci. 2019;40(8):577–591. doi:10.1016/j.tips.2019.05.005
65. Zhang B, Zhang L, Chen Q, Jin Z, Liu S, Zhang S. Harnessing artificial intelligence to improve clinical trial design. Commun Med. 2023;3(1). doi:10.1038/s43856-023-00425-3
66. Saltonstall PL, Ross H, Kim PT. The orphan drug act at 40: legislative triumph and the challenges of success. Milbank Q. 2023;102(1):83–96. doi:10.1111/1468-0009.12680
67. Niazi SK. The coming of age of AI/ML in drug discovery, development, clinical testing, and manufacturing: the FDA perspectives. Drug Des Devel Ther. 2023;2691–2725. doi:10.2147/dddt.s424991
68. Raju KV, Rehana S, Seethamraju SM, Nori LP. Revolutionizing medicine: unleashing the power of real-world data and AI in advancing clinical trials. Braz J Pharm Sci. 2024;60. doi:10.1590/s2175-97902024e23980
69. Agarwal R, Bjarnadóttir MV, Rhue LA, et al. Addressing algorithmic bias and the perpetuation of health inequities: an AI bias aware framework. Health Policy Technol. 2022;12(1):100702. doi:10.1016/j.hlpt.2022.100702
70. Chien I, Deliu N, Turner RE, Weller A, Villar SS, Kilbertus N. Multi-disciplinary fairness considerations in machine learning for clinical trials. ArXiv Cornell Univ. 2022. doi:10.48550/arxiv.2205.08875
71. Farnadi G, Havaei M, Rostamzadeh N. Position: cracking the code of cascading disparity towards marginalized communities. ArXiv Cornell Univ. 2024. doi:10.48550/arxiv.2406.01757
72. Hatem R, Simmons B, Thornton J. A call to address AI “Hallucinations” and how healthcare professionals can mitigate their risks. Cureus. 2023. doi:10.7759/cureus.44720
73. Samala AD, Rawas S. Bias in artificial intelligence: smart solutions for detection, mitigation, and ethical strategies in real-world applications. IAES Int J Artif Intell. 2024;14(1):32. doi:10.11591/ijai.v14.i1.pp32-43
74. Feldman T, Peake A. End-to-end bias mitigation: removing gender bias in deep learning. ArXiv Cornell Univ. 2021. doi:10.48550/arxiv.2104.02532
75. Nazer L, Zatarah R, Waldrip S, et al. Bias in artificial intelligence algorithms and recommendations for mitigation. PLOS Digit Health. 2023;2(6):e0000278. doi:10.1371/journal.pdig.0000278
76. Hort M, Chen Z, Zhang JM, Harman M, Sarro F. Bias mitigation for machine learning classifiers: a comprehensive survey. ACM J Responsible Comput. 2023;1(2):1–52. doi:10.1145/3631326
77. Vokinger KN, Feuerriegel S, Kesselheim AS. Mitigating bias in machine learning for medicine. Commun Med. 2021;1(1). doi:10.1038/s43856-021-00028-w
78. Pye V, Taylor N, Clay-Williams R, Braithwaite J. When is enough, enough? Understanding and solving your sample size problems in health services research. BMC Res Notes. 2016;9(1). doi:10.1186/s13104-016-1893-x
79. Rost DH. Effect strength vs. Statistical significance: a warning against the danger of small samples. Eur J High Abil. 1991;2(2):236–243. doi:10.1080/0937445910020212
80. Nakada R, Xu Y, Li L, Zhang L. Synthetic oversampling: theory and a practical approach using llms to address data imbalance. ArXiv Cornell Univ. 2024. doi:10.48550/arxiv.2406.03628
81. Yu L, Zhou N. Survey of imbalanced data methodologies. ArXiv Cornell Univ. 2021. doi:10.48550/arxiv.2104.02240
82. Yang Y, Khorshidi HA, Aickelin U. A review on over-sampling techniques in classification of multi-class imbalanced datasets: insights for medical problems. Front Digit Health. 2024;6. doi:10.3389/fdgth.2024.1430245
83. Seiffert C, Khoshgoftaar TM, Hulse JV. Hybrid sampling for imbalanced data. 2008:202–207. doi:10.1109/iri.2008.4583030
84. Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: synthetic minority over-sampling technique. J Artif Intell Res. 2002;16:321–357. doi:10.1613/jair.953
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.