Back to Journals » Cancer Management and Research » Volume 12
BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects
Authors Wong W, Raufi AG, Safyan RA, Bates SE ![]() , Manji GA
, Manji GA ![]()
Received 14 January 2020
Accepted for publication 4 April 2020
Published 23 April 2020 Volume 2020:12 Pages 2731—2742
DOI https://doi.org/10.2147/CMAR.S211151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kenan Onel
Winston Wong,1 Alexander G Raufi,1,2 Rachael A Safyan,1 Susan E Bates,1,3 Gulam A Manji1,4
1Division of Hematology and Oncology, Columbia University Medical Center and New York Presbyterian Hospital Herbert Irving Pavilion, New York, NY 10032, USA; 2Division of Hematology-Oncology, Lifespan Cancer Institute, Warren-Alpert Medical School of Brown University, Providence, RI, USA; 3Division of Hematology and Oncology, James J. Peters Veterans Affairs Medical Center, The Bronx, NY 10468, USA; 4Herbert Irving Comprehensive Cancer Center, Columbia University Medical Center and New York Presbyterian Hospital Herbert Irving Pavilion, New York, NY 10032, USA
Correspondence: Winston Wong
Division of Hematology and Oncology, Columbia University Medical Center and New York Presbyterian Hospital, Milstein Hospital Building, 6 Garden North, Rm 6-435 177 Fort Washington Ave, New York, NY 10032, USA
Tel +646-675-8254
Email [email protected]
Abstract: Pancreatic ductal adenocarcinoma (PDAC) remains a challenging disease to treat. Despite advances in surgical techniques, radiation, and medical therapies, the 5-year survival rate remains below 9%. Over the past decade, the genomic landscape of PDAC has been well studied and BRCA mutations have emerged as a target for the development of more effective therapies. Alterations in germline BRCA and PALB2 are detected in approximately 5– 9% of patients with PDAC and can lead to homologous repair deficiency (HRD). PDAC with HRD is more susceptible to cytotoxic agents, such as platinum salts and topoisomerase inhibitors, that cause DNA damage. Furthermore, PARP inhibitors have emerged as an effective non-cytotoxic approach to treating HRD-PDAC. In addition to BRCA and PALB2, germline mutations in other genes involved in the homologous DNA repair pathway – such as ATM and RAD51 – are potential targets, as are patients with the “BRCAness” phenotype and somatic mutations in the DNA repair pathway. Given the clinical implications of germline mutation related HRD in PDAC, universal germline testing is now recommended. In this review, we will discuss current and emerging biomarkers for HRD in PDAC, treatments, and the challenges associated with them.
Keywords: pancreas cancer, clinical trials, BRCA
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a dismal 5-year overall survival rate of 9%, and by 2030 it is projected to become the 2nd leading cause of cancer-related death in the United States.1,2 Currently, surgery is the only curative measure, but only 15–20% of patients are diagnosed with resectable disease.3 Even if an R0 resection is achieved, 75% of patients will experience disease recurrence within 5 years. Increasingly, we have begun to understand that pancreas cancer is fundamentally a systemic disease at presentation. For patients who present with metastatic or unresectable disease, FOLFIRINOX and gemcitabine with nab-paclitaxel form the mainstay of treatment, but only improves survival by several months indicating a need for novel therapies.2
Familial pancreas cancer (FPC) and genetic predisposition syndromes have become an area of interest due to the potential clinical implications of targeted therapies. It is estimated that approximately 10 to 15% of pancreas cancers are attributed to a genetic cause.4–8 Of these hereditary predisposition syndromes, Breast cancer type 1 susceptibility protein (BRCA1) and BRCA2 have been the most clinically relevant in pancreas cancer to date. BRCA1 and BRCA2 are tumor suppressor proteins involved in repairing double strand DNA breaks via the homologous DNA repair mechanism. Deleterious mutations within BRCA1 and BRCA2 were first implicated as a risk factor for the development of breast and ovarian cancer in the mid-1990s through the work of Miki et al (1994), and Wooster et al (1995), respectively.9,10 These deleterious mutations are now known to be a risk factor for the development of PDAC.
Until recently, identifying patients with familial PDAC has had little impact on clinical outcomes. However, this changed with the development of treatments, such as the poly-ADP ribose polymerase (PARP) inhibitors, which are capable of exploiting homologous repair deficiency (HRD) in BRCA-mutated tumor cells. The importance of deleterious germline mutations in BRCA1/2 have led to further evaluation of other germline mutations intimately involved in the homologous repair process such as partner and localizer of BRCA2 (PALB2) and ataxia telangiectasia mutated (ATM). Furthermore, somatic mutations in the homologous recombination pathway that mimic loss of germline BRCA1/2 are now collectively labeled as “BRCAness” genes.11,12 Whether treatments that benefit patients with germline BRCA1/2-mutated PDAC will also be effective in patients with PDAC associated with other forms of HRD or those with somatic mutations within the homologous repair pathway remains to be determined.13–16 In this review, we will discuss the biology, current status, and future prospect of BRCA1/2 mutations in the context of PDAC and how it could influence current management and treatment.
BRCA and Homologous Repair Deficiency
DNA double strand breaks occur commonly in eukaryotic cells as a result of endogenous and exogenous factors. They are repaired by two major pathways: homologous recombination and non-homologous end joining repair. Homologous recombination repairs double strand breaks that arise from single strand breaks typically caused by DNA damaging agents such as ionizing radiation and reactive oxygen species. This is a complex and tightly regulated mechanism involving many proteins including BRCA1/2, PALB2, ATM and RAD50. DNA double strand breaks initiate recruitment of ATM by NBS1, a component of the MRN complex, which also consists of Mre11 and Rad50, to the double strand break sites. The MRN complex activates ATM kinase, which, along with ATR (ataxia telangiectasia and rad3 related protein), recruits BRCA1 to displace the p53-binding protein 1 at the site of the DNA double stand break. This in turn recruits CtIP and the MRN complex resulting in resection of the ends of the DNA strands. This step is essential for RAD51 to bind to the DNA strand, catalyzed by BRCA2, which is dependent on PALB2. PALB2 co-localizes with BRCA2 and allows for intra-nuclear accumulation and stabilization of BRCA2. RAD51 then forms the homo-polymers, which are required for sister chromatid invasion and final recombination.17,18
Epidemiology of Germline Mutations in PDAC and Screening
The incidence of targetable deleterious germline mutations in BRCA1/2 and PALB2 in patients with PDAC is estimated to be about 5–9%.19–21 Deleterious germline mutations in BRCA1 and BRCA2 have been described in patients with both FPC and non-familial PDAC.19,22-27 In patients with FPC, the frequency of these mutations, specifically BRCA2, may be up to 17%.23,28,29 In fact, harboring a germline mutation in BRCA2 is associated with a relative risk of 3.5 to 10 for developing PDAC as compared to non-carriers and is inherited in an autosomal dominant fashion with incomplete penetrance.30–32 The relative risk of developing PDAC in patients with BRCA1 as compared to non-carriers is reported to be approximately 2.26 to 3 in one’s lifetime.20,33,34 Within the Ashkenazi Jewish population, up to 21% of patients with PDAC harbor a germline BRCA1 or BRCA2 mutation.19,24-27 Genome sequencing has identified other germline alterations in the DNA repair pathway, such as ATM and PALB2, as susceptibility genes for FPC.35,36 Germline ATM mutations have been reported at a prevalence of approximately 2.4% in FPC and have an estimated relative risk of 2.4 for the development of pancreas cancer.35,37 Germline PALB2 mutations have a prevalence of 1 to 4.9% in FPC families and carriers of the mutation are diagnosed with PDAC at a median age of 51 years old as compared to 63 years old for those who are non-carriers.38–42 Table 1 summarizes these statistics.
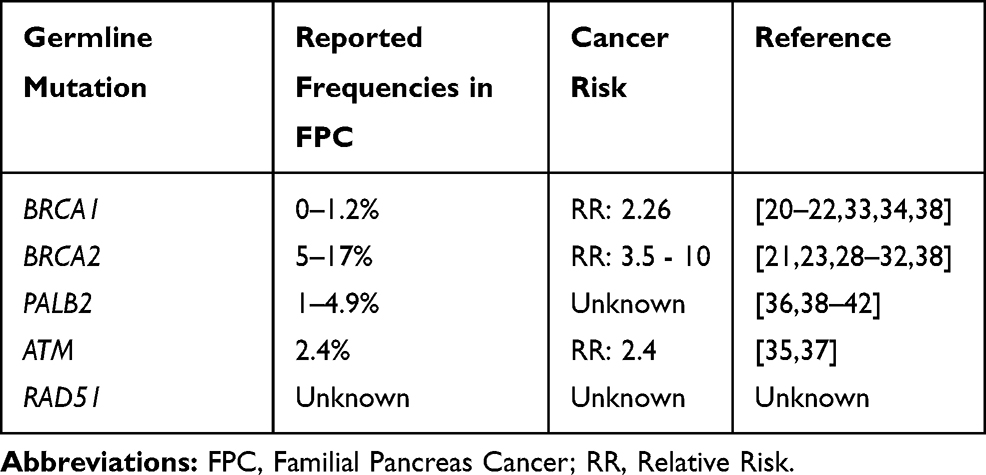 |
Table 1 DNA Repair Genes and PDAC |
Historically, screening for BRCA and other germline mutations has been limited to those patients with PDAC and a family history suggestive of FPC. However, this strategy fails to capture a significant proportion of patients with germline BRCA1/2 mutations and given the significant treatment implications this may have, the National Comprehensive Cancer Network (NCCN) guidelines now recommends universal germline testing of all patients with PDAC.19,24-27
In addition to screening for germline mutations, family members of patients with PDAC should also be counseled regarding screening as the risk of developing PDAC in carriers is increased anywhere between 1.5 to 13% depending on the number of affected blood relatives.29,34-36,43–46 There is growing consensus that patients, with relatives with pancreas cancer, who are at high risk for developing pancreas cancer should be evaluated for screening to identify early stage disease amenable for curative surgery. Currently, there is no clear consensus on the optimal screening modality (magnetic resonance cholangiopancreatography (MRCP) or endoscopic ultrasound (EUS)), age to initiate and terminate screening, interval duration between screening, and ways to manage patients with detected lesions. The International Cancer of the Pancreas Screening (CAPS) Consortium considers high-risk patients as those who meet one or more of the following criteria: first-degree relatives of a patient with FPC, as defined by kindreds with at least two first-degree relatives with pancreas cancer; those with Peutz-Jeghers syndrome; those with a p16/CDKN2A mutation; and those who harbor a BRCA2 mutation or are diagnosed with hereditary non-polyposis colorectal cancer (HNPCC) and have one or more first-degree relatives with pancreas cancer. These patients should participate in screening with EUS or MRI through clinical trials at high-volume pancreas cancer centers and should have their case discussed at a multidisciplinary conference.5 The American College of Gastroenterology (ACG) recommends screening of high-risk individuals with EUS and/or MRI annually beginning at age 50 or 10 years prior to the earliest age of pancreas cancer diagnosis within the family. The ACG considers patients to be high-risk if they: are first-degree relatives of a patient with FPC; have Peutz-Jeghers syndrome; or harbor mutations in BRCA1/2, ATM, PALB2, or Lynch syndrome genes and have a first or second-degree relatives with pancreas cancer.47 See Table 2 for summarized recommendations for screening.
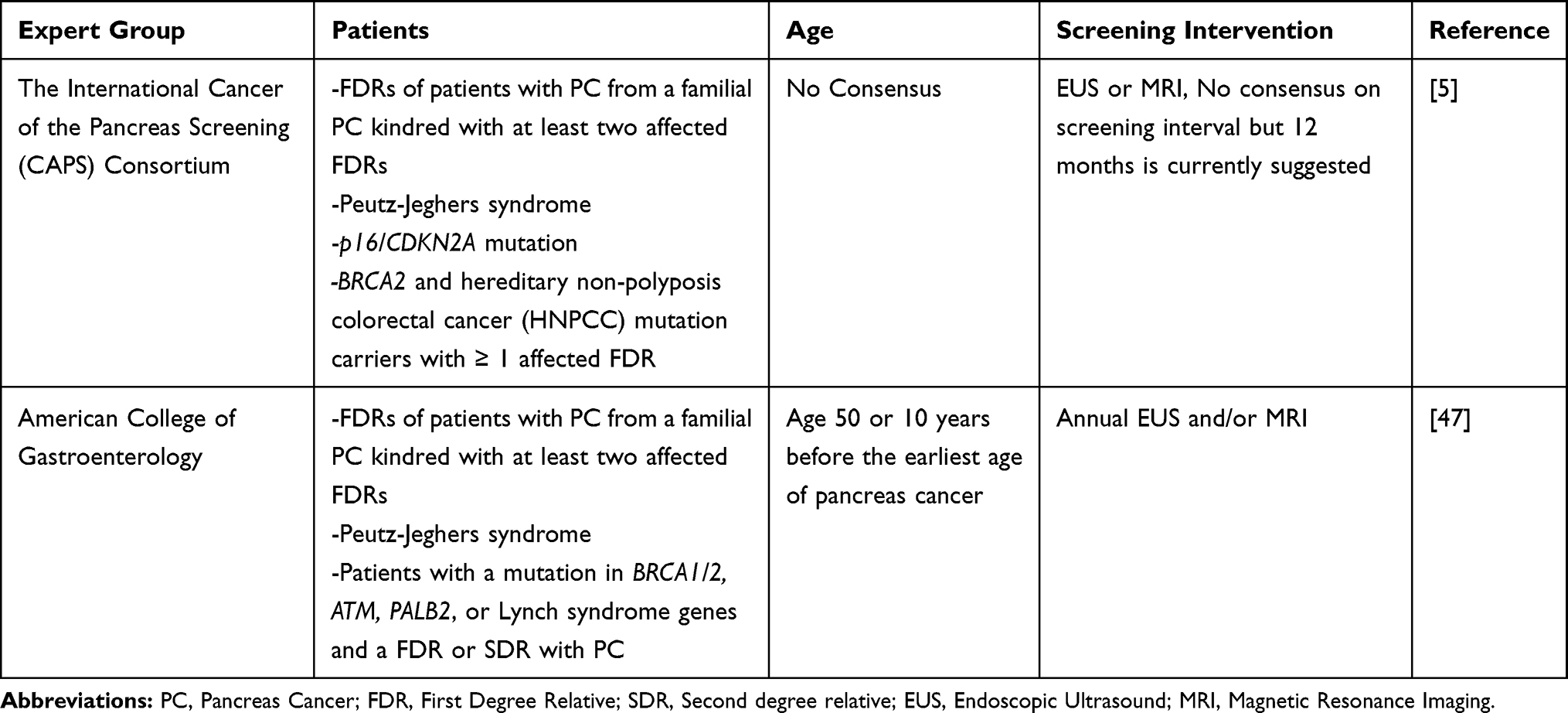 |
Table 2 Recommendations for Screening of High Risk Individuals |
Therapeutic Approaches
DNA Damaging Agents
Cancers harboring BRCA mutations and HRD are considered more vulnerable to DNA damage and are especially susceptible to drugs that induce double strand breaks in DNA. This serves as the rationale for use of platinum-based chemotherapy. The efficacy of platinum therapy in patients with germline BRCA mutations has been established in clinical trials for breast and ovarian cancers and there is a growing consensus and data that HRD-PDAC also benefits.
Preclinical data in BRCA1/2-mutant PDAC xenografts demonstrated susceptibility to platinum-based therapy when compared to that of BRCA1/2 wild type xenografts. After treatment with cisplatin, mice with BRCA1/2-mutant PDAC xenografts demonstrated increased DNA damage, tumor shrinkage, and improved overall survival as compared to control mice bearing BRCA1/2 wild type xenografts.48 Several case reports have also detailed robust clinical responses of BRCA1/2-mutated PDAC to platinum-based therapy. Shimmura et al (2019) reported a case of a 47-year-old patient with germline BRCA2 mutation with metastatic PDAC to the liver who experienced a near complete response to FOLFIRINOX allowing for pancreaticoduodenectomy.49 On pathological review of the resected specimen less than 2.5mm of tumor foci remained. Similarly, Sonnenblick et al (2011) reported a case of a 60-year-old patient with germline BRCA2 mutation who experienced a complete radiographic and biochemical response after addition of cisplatin to gemcitabine therapy.50 The profound treatment effect of BRCA-mutated PDAC is not limited to platinum therapy as similar responses have been observed in patients receiving alternative DNA damaging agents such as irinotecan (topoisomerase inhibitor) or mitomycin C (non-platinum alkylators).51,52
Several retrospective series have described anti-tumor activity of DNA damaging agents in patients with BRCA1/2-mutated pancreas cancer. Lowery et al (2011) reported impressive outcomes in which five of the six patients with pancreas cancer and germline BRCA mutation treated with platinum therapy experienced an objective response, one of which was a complete response.53 Wattenberg et al (2019) found a significantly greater objective response rate (58% vs 21%, p=0.0022) and improved progression free survival (10.1 months vs 6.9 months, p= 0.0068) in 26 patients with BRCA1/2 or PALB2-mutated PDAC treated with platinum-based therapy as compared to patients with non-BRCA1/2 or PALB2-mutated PDAC.54 In addition to objective response, survival benefits have also been reported in retrospective studies. Golan et al (2014) reported a series of 71 patients with deleterious germline BRCA1/2 mutation and advanced pancreas cancer who, when treated with platinum-based therapy, experienced an improved overall survival (OS) as compared to those who were treated with non-platinum based regimens (22 months vs 9 months p= 0.039).55 Similarly, a study by Reiss et al (2018) also reported a survival benefit in patients with germline BRCA1/2 or PALB2-mutated PDAC when they were treated with platinum therapy in comparison to patients without germline alterations (21.8 months vs 8.1 months, p<0.001).56 Finally, Yu et al (2018) reported a trend towards improved median OS with neoadjuvant platinum-based therapy in patients with resectable PDAC and pathogenic germline mutations in BRCA1/2 or PALB2 as compared to patients without pathogenic germline mutations (not met vs 23.1 months, p=0.07).57
In a prospective Phase IB/II trial, Jameson et al (2019) studied gemcitabine, nab-paclitaxel, and cisplatin in an unselected cohort of 25 patients that included three patients with BRCA2, two with ATM and one with RAD51.58 The median OS of the three patients harboring BRCA2 ranged from 39.8 to 45.3 months and all three patients experienced a partial radiographic response. Furthermore, one patient with germline ATM alteration and concomitant germline MUTYH alteration experienced a complete response to the triplet therapy. Recently, O’Reilly et al (2020) evaluated combination gemcitabine and cisplatin with or without veliparib in patients with germline BRCA1/2 or PALB2-mutated PDAC via a prospective randomized Phase II clinical trial. They reported an impressive response rate of 65.2% and a disease control rate of 78.3% with gemcitabine and cisplatin indicating significant activity and effectively establishing the combination, according to the authors, as current standard of care in advanced PDAC with pathogenic germline BRCA1/2 or PALB2 mutation.59 It is unclear however, whether or not gemcitabine with cisplatin is superior to FOLFOX or FOLFIRINOX in this patient population. Pre-clinical data suggests that cisplatin may be more effective than oxaliplatin in BRCA-mutated and HRD PDAC.60 Ultimately, additional prospective randomized control studies testing distinct platinum combinations are needed.
PARP Inhibition as Maintenance
In recent years, PARP inhibitors (PARPi) have emerged as a novel class of targeted therapy with significant activity in breast, ovarian, and HRD-PDAC.61–64 These agents interfere with base excision repair by binding to the catalytic domain of PARP, which prevents PARylation and thus traps PARP to the single-strand DNA break. This prevents repair and leads to an accumulation of single-strand DNA lesions, which degenerate into DNA double strand breaks.65,66 HRD tumor cells, including those with loss of function BRCA mutations, therefore undergo cell cycle arrest and apoptosis when exposed to these agents.
Efficacy of PARPi was initially demonstrated in BRCA-mutated breast and ovarian cancers and these agents have since been applied to PDAC. An early retrospective series by Lowery et al (2011) described three patients with BRCA-mutated PDAC who achieved a partial radiographic response to treatment with a PARPi, either alone or in combination with gemcitabine.53 Olaparib, a PARPi, has been studied in a phase II clinical trial in 23 patients with germline BRCA-mutated advanced PDAC in the second line setting after failure to gemcitabine. This treatment resulted in a median progression free survival (PFS) and OS of 4.6 and 9.8 months, respectively.67 Another PARPi, veliparib, was tested in the first-line setting in 16 patients with germline BRCA1/2 or PALB2-mutated advanced PDAC in a single arm phase II study. One patient experienced a partial response (6%), four patients had stable disease (25%), and the remainder experienced disease progression. The overall median PFS for these patients was 52 days.68 More recently, a phase II study of yet another PARPi, rucaparib, in 16 patients with BRCA1/2-mutated PDAC demonstrated a disease control rate of 31.6%. Two patient experienced a complete response (12.5%), two patients achieved a partial response (12.5%), and two experienced stable disease (12.5%). Interestingly, three of these patients had BRCA2 somatic mutations while the remainder had germline BRCA1/2 mutations.14 An additional phase II clinical trial is studying rucaparib as maintenance monotherapy in patients with advanced PDAC and pathogenic germline or somatic mutations in BRCA1/2 or PALB2 who have not progressed on first line platinum therapy. Preliminary data presented at American Association for Cancer Research (2019) showed an ORR of 36.8% (six partial response and one complete response) and a disease control rate of 89.5% for at least eight weeks. At the time of report, eight of 24 patients had been on treatment for six months and two patients remained on treatment for greater than one year, indicative of prolonged benefit.16
In July of 2019, results of the POLO trial were reported. In this study, patients with metastatic germline BRCA1/2-mutated PDAC who had achieved at least stable disease after four months of platinum-based chemotherapy were randomized to receive either maintenance olaparib or placebo. Of the 3315 patients who were screened for the study, 154 patients underwent randomization and efficacy analysis. This was a highly selected group of patients who were thought to respond to PARPi based on a dual selection criteria. Patients could not have had disease progression for at least 16 weeks of platinum-based chemotherapy and had to harbor a germline BRCA mutation. Maintenance olaparib improved median PFS over placebo (7.4 vs 3.8 months, respectively), but it did not extend OS by the time the data was reported (18.9 vs 19.1 months). Only 46% of the participants had met the endpoint at the time of data report.69 The reason for the lack of OS benefit was likely due to resumption of platinum-based chemotherapy in the placebo group as well as a subset of patients receiving off label PARPi. A criticism of the study was the cessation of chemotherapy in the control arm, which is generally not standard practice. Despite the lack of an interim overall survival benefit, this trial clearly demonstrates that PARPi are effective agents in BRCA-mutated pancreas cancer, which led to the FDA approval of olaparib on 12/27/2019 in PDAC with known BRCA germline mutation. This trial highlights the potential of PARPi as a chemotherapy-sparing agent. Refer to Table 3 for ongoing clinical trials of other PARPi as monotherapy in PDAC with HRD.
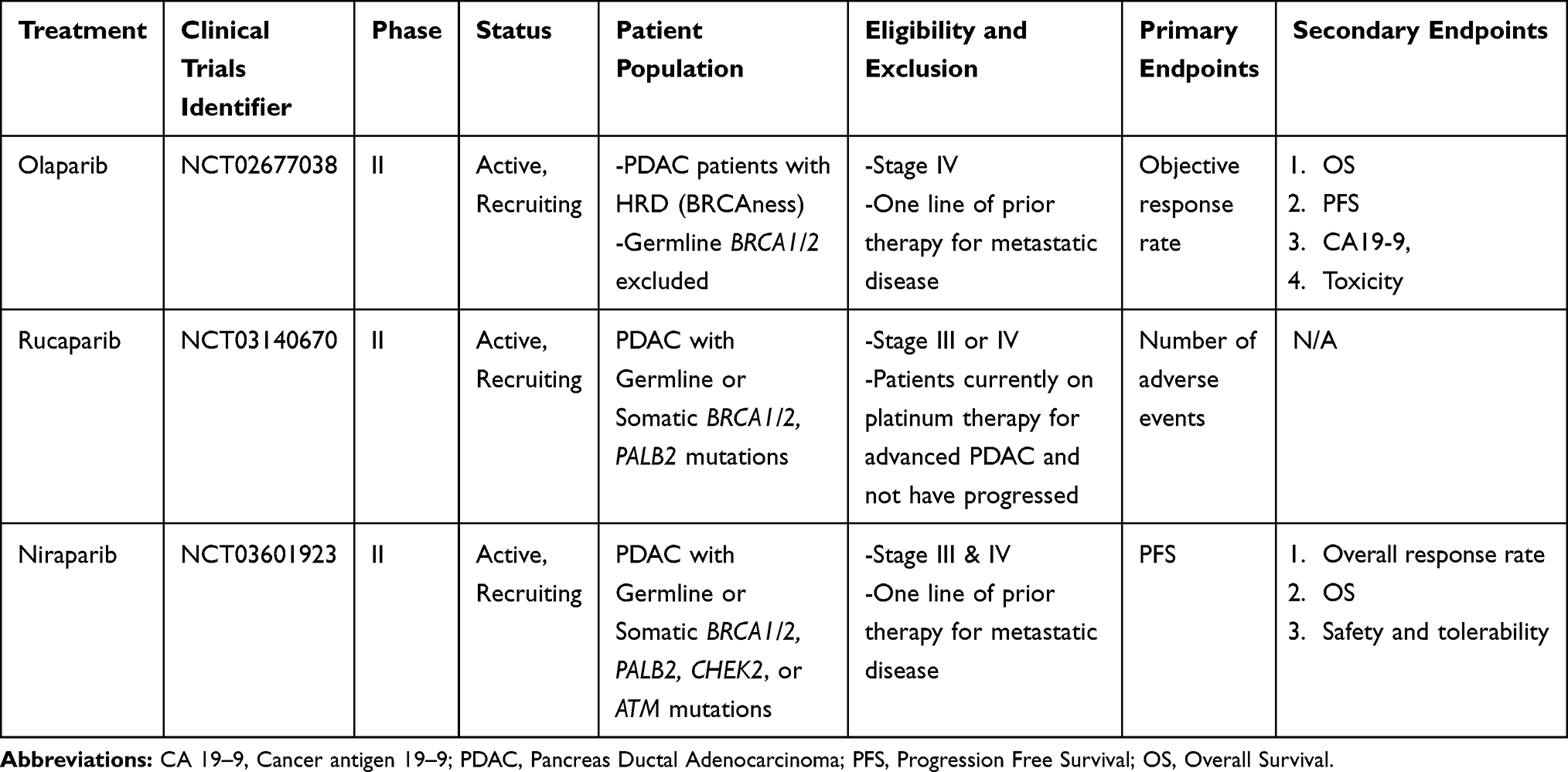 |
Table 3 Clinical Trials Utilizing PARPi Monotherapy |
PARP Inhibition in Combination Therapy
Given that PARPi and platinum agents act on distinct DNA repair pathways, it has been hypothesized that the combination therapy may represent a synthetically lethal and synergistic therapeutic strategy in HRD-PDAC. O’Reilly et al (2018) reported the results of a Phase I trial testing the combination of veliparib, gemcitabine, and cisplatin in patients with germline BRCA1/2 mutated and wild-type (WT) PDAC. They reported an objective response in seven out of nine patients with BRCA1/2-mutated PDAC (77.8%) with a median OS of 23.3 months as compared to no objective responses and a median OS of 11 months in patients with BRCA-WT PDAC.70 A follow-up randomized phase II trial by O’Reilly et al (2020) studied gemcitabine and cisplatin with or without veliparib in patients with advanced germline BRCA1/2 or PALB2-mutated PDAC. The authors found a non-significant benefit in response rate between the two arms (74.1% (with veliparib) vs 65.2% (without veliparib), p=0.55). Additionally, no significant OS or PFS benefit was seen with addition of veliparib to chemotherapy. The results of this trial established gemcitabine and cisplatin’s efficacy in patients with advanced PDAC harboring germline BRCA1/2 or PALB2 alterations. However, addition of PARPi concurrently with chemotherapy failed to demonstrate clinical benefit in this study, possibly due to increased hematologic toxicity leading to a greater number of dose reductions.59 At this time, a maintenance strategy with single agent PARPi continues to be the current standard given the lack of survival benefit demonstrated by this trial.
Several clinical trials testing combination PARPi with a DNA damaging agents have completed accrual, but results are pending (Table 4). It is not yet clear what agent(s) are ideal to combine with PARPi. Preclinical data suggests that topoisomerase inhibitors may be more synergistic with PARPi than platinum agents due to increased catalytic PARP inhibition.71 This effect may lead to increased toxicity in the clinical setting and as a result, topoisomerase and PARP inhibitors may have been more difficult to combine though attempts are ongoing (Table 4). Neoadjuvant and adjuvant studies evaluating DNA damaging agents with PARPi are likely to follow based on efficacy established in the metastatic setting. Since platinum-based therapy likely affords an overall response, it is likely to remain the backbone for neo-adjuvant and adjuvant therapies.
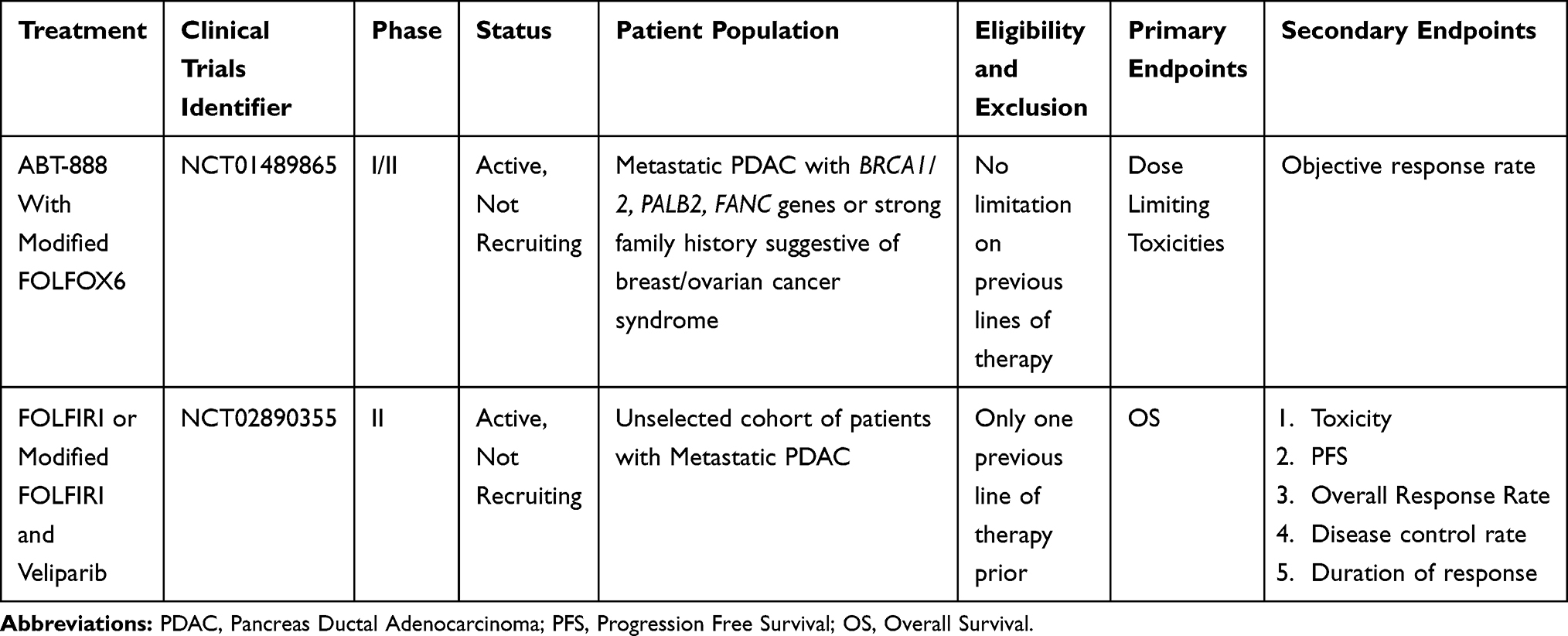 |
Table 4 Clinical Trials Utilizing Combination of DNA Damaging Agents with PARPi |
Finally, other combinations treatments utilizing PARPi are currently being tested in other malignancies, including lung, breast and prostate. Combining PARPi with other DNA repair pathway inhibitors (ATR, WEE1), targeted therapies aimed at oncogenes that directly or indirectly influence the homologous recombination pathway (RAS, PI3K), and PD-1/PD-L1 blockade all have clear rationale and are being actively studied in the pre-clinical and clinical setting.72 However, these combination treatments have yet to be thoroughly tested in PDAC as of yet and warrants further study.
Treatment Challenges
Toxicity
While effective, platinum-based chemotherapy regimens are often limited by cumulative dose-limiting toxicities such as sensory neuropathy, nephrotoxicity and bone marrow suppression. In advanced PDAC, FOLFIRINOX is considered the regimen of choice for patients with good performance status (ECOG 0–1) based on data published by Conroy et al (2011).73 The authors observed a significantly greater proportion of patients treated with FOLFIRINOX experiencing grade 3 or 4 neutropenia (45.7% vs 21%), febrile neutropenia (5.4% vs 1.2%), thrombocytopenia (9.1% vs 3.6%), diarrhea (12.7 vs 1.8%), and sensory neuropathy (9.0% vs 0%) in comparison to patients treated with gemcitabine alone. Gemcitabine-based platinum regimens appear more tolerable but afford a greater adverse event profile compared to gemcitabine alone. Heineman et al (2006) observed increased nausea and emesis with combination gemcitabine and cisplatin compared to gemcitabine alone (22.2% vs 5.9%). However, they found that less than 15% of their overall patient cohort experienced a grade 3 to 4 hematologic toxicity suggesting tolerability.74 In a phase I/IIb trial, Jameson et al (2019) tested the combination of gemcitabine, nab-paclitaxel and cisplatin and observed that 12 (48%) and nine (36%) patients experienced a grade 3 or 4 toxicity, respectively.58 The majority of these grade 3 or 4 toxicities were hematologic with thrombocytopenia and anemia being the most common. Two patients experienced a grade 5 adverse event that was considered treatment related. Despite the adverse events, of the 22 patients treated with the combination at the recommended phase II dose (gemcitabine at 1000 mg/m2, nab-paclitaxel at 125mg/m2, and cisplatin at 25mg/m2), 14 (64%) patients completed three or more cycles, eight (36%) completed six or more cycles, and four (18%) completed nine or more cycles suggesting durable tolerability of the regimen.
In comparison to combination cytotoxic therapy, single agent PARPi’s appear to be generally better tolerated. Anemia (11%), fatigue or asthenia (5%), and anorexia (3%) were the most common grade 3 or greater side-effects experienced by patients receiving olaparib in the POLO trial. Only 5% of patients required permanent discontinuation of the drug as compared to 2% in the placebo arm due to adverse event. Only 35% and 16% of patients required dose interruptions and dose reductions of olaparib due to adverse event, respectively.69
Expected hematologic toxicity of combination PARPi and platinum-based chemotherapy is likely to result in dose-limiting toxicity. In a recent phase I trial where 17 patients with PDAC were treated with the combination of gemcitabine, cisplatin and veliparib, two patients experienced grade 4 neutropenia and three experienced grade 4 thrombocytopenia beyond the initial dose-limiting period. Two grade 5 adverse events were also noted on study from likely treatment related AML and non-treatment related colonic perforation.70 The follow up Phase 2 trial studying gemcitabine, cisplatin, with or without veliparib, in patients with advanced PDAC demonstrated twice as many grade 3 and 4 hematologic adverse events in the combination arm as compared to the chemotherapy only arm. They found that 14 (52%) and 15 (55%) of their patients receiving combination therapy experienced grade 3 to 4 anemia and thrombocytopenia, respectively. Eight (35%) and two (9%) patients in the chemotherapy only arm experienced grade 3 anemia and thrombocytopenia, respectively. No grade 4 hematologic toxicity was seen in the chemotherapy only arm. Notably, 20 (74%) patients receiving combination PARPi and chemotherapy required dose reduction or discontinuation due to toxicity as compared to six (26%) patients in the chemotherapy only arm.59 This trial clearly highlights the poorer tolerability and increased toxicity of combination therapy. Given the potential superior efficacy of combination therapy, further study design to identify better tolerated doses and schedules, which remain efficacious, is warranted.
Resistance
Acquired resistance to both platinum-based chemotherapy and PARPi is well described in patients with BRCA1/2-related cancers. Several mechanisms have been proposed, including epigenetic changes, accumulation of somatic mutations that restore the homologous repair functions of BRCA1/2, upregulation of drug efflux pumps, and down regulation of drug influx pumps.75–77 Preclinical and clinical studies have suggested that these acquired and intrinsic mechanisms provide overlapping resistance to both PARPi and platinum therapy.78–80 In addition, prolonged exposure to platinum salts and/or PARPi are thought to exert selective pressure on clones with secondary mutations. Clearly, studies to both detect the emergence of primary and secondary resistance mechanisms and investigate options to circumvent them are needed.
Non-Germline BRCA Biomarkers
Non-BRCA Germline Mutations
As additional treatment options become available, there is tremendous interest in identifying predictive biomarkers to help guide therapy selection. Germline mutations that are intimately involved with BRCA1/2 and the homologous repair pathway such as ATM, PALB2, ATR, RAD 51, CHEK2, FANCA, and BRIP1 have also been implicated as potential targets for both DNA damaging agents and PARPi. Preclinical and clinical data have suggested that both platinum and PARPi may have activity in a number of germline mutations that confer HRD.81–84 For example, Villaroel et al (2011) reported a 61-year-old patient whose patient derived xenograft failed to respond to single agent gemcitabine but responded to mitomycin-c (MMC). The patient was found to have a germline PALB2 alteration and complete sequencing of the patient’s tumor revealed bi-allelic inactivation of PALB2. He underwent treatment with MMC for 22 months, which resulted in a radiographic response and normalization of cancer antigen 19–9 (CA19-9).84 In another case, Chan et al (2015) reported a patient with PDAC and coexisting low grade neuroendocrine pancreas tumor with a PALB2 mutation who experienced clinical improvement and normalization of CA 19–9 levels after treatment with gemcitabine and cisplatin.85 PALB2 is currently considered an equivalent HRD biomarker to BRCA1/2 germline alterations.54,56,57,59 Whether other germline mutations in the HRD pathway are equivalent HRD biomarkers to BRCA1/2 remains unknown. Germline mutations in PALB2, ATM, CHEK2 are currently being investigated in clinical studies testing PARP inhibitors (Table 3).
Somatic Mutations
Somatic BRCA1/2 and other HRD gene mutations are increasingly being detected with the use of widespread genomic testing. Somatic mutations in BRCA1 and BRCA2 have been reported in up to 9% of unselected patients with PDAC.14,15 There is growing evidence demonstrating that ovarian and prostate tumors with somatic BRCA1/2 mutations respond to treatment with PARPi and DNA damaging agents.82,86-91 Furthermore, outcomes for patients with somatic BRCA mutations treated with PARPi appear be similar to that of patients with germline BRCA mutations.86,92 Shroff et al (2018) tested rucaparib in a phase II study in patients with PDAC harboring germline or somatic BRCA1/2 mutations. Three of 19 patients harboring a somatic BRCA2 mutations were treated and two of these patients experience an objective radiographic response, one of which was a complete response.14 Lowery et al (2017) reported results from 50 patients with somatic mutation in one or more genes associated with DNA damage response including BRCA1/2, FANCA, ATM and ATR of whom 17 patients (34%) experienced a partial response to platinum-based therapy. However, the authors concluded that the presence of these genes failed to enrich patient’s response to platinum-based chemotherapies.15 This is perhaps due to the heterogeneity and mosaicism of tumor HRD that could be present in the setting of these somatic mutations. Clearly, further studies are necessary to elucidate the degree of HRD these somatic mutations confer and who would benefit from HRD direct treatments. Table 3 details studies evaluating PARPi in patients with PDAC and HRD-related somatic genes.
BRCAness
Recently, the term “BRCAness” has entered use in clinical practice as biomarker for tumor HRD. It is used to describe sporadic cancers that share molecular features with germline BRCA1/2 mutated cancers and denotes a set of characteristics that reflect distinct consequences of mutations in the homologous repair pathway.11,12,93 Whole genome sequencing in PDAC has identified a subset of tumors with the BRCAness phenotype suggesting that these tumors may respond to DNA damaging agents and PARPi. It is thought that a variety of somatic mutations in several of the homologous repair genes, such as BRCA1/2, ATM, PALB2, CHEK1, RAD51, and FANCA can contribute to the BRCAness phenotype.11,12 The frequency of each of these individual gene mutations within the DNA repair gene family are thought to be less than <5%, slightly greater in BRCA1/2, however, collectively these genes affect approximately 14% of PDAC.15,55,94 The key advantage of using BRCAness as a biomarker over individual somatic mutations in the homologous repair pathway is that it takes into account the degree to which a tumor is HRD. PARPi have been studied in the context of somatic mutations but why some patients respond and others do not maybe due to the BRCAness of the tumor. At this time, there is an ongoing phase II trial studying the effectiveness of olaparib in PDAC demonstrating the BRCAness phenotype in the absence of germline HRD genes. Preliminary data from this trial reported that of the 32 patients treated, 2 and 11 patients experienced a partial response and stable disease for at least 16 weeks, respectively (NCT02677038).13
Conclusions
Recent advances in treatments have made identifying patients with tumor HRD through mutations within the homologous repair pathway, such as BRCA1/2, critically important. This underlies the current NCCN recommendation that all patients with PDAC be tested for germline mutations. Treatments aimed at targeting HRD such as platinum agents and PARPi may greatly improve survival. Furthermore, single agent PARPi as maintenance therapy in patients with BRCA1/2-mutated PDAC has demonstrated benefit and affords a reduced toxicity compared to standard combination cytotoxic therapy. In patients with good functional status, combination therapy with platinum and PARPi may represent a new treatment option though toxicity may be a concern.
The frequency of germline mutations in the homologous repair pathway like BRCA1/2 represents a small proportion of patients with PDAC. However, there is evidence that germline BRCA1/2 mutations represent only a subset of PDAC that harbor HRD and that several other distinct biomarkers exist to elucidate these HRD tumors. Somatic mutations within the HRD pathway and the BRCAness characteristics of tumors are likely to further expand the group of patients with PDAC who will potentially derive benefit from HRD-directed therapies. Elucidating these nuances will help to broaden the scope of these targeted treatments and improve the outcomes of patients with HRD-PDAC.
Disclosure
Gulam A Manji, MD, PhD, has research funding from Roche/Genentech, Bio-Line, and Regeneron; grants from Merck, outside the submitted work. He also serves as an advisory board member for Roche. Rachael A Safyan, MD, has research funding from Merck. The authors report no other conflicts of interest in this work.
References
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–2921. doi:10.1158/0008-5472.CAN-14-0155
2. SEER Cancer Stat Facts: Pancreatic Cancer; 2019.
3. Dal Molin M, Zhang M, de Wilde RF, et al. Very long-term survival following resection for pancreatic cancer is not explained by commonly mutated genes: results of whole-exome sequencing analysis. Clin Cancer Res. 2015;21(8):1944–1950. doi:10.1158/1078-0432.CCR-14-2600
4. Klein AP. Genetic susceptibility to pancreatic cancer. Mol Carcinog. 2012;51(1):14–24. doi:10.1002/mc.20855
5. Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62(3):339–347. doi:10.1136/gutjnl-2012-303108
6. Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64(7):2634–2638. doi:10.1158/0008-5472.CAN-03-3823
7. Hiripi E, Lorenzo Bermejo J, Li X, Sundquist J, Hemminki K. Familial association of pancreatic cancer with other malignancies in Swedish families. Br J Cancer. 2009;101(10):1792–1797. doi:10.1038/sj.bjc.6605363
8. Schneider R, Slater EP, Sina M, et al. German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer. 2011;10(2):323–330. doi:10.1007/s10689-010-9414-x
9. Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. doi:10.1126/science.7545954
10. Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–792. doi:10.1038/378789a0
11. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. doi:10.1038/nrc1457
12. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–120. doi:10.1038/nrc.2015.21
13. Golan T, Varadhachary GR, Sela T, et al. Phase II study of olaparib for BRCAness phenotype in pancreatic cancer. J Clin Oncol. 2018;36(4_suppl):297. doi:10.1200/JCO.2018.36.4_suppl.297
14. Shroff RT, Hendifar A, McWilliams RR, et al. Rucaparib monotherapy in patients with pancreatic cancer and a known deleterious BRCA mutation. JCO Precis Oncol. 2018;2018:1–5.
15. Lowery MA, Jordan EJ, Basturk O, et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: potential actionability and correlation with clinical phenotype. Clin Cancer Res. 2017;23(20):6094–6100. doi:10.1158/1078-0432.CCR-17-0899
16. Binder KAR, Mick R, O’Hara M, et al. A phase II, single arm study of maintenance rucaparib in patients with platinum-sensitive advanced pancreatic cancer and a pathogenic germline or somatic mutation in BRCA1, BRCA2 or PALB2 :
17. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol Cancer Ther. 2016;15(8):1781–1791. doi:10.1158/1535-7163.MCT-15-0945
18. Chartron E, Theillet C, Guiu S, Jacot W. Targeting homologous repair deficiency in breast and ovarian cancers: biological pathways, preclinical and clinical data. Crit Rev Oncol Hematol. 2019;133:58–73. doi:10.1016/j.critrevonc.2018.10.012
19. Lowery MA, Wong W, Jordan EJ, et al. Prospective evaluation of germline alterations in patients with exocrine pancreatic neoplasms. J Natl Cancer Inst. 2018;110(10):1067–1074. doi:10.1093/jnci/djy024
20. Lynch HT, Deters CA, Snyder CL, et al. BRCA1 and pancreatic cancer: pedigree findings and their causal relationships. Cancer Genet Cytogenet. 2005;158(2):119–125. doi:10.1016/j.cancergencyto.2004.01.032
21. Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005;7(2):60.
22. Thompson D, Easton DF, Consortium BCL. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–1365. doi:10.1093/jnci/94.18.1358
23. Couch FJ, Johnson MR, Rabe KG, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16(2):342–346. doi:10.1158/1055-9965.EPI-06-0783
24. Hu C, Hart SN, Polley EC, et al. Association between inherited germline mutations in cancer predisposition genes and risk of pancreatic cancer. JAMA. 2018;319(23):2401–2409. doi:10.1001/jama.2018.6228
25. Hu C, Hart SN, Bamlet WR, et al. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol Biomarkers Prev. 2016;25(1):207–211. doi:10.1158/1055-9965.EPI-15-0455
26. Salo-Mullen EE, O’Reilly EM, Kelsen DP, et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer. 2015;121(24):4382–4388. doi:10.1002/cncr.29664
27. Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35(30):3382–3390. doi:10.1200/JCO.2017.72.3502
28. Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%.. Cancer Res. 2002;62(13):3789–3793.
29. Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95(3):214–221. doi:10.1093/jnci/95.3.214
30. van Asperen CJ, Brohet RM, Meijers-Heijboer EJ, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005;42(9):711–719. doi:10.1136/jmg.2004.028829
31. Consortium BCL. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91(15):1310–1316. doi:10.1093/jnci/91.15.1310
32. Iqbal J, Ragone A, Lubinski J, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107(12):2005–2009. doi:10.1038/bjc.2012.483
33. Brose MS, Rebbeck TR, Calzone KA, Stopfer JE, Nathanson KL, Weber BL. Cancer risk estimates for BRCA1 mutation carriers identified in a risk evaluation program. J Natl Cancer Inst. 2002;94(18):1365–1372. doi:10.1093/jnci/94.18.1365
34. Thompson D, Easton DF. Breast cancer linkage C. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94(18):1358–1365.
35. Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–46. doi:10.1158/2159-8290.CD-11-0194
36. Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324(5924):217. doi:10.1126/science.1171202
37. Thompson D, Duedal S, Kirner J, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97(11):813–822. doi:10.1093/jnci/dji141
38. Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2014;17(7):569–577. doi:10.1038/gim.2014.153
39. Slater EP, Langer P, Niemczyk E, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78(5):490–494. doi:10.1111/j.1399-0004.2010.01425.x
40. Tischkowitz MD, Sabbaghian N, Hamel N, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology. 2009;137(3):1183–1186. doi:10.1053/j.gastro.2009.06.055
41. Hofstatter EW, Domchek SM, Miron A, et al. PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer. 2011;10(2):225–231. doi:10.1007/s10689-011-9426-1
42. Borecka M, Zemankova P, Vocka M, et al. Mutation analysis of the PALB2 gene in unselected pancreatic cancer patients in the Czech Republic. Cancer Genet. 2016;209(5):199–204. doi:10.1016/j.cancergen.2016.03.003
43. Rebours V, Boutron-Ruault MC, Jooste V, et al. Mortality rate and risk factors in patients with hereditary pancreatitis: uni- and multidimensional analyses. Am J Gastroenterol. 2009;104(9):2312–2317. doi:10.1038/ajg.2009.363
44. Rebours V, Boutron-Ruault MC, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103(1):111–119. doi:10.1111/j.1572-0241.2007.01597.x
45. Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2(3):252–261. doi:10.1016/S1542-3565(04)00013-8
46. Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International hereditary pancreatitis study group. J Natl Cancer Inst. 1997;89(6):442–446. doi:10.1093/jnci/89.6.442
47. Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110(2):223–262. doi:10.1038/ajg.2014.435
48. Lohse I, Borgida A, Cao P, et al. BRCA1 and BRCA2 mutations sensitize to chemotherapy in patient-derived pancreatic cancer xenografts. Br J Cancer. 2015;113(3):425–432. doi:10.1038/bjc.2015.220
49. Shimmura H, Kuramochi H, Jibiki N, Katagiri S, Nishino T, Araida T. Dramatic response of FOLFIRINOX regimen in a collision pancreatic adenocarcinoma patient with a germline BRCA2 mutation: a case report. Jpn J Clin Oncol. 2019;49(11):1049–1054. doi:10.1093/jjco/hyz141
50. Sonnenblick A, Kadouri L, Appelbaum L, et al. Complete remission, in BRCA2 mutation carrier with metastatic pancreatic adenocarcinoma, treated with cisplatin based therapy. Cancer Biol Ther. 2011;12(3):165–168. doi:10.4161/cbt.12.3.16292
51. Chalasani P, Kurtin S, Dragovich T. Response to a third-line mitomycin C (MMC)-based chemotherapy in a patient with metastatic pancreatic adenocarcinoma carrying germline BRCA2 mutation. JOP. 2008;9(3):305–308.
52. James E, Waldron-Lynch MG, Saif MW. Prolonged survival in a patient with BRCA2 associated metastatic pancreatic cancer after exposure to camptothecin: a case report and review of literature. Anticancer Drugs. 2009;20(7):634–638. doi:10.1097/CAD.0b013e32832b511e
53. Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist. 2011;16(10):1397–1402. doi:10.1634/theoncologist.2011-0185
54. Wattenberg MM, Asch D, Yu S, et al. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br J Cancer. 2020;122(3):333–339. doi:10.1038/s41416-019-0582-7
55. Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111(6):1132–1138. doi:10.1038/bjc.2014.418
56. Reiss KA, Yu S, Judy R, Symecko H, Nathanson KL, Domchek SM. Retrospective survival analysis of patients with advanced pancreatic ductal adenocarcinoma and germline BRCA or PALB2 mutations. JCO Precis Oncol. 2018;2:1–9. doi:10.1200/PO.17.00152
57. Yu S, Agarwal P, Mamtani R, et al. Retrospective survival analysis of patients with resected pancreatic ductal adenocarcinoma and a germline BRCA or PALB2 mutation. JCO Precis Oncol. 2019;3:1–11. doi:10.1200/PO.18.00271
58. Jameson GS, Borazanci E, Babiker HM, et al. Response rate following albumin-bound paclitaxel plus gemcitabine plus cisplatin treatment among patients with advanced pancreatic cancer: a phase 1b/2 Pilot clinical trial. JAMA oncol. 2019;6:125–132.
59. O’Reilly EM, Lee JW, Zalupski M, et al. Randomized, multicenter, phase II trial of gemcitabine and cisplatin with or without veliparib in patients with pancreas adenocarcinoma and a germline BRCA/PALB2 mutation. J Clin Oncol. 2020:JCO1902931. doi:10.1200/JCO.19.02931
60. Bruno PM, Liu Y, Park GY, et al. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med. 2017;23(4):461–471. doi:10.1038/nm.4291
61. Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. doi:10.1016/S0140-6736(10)60893-8
62. Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. doi:10.1016/S0140-6736(10)60892-6
63. Lowery, M.A., Lee, A, Tobias E, et al. Evaluation of PARP inhibition as a platinum sparing strategy in Brca2-deficient pancreatic tumors. J Clin Oncol. 2014;32:e15237. doi:10.1200/jco.2014.32.15_suppl.e15237
64. McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2-deficient CAPAN-1 cells are extremely sensitive to the inhibition of Poly (ADP-Ribose) polymerase: an issue of potency. Cancer Biol Ther. 2005;4(9):934–936. doi:10.4161/cbt.4.9.2141
65. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. doi:10.1038/nature03445
66. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. doi:10.1038/nature03443
67. Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. doi:10.1200/JCO.2014.56.2728
68. Lowery MA, Kelsen DP, Capanu M, et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2017;89:19–26. doi:10.1016/j.ejca.2017.11.004
69. Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline. N Engl J Med. 2019;381(4):317–327. doi:10.1056/NEJMoa1903387
70. O’Reilly EM, Lee JW, Lowery MA, et al. Phase 1 trial evaluating cisplatin, gemcitabine, and veliparib in 2 patient cohorts: germline BRCA mutation carriers and wild-type BRCA pancreatic ductal adenocarcinoma. Cancer. 2018;124(7):1374–1382. doi:10.1002/cncr.31218
71. Murai J, Zhang Y, Morris J, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349(3):408–416. doi:10.1124/jpet.113.210146
72. Yap TA, Plummer R, Azad NS, Helleday T. The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Edu. 2019;39:185–195. doi:10.1200/EDBK_238473
73. Conroy T, Desseigne F, Ychou M, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–1825. doi:10.1056/NEJMoa1011923
74. Heinemann V, Quietzsch D, Gieseler F, et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J Clin Oncol. 2006;24(24):3946–3952. doi:10.1200/JCO.2005.05.1490
75. Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–1120. doi:10.1038/nature06633
76. Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68(8):2581–2586. doi:10.1158/0008-5472.CAN-08-0088
77. Norquist B, Wurz KA, Pennil CC, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–3015. doi:10.1200/JCO.2010.34.2980
78. Chiarugi A. A snapshot of chemoresistance to PARP inhibitors. Trends Pharmacol Sci. 2012;33(1):42–48. doi:10.1016/j.tips.2011.10.001
79. Johnson N, Johnson SF, Yao W, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110(42):17041–17046. doi:10.1073/pnas.1305170110
80. Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. doi:10.1200/JCO.2009.26.9589
81. McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. doi:10.1158/0008-5472.CAN-06-0140
82. Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
83. Golmard L, Caux-Moncoutier V, Davy G, et al. Germline mutation in the RAD51B gene confers predisposition to breast cancer. BMC Cancer. 2013;13:484. doi:10.1186/1471-2407-13-484
84. Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10(1):3–8. doi:10.1158/1535-7163.MCT-10-0893
85. Chan D, Clarke S, Gill AJ, et al. Pathogenic PALB2 mutation in metastatic pancreatic adenocarcinoma and neuroendocrine tumour: a case report. Mol Clin Oncol. 2015;3(4):817–819. doi:10.3892/mco.2015.533
86. Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. doi:10.1056/NEJMoa1611310
87. Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. doi:10.1016/S1470-2045(16)30559-9
88. Clarke N, Wiechno P, Alekseev B, et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018;19(7):975–986. doi:10.1016/S1470-2045(18)30365-6
89. Hussain M, Mateo J, Fizazi K, et al. Phase III study of olaparib versus enzalutamide or abiraterone for metastatic castration-resistant prostate cancer (mCRPC) with homologous recombination repair (HRR) gene alterations (LBA12_PR).
90. Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. doi:10.1056/NEJMoa1506859
91. Mateo J, Porta N, McGovern B, et al. TOPARP-B: a phase II randomized trial of the poly(ADP)-ribose polymerase (PARP) inhibitor olaparib for metastatic castration resistant prostate cancers (mCRPC) with DNA damage repair (DDR) alterations (abstract). J Clin Oncol. 2019;37:5005. doi:10.1200/JCO.2019.37.15_suppl.5005
92. Dougherty BA, Lai Z, Hodgson DR, et al. Biological and clinical evidence for somatic mutations in BRCA1 and BRCA2 as predictive markers for olaparib response in high-grade serous ovarian cancers in the maintenance setting. Oncotarget. 2017;8(27):43653–43661. doi:10.18632/oncotarget.17613
93. Pihlak R, Valle JW, McNamara MG. Germline mutations in pancreatic cancer and potential new therapeutic options. Oncotarget. 2017;8(42):73240–73257. doi:10.18632/oncotarget.17291
94. Singhi AD, George B, Greenbowe JR, et al. Real-time targeted genome profile analysis of pancreatic ductal adenocarcinomas identifies genetic alterations that might be targeted with existing drugs or used as biomarkers. Gastroenterology. 2019;156(8):2242–2253. doi:10.1053/j.gastro.2019.02.037
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.