Back to Journals » International Journal of Nanomedicine » Volume 15
Brain Targeted Gold Liposomes Improve RNAi Delivery for Glioblastoma
Authors Grafals-Ruiz N ![]() , Rios-Vicil CI, Lozada-Delgado EL
, Rios-Vicil CI, Lozada-Delgado EL ![]() , Quiñones-Díaz BI, Noriega-Rivera RA, Martínez-Zayas G
, Quiñones-Díaz BI, Noriega-Rivera RA, Martínez-Zayas G ![]() , Santana-Rivera Y, Santiago-Sánchez GS, Valiyeva F, Vivas-Mejía PE
, Santana-Rivera Y, Santiago-Sánchez GS, Valiyeva F, Vivas-Mejía PE
Received 4 December 2019
Accepted for publication 27 March 2020
Published 23 April 2020 Volume 2020:15 Pages 2809—2828
DOI https://doi.org/10.2147/IJN.S241055
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Nilmary Grafals-Ruiz,1,2 Christian I Rios-Vicil,2,3 Eunice L Lozada-Delgado,2,4 Blanca I Quiñones-Díaz,2,5 Ricardo A Noriega-Rivera,2,5 Gabriel Martínez-Zayas,2,6 Yasmarie Santana-Rivera,2 Ginette S Santiago-Sánchez,2,5 Fatma Valiyeva,2 Pablo E Vivas-Mejía2,5
1Department of Physiology, University of Puerto Rico, San Juan, Puerto Rico; 2Comprehensive Cancer Center, University of Puerto Rico, San Juan, Puerto Rico; 3Department of Neurosurgery, University of Puerto Rico, San Juan, Puerto Rico; 4Department of Biology, University of Puerto Rico, San Juan, Puerto Rico; 5Department of Biochemistry, University of Puerto Rico, San Juan, Puerto Rico; 6Department of Chemistry, University of Puerto Rico, San Juan, Puerto Rico
Correspondence: Pablo E Vivas-Mejía
Comprehensive Cancer Center, University of Puerto Rico, Medical Sciences Campus, San Juan 00935, Puerto Rico
Tel +1787 772 8300, ext 1114
Fax +1787 758 2557
Email [email protected]
Introduction: Glioblastoma (GBM) is the most common and lethal of the central nervous system (CNS) malignancies. The initiation, progression, and infiltration ability of GBMs are attributed in part to the dysregulation of microRNAs (miRNAs). Thus, targeting dysregulated miRNAs with RNA oligonucleotides (RNA interference, RNAi) has been proposed for GBM treatment. Despite promising results in the laboratory, RNA oligonucleotides have clinical limitations that include poor RNA stability and off-target effects. RNAi therapies against GBM confront an additional obstacle, as they need to cross the blood-brain barrier (BBB).
Methods: Here, we developed gold-liposome nanoparticles conjugated with the brain targeting peptides apolipoprotein E (ApoE) and rabies virus glycoprotein (RVG). First, we functionalized gold nanoparticles with oligonucleotide miRNA inhibitors (OMIs), creating spherical nucleic acids (SNAs). Next, we encapsulated SNAs into ApoE, or RVG-conjugated liposomes, to obtain SNA-Liposome-ApoE and SNA-Liposome-RVG, respectively. We characterized each nanoparticle in terms of their size, charge, encapsulation efficiency, and delivery efficiency into U87 GBM cells in vitro. Then, they were administered intravenously (iv) in GBM syngeneic mice to evaluate their delivery efficiency to brain tumor tissue.
Results: SNA-Liposomes of about 30– 50 nm in diameter internalized U87 GBM cells and inhibited the expression of miRNA-92b, an aberrantly overexpressed miRNA in GBM cell lines and GBM tumors. Conjugating SNA-Liposomes with ApoE or RVG peptides increased their systemic delivery to the brain tumors of GBM syngeneic mice. SNA-Liposome-ApoE demonstrated to accumulate at higher extension in brain tumor tissues, when compared with non-treated controls, SNA-Liposomes, or SNA-Liposome-RVG.
Discussion: SNA-Liposome-ApoE has the potential to advance the translation of miRNA-based therapies for GBM as well as other CNS disorders.
Keywords: glioblastoma, GBM, central nervous system, CNS, microRNAs, RNA interference, spherical nucleic acids, liposomes
Introduction
Glioblastoma (GBM) is the most common and malignant form of all primary brain tumors. It is responsible for over 14,000 annual deaths in the US alone (National Cancer Institute). The current standard of care for GBM patients consists of maximal safe resection in combination with radiotherapy and temozolomide (TMZ) chemotherapy. Following this treatment regimen, GBM patients usually survive two years or less after the initial diagnosis, making it a universally fatal disease with no cure. Despite a robust arsenal of diagnostic and treatment modalities available for GBM treatment, the overall survival of GBM patients has barely improved over the last 20 years.1–3 Therefore, novel and more effective therapies against GBM are urgently needed.
MiRNAs are endogenous small non-coding RNAs (22 nucleotides in length) that regulate gene expression at the post-transcriptional level.4,5 Evidence indicates that miRNA dysregulation contributes to GBMs’ initiation, progression, and infiltration ability.6–9 Thus, miRNAs are potential targets for GBM treatment.6 MiRNA-based therapies use oligonucleotide miRNA inhibitors (OMIs) against upregulated miRNAs or oligonucleotide miRNA mimics (OMMs) to replace downregulated miRNAs. Despite promising outcomes in the laboratory, the clinical translation of RNAi-based therapies is developing slowly due to encountered hurdles like fast renal clearance, propensity for nuclease degradation, low incorporation into cancer cells, and activation of immune responses.10–13
Nanoparticles have been designed to address these concerns. As drug carriers, nanoparticles improve RNAi’s circulatory stability, decrease their fast renal clearance, reduce their immune responses, and increase their cellular uptake. Nanoparticles can be synthesized with organic or inorganic materials,14–17 and they can transport both hydrophilic and lipophilic molecules.18 Also, nanoparticles of 10–100 nm in size can accumulate in tumor tissues due to incomplete vascularization—a trademark known as the enhanced permeability retention effect (EPR).19 Examples of nanoparticles for RNA and DNA delivery into cancer cells include poly (amidoamine) (PAMAM), polyethyleneimine (PEI)-complexed nanoparticles, liposomes, and spherical nucleic acids (SNAs), among others.20–25
Liposomes are the most studied nanoparticles for cancer therapeutics. Generally, the lipids used for liposome preparation are biocompatible, biodegradable, and of low toxicity.23,26 The phospholipid portions of the liposomes can be linked with polyethylene glycol (PEG) or PEI molecules to increase liposome blood stability. Liposomes can acquire tissue selectivity by modifying PEG and PEI molecules with amines, carboxylic acids, or maleimide functional groups.14,27,28 These modifications facilitate the conjugation of ligands, peptides, or antibodies against targeted tissues.14,27,28 Due to successful results in preclinical and clinical studies, liposomes are currently the only nanoparticles approved by the FDA as drug delivery carriers.16,17,29-31
More recently, gold nanoparticles (AuNPs) have also gained acceptance as suitable drug delivery vehicles.29,32-34 AuNPs are biologically inert, easily synthesized, commercially available, and highly stable composites.35,36 They possess feasible characteristics that enable their application in diagnostics, imaging, and therapy.16,32,36,37 The surface of AuNPs can enable multiple coupling with drugs, CNS-specific ligands, and oligonucleotides.26,38,39 Because of their high surface area and spherical shape, oligonucleotides can conjugate to the AuNP’s core leading to a 3D structure, commonly known as spherical nucleic acids (SNAs).25,40 SNAs internalize more than 50 types of human tissue cells through class A scavenger receptors.41 SNAs carrying siRNAs against Bcl2L12 oncogene, named UN-0129, are under Phase I clinical trial for gliosarcoma treatment (NCT03020017).
Most nanoparticles designed to treat brain-related diseases encounter an additional challenge as they need to cross the blood-brain barrier (BBB). The BBB is an anatomical semipermeable barrier that protects the brain from foreign substances and regulates molecular transit from the blood into the brain and vice versa.42 More than 98% of potential drugs against CNS malignancies fail to cross the BBB.42 Therefore, creating nanoparticles that enable efficient delivery of RNAi-based therapies through the BBB and into GBM tumors has clinical significance. Apolipoprotein E (ApoE) and rabies virus glycoprotein (RVG) peptides have proven to enable the transport of molecules from the circulation to the brain.43–47
ApoE is a specific ligand for the low-density lipoprotein (LDL) receptors, highly expressed in brain vascular endothelial cells and GBM cells.48,49 This peptide enables the transport of molecules in circulation across the BBB by interacting with LDL receptors of brain capillary endothelial cells, receptor-mediated transcytosis (RMT).50–52 Published data from Böckenhoff and collaborators demonstrated that ApoE, in comparison to other BBB permeable peptides, Angiopep, apolipoprotein B (ApoB), and Transactivator of Transcription (TAT), had the highest delivery of the lysosomal enzyme Arylsulfatase A (ASA) to the brain.43 RVG, on the other hand, is a peptide sequence from a neurotropic virus, known to reach the brain by targeting nicotinic acetylcholine receptors of neuromuscular junctions.53 Brain endothelium, neurons, and GBM cells also express these receptors.46,53-55 Previous studies by Kumur and collaborators showed that RVG peptide significantly increased oligonucleotide delivery to the brain (p=0.001) in comparison to other organs (liver and spleen), making it an excellent candidate to improve RNA delivery to the brain. Moreover, both peptides have proven to facilitate the delivery of different nanoparticles into GBM cells in vitro and in vivo.54,56-60
In this study, we synthesized nanoparticles capable of delivering OMIs inside GBM cells in vitro and in vivo. We generated SNAs by chemically functionalizing OMIs to AuNPs. Then, we encapsulated SNAs inside ApoE or RVG peptides-conjugated liposomes. Our results showed that SNA-Liposome-ApoE and SNA-Liposome-RVG efficiently deliver OMIs in GBM cell lines and brain tumors of GBM syngeneic mice.
Materials and Methods
Chemicals and Reagents
AuNPs, Dulbecco’s Phosphate Buffer Saline (PBS), DL-Dithiothreitol (DTT), Tert-butanol, and 50 kDa microdialysis membranes were purchased from Sigma-Aldrich (St. Louis, MO). Nap-10 G-25 Sephadex Columns were obtained from GE Lifesciences (Pittsburgh, PA). DSPE-PEG-2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]), DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), and cholesterol were purchased at Avanti Polar Lipids (Alabaster, AL). DSPE-PEG-Maleimide and mPEG-SH (SH=thiol or sulfhydryl group) were purchased from Nanocs (New York, NY). RVG (YTIWMPENPRPGTPCDIFTNSRGKRASNG) was purchased from Anaspec (Freemont, CA), ApoE (CGRLVQYRG-EVQAMLGQSTEELRVRLASHLRKLRKRLLRD) was purchased from Lifetein (Somerset, NJ). mirVana oligonucleotide miRNA inhibitors (OMIs) were purchased from Thermo Fisher (Waltham, MA) which includes the Negative Control #1, SH-Negative Control #1 (5ʹ-sequence-SH-3ʹ), OMIs-miR-92b, SH-OMIs-miR-92b (MIMAT0003218), Negative Control #1-Alexa-Fluor 647, and SH-Negative Control #1-Alexa-Fluor 647 (5ʹ-Alexa-Fluor 647-sequence-SH-3ʹ). 2-mercaptoethanol (2-ME) was purchased from Bio-Rad (Berkeley, CA). The “Measure-IT Thiol Assay Kit” was purchased from Thermo Fisher, and the “Maleimide Quantification Assay Kit” was purchased from Abcam (Cambridge, UK).
Cell Line and Culture Conditions
The U87 GBM cell line was purchased from the American Type Culture Collection (ATCC, VA). GL261 cells (mouse astrocytomas) were a gift from Dr. Lilia Y Kucheryavykh. Both cell lines were grown as adherent cells and maintained in DMEM/F12 Media from HyClone Lab (Logan, UT) supplemented with 10% of fetal bovine serum (FBS) (Thermo Fisher) and 0.1% of penicillin/streptomycin (Thermo Fisher) at 37°C in a humid atmosphere with 5% CO2 (normal cell conditions). Cells used for in vitro experiments had confluences of 75–85%.
Spherical Nucleic Acid (SNA) Synthesis
A diagram showing SNA synthesis is depicted in Figure 1A. We used the PEG and Tween-20 stabilization method described by Jiuxing Li et al with some modifications (Figure 1A).61 First, SH-OMIs were reduced with 50 mM of DTT during 1.5 hours at room temperature and purified with Nap-10 G25 Sephadex columns before the conjugation reaction. Citrate capped 15 nm AuNPs (Sigma) resuspended in deionized water (ddH2O) were mixed with mPEG(2000)-SH and Tween-20 for 30 minutes at room temperature. The filtered SH-OMIs were added to the mixture and vortexed. The final concentrations of each of the four components were as follows: 1.5 nM for AuNPs, 100 nM for mPEG-SH, 1 mM for Tween-20, and 450 nM for SH-OMIs. NaCl was added to a final concentration of 1M and incubated for 1.5 hours while rotating at room temperature (RT). Salt and reagent excess was removed by two sequential centrifugations of 17,000 RCF at 4°C for 30 minutes and resuspended with PBS 1X. The obtained product consisted of SH-OMIs conjugated to AuNPs, forming spherical nucleic acid, commonly known as SNAs. SNA’s concentration was calculated with the Beer-Lambert Equation using AuNP’s absorbance at 520 nm UV-visible and an extinction coefficient of 3.67 X 108 M−1 cm−1.
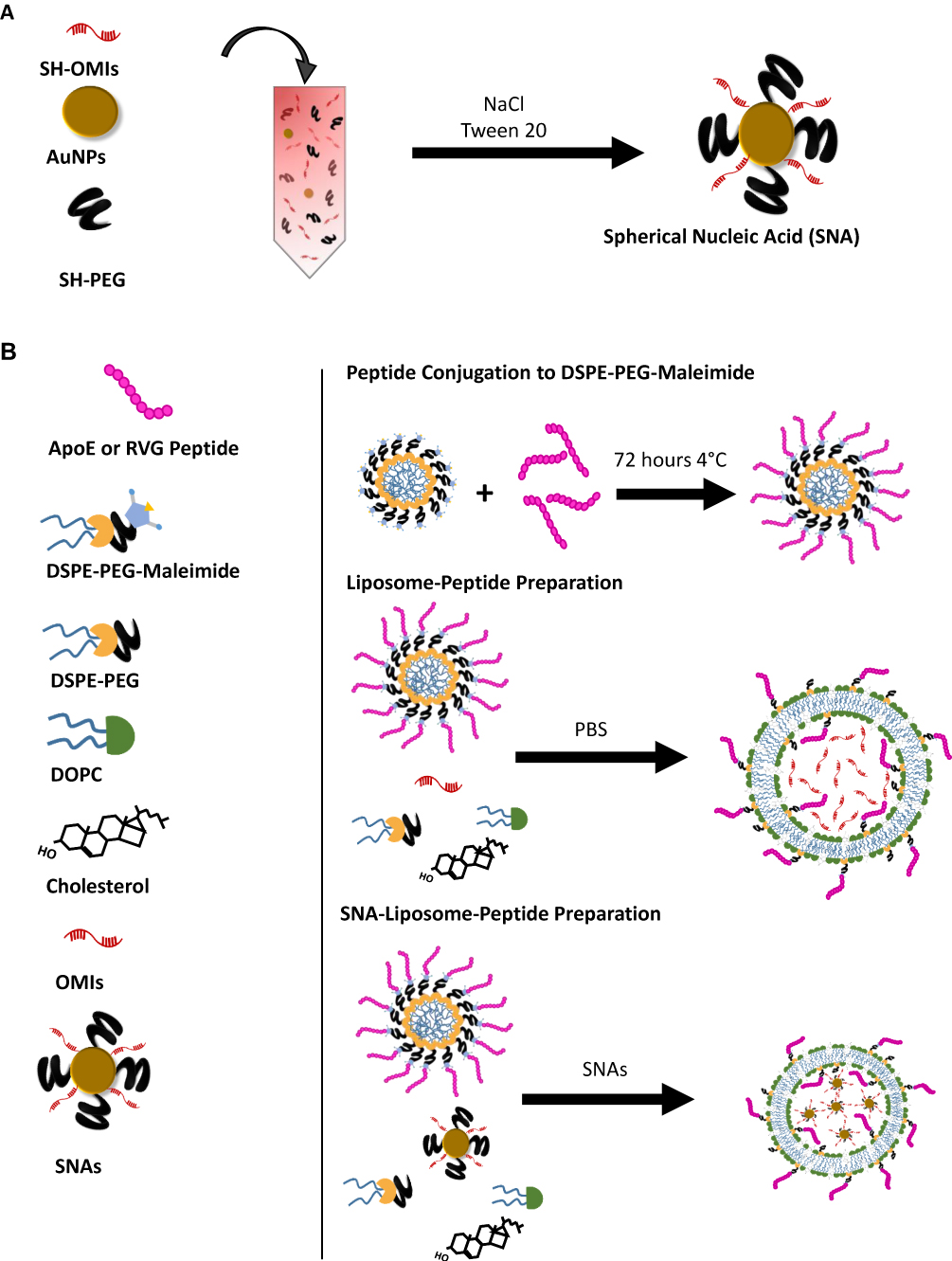 |
Figure 1 Schematic representation of the peptide-conjugated gold-Liposomes’ synthesis. Notes: (A) Spherical nucleic acids (SNAs) preparation. (B) Synthesis of Liposome-Peptide and SNA-Liposome-peptide nanoparticles. |
SNA’s Oligonucleotide Content Measurements
To quantify the RNA/AuNP molar ratio, we reacted SNAs (containing SH-OMIs-Alexa-Fluor 647) with 2-ME (20 mM final concentration) while shaking at RT for 5 hours. After centrifugation at 17,000 RCF (4°C) for 30 minutes, the supernatant with detached SH-OMIs-Alexa-Fluor 647 was collected, transferred to 96 well Nunc-Optical bottom plates (Rochester, NY) and analyzed for fluorescence measurement at 650/665 excitation and emission spectra with the Varioskan Flash Spectral Scan Multimode Reader from Thermo Fisher. The concentration was determined using a standard curve with fluorescence intensities of SH-OMIs-Alexa-Fluor 647. The molar concentrations were expressed as [RNA]/[AuNP] ratios.
SNA’s in vitro Toxicity Assay
The Alamar blue assay from Thermo Fisher was used, following the manufacturer’s instructions, to measure in vitro toxicity in GBM. Here, U87 cells (6 X 104 cells/mL) were seeded into 96-well cell culture plates (Eppendorf, Hamburg, Germany) and incubated for 24 hours at normal cell conditions. The next day, cells were treated with different concentrations (10 nM, 5 nM, 2.5 nM, 1.25 nM, 0.625 nM, and 0.313 nM final concentrations) of SNAs carrying non-targeted negative control OMIs. Seventy-two hours after incubation at 37ºC, cell viability was determined by adding 95 µL of Alamar blue dye. Three hours later, optical density (OD) values were measured with the Bio-Rad XMark Microplate Spectrophotometer. Cell viability was expressed in percentage using the OD of non-treated (NT) cells as 100% of viability.
Preparation of Peptide-Labeled Gold-Liposome Nanoparticles
A schematic representation of the liposome preparation method is shown in Figure 1B. For targeted liposomes, DSPE-PEG(2000)-Maleimide micelles (previously resuspended in 1X PBS, Mg2+ and Ca2+-free) were reacted with either ApoE (containing one cysteine at the end of the amino acid chain) or RVG peptides (containing one cysteine at the middle of the amino acid chain) at a 1:2 molar ratio (DSPE-PEG-Maleimide: Peptide) for 72 hours at 4°C. The conjugation efficiency was monitored by quantifying free thiol groups or maleimide groups with the “Measure- IT Thiol Assay Kit” and the “Maleimide Quantification Assay Kit,” respectively. DSPE-PEG-Mal-Peptide (ApoE or RVG) conjugates were purified with the Pur-A-Lyzer Maxi 50,000 Dialysis Kit (Sigma) and recovered in ddH2O for nanoparticle preparation.
Liposomes were prepared as previously described by Reyes-Gonzalez et al.62 Supplementary Table 1 lists the lipid amount used for each nanoparticle. To prepare untargeted OMIs-containing liposomes (without AuNPs), OMIs were mixed with DOPC (DOPC/OMIs in a 10:1 w/w ratio), cholesterol (DOPC/cholesterol in a 4:1 w/w ratio), DSPE-PEG(2000) (5% of DOPC), and excess of tert-butanol. To prepare Liposome-ApoE and Liposome-RVG nanoparticles, OMIs were mixed with DOPC (DOPC/OMIs in a 10:1 w/w ratio), cholesterol (DOPC/cholesterol in a 4:1 w/w ratio), DSPE-PEG(2000)-Mal-Peptide conjugates (4% of DOPC), DSPE-PEG(2000) (1% of DOPC), and excess of tert-butanol. The lipid mixture of each OMIs-containing liposomes (Liposome, Liposome-ApoE, and Liposome-RVG) was lyophilized and stored at −20°C until used. The lyophilized material was rehydrated with 1X PBS (Mg2+ and Ca2+-free), vortexed for 5 minutes and sonicated for 10 minutes.
 |
Table 1 Physicochemical Characteristics of SNAs |
To prepare untargeted SNA-containing liposomes (SNA-Liposomes), DOPC (DOPC/OMIs in a 10:1 w/w ratio), cholesterol (DOPC/cholesterol in a 4:1 w/w ratio), DSPE-PEG(2000) (5% of DOPC), and excess of tert-butanol were mixed and lyophilized before SNA encapsulation. To prepare SNA-Liposome-ApoE and SNA-Liposome-RVG, DOPC (DOPC/RNA in a 10:1 w/w ratio), cholesterol (DOPC/cholesterol in a 4:1 w/w ratio), purified DSPE-PEG-Mal-Peptide conjugates (4% of DOPC), DSPE-PEG(2000) (1% of DOPC), and excess of tert-butanol were mixed; followed by their lyophilization and storage at −20 °C until used. The lyophilized material was resuspended with SNAs pre-diluted in 1x PBS (Mg2+ and Ca2+-free). The mixture was then vortexed for 5 minutes and sonicated for 10 minutes.
Liposome Encapsulation Efficiency Studies
OMIs containing liposomes (Liposome, Liposome-ApoE, and Liposome-RVG) were dialyzed with Pur-A-Lyzer™ Maxi 50,000 dialysis tubes. Water (ddH2O) was changed every 30 minutes for a total period of 8 hours. Afterward, 2% of Triton X-100 was added to the dialysis tubes, and the amount of OMIs was quantified with a Qubit microRNA Assay Kit in a Qubit 3.0 Fluorometer (Thermo Fisher), according to manufacturer’s specifications. Relative Fluorescent Unit values and OMIs concentrations were determined by using a standard curve corrected with 2.0% of Triton-X.63 The amount of encapsulated OMIs (encapsulation efficiency) was calculated as the percentage amount of OMI inside the membrane divided by the amount of OMI before dialysis.
To determine the encapsulation efficiency of SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG, samples were transferred to Nanosep 300K filter columns (Pall Corporation; NY) and centrifuged at 7500 RPM for 10 minutes. The first filtrate (non-encapsulated SNAs) was collected. The columns (containing SNA-Liposomes) were transferred to fresh new microtubes and treated with 2% of Triton X-100, followed by additional centrifugation at 7500 RPM for 10 minutes. AuNP concentrations before centrifugation, and after first and second centrifugation (treated with Triton X-100) were calculated as described above.
Nanoparticle Size and Zeta Potential Measurements
The nanoparticle’s hydrodynamic diameter, charge, and polydispersity were measured by Dynamic Light Scattering (DLS) with a Mobius instrument (Wyatt Technologies, Santa Barbara, CA).62 Each sample that was analyzed had the same concentration of OMIs (12.5 µg/mL) dispersed in 1X PBS (Mg2+ and Ca2+-free) at room temperature.
Nanoparticle Uptake into GBM Cells Assessment
U87 cells (3.5 X 104 cells/mL) were plated into Lab-Tek Chamber Slides (Thermo-Fisher) and incubated overnight at 37°C, 5% CO2 in a humid atmosphere (normal cell conditions). The next day cells were treated with each nanoparticle (Liposome, Liposome-ApoE, Liposome-RVG, SNA, SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG) diluted in Opti-MEM (Thermo Fisher) with a final concentration of 100 nM. In this experiment, OMIs were labeled with the Alexa-Fluor 647 fluorescent dye. A non-treated control and a Lipofectamine RNAimax (OMI: Lipofectamine ratio of 1:1 v/v) (Thermo Fisher) positive control were included.64 Cells were incubated with each treatment for 6 hours at normal cell conditions. The media was removed, cells were washed with 1X PBS, and then fixed with ethanol 100%; nuclei were counterstained with DAPI (1:5000), and slides were mounted with Permafluor Mountant (Thermo Fisher). Cells were observed under a Nikon Eclipse E400 fluorescent microscope, and pictures were taken with the Nikon DS-Qi2 Camera. The amount of fluorescence inside cells was quantified with NIS-Element Microscope Imaging Software. Mean intensities were evaluated by normalizing Alexa-Fluor 647 fluorescence with DAPI fluorescence.
In vitro miR-92b Downregulation Studies
U87 cells (3.5 X 104 cells/mL) were seeded into 6-well plates (Eppendorf) and incubated overnight at normal cell conditions. The next day, cells were treated with each nanoparticle formulation (Liposome, Liposome-ApoE, Liposome-RVG, SNAs, SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG) containing 100 nM of either negative control OMIs (NC-OMIs) or OMIs that targeted miR-92b (miR92b-OMIs) in Opti-Mem media. A Lipofectamine RNAimax (OMI: Lipofectamine ratio of 1:1 v/v) (Thermo Fisher) positive control was also included. Seven-hours later, Opti-MEM was replaced with DMEM/F12, and cells were incubated at normal cell conditions overnight. The next day, cells were detached with trypsin (0.25%), collected, washed with PBS 1X, pelleted, and stored at −80ºC until used.
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Total RNA (including miRNAs) was isolated with the mirVana miRNA Isolation Kit (Thermo Fisher) as per manufacturer’s instructions. Ten nanograms (ng) of RNA were reversed transcribed to cDNA with the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher) in an Applied Biosystems Veriti 96 well Thermal Cycler (16°C for 30 minutes, 42°C for 30 minutes, 85°C for 5 minutes, and 4°C for 15 minutes). One µL of cDNA was added to TaqMan Universal Master Mix II, with UNGs (Thermo Fisher) and primers for miR-92b or U48 (internal control).64,65 The PCR was performed on a StepOnePlus Real-Time PCR System (Thermo Fisher), and data were processed with the StepOne V2.3 Analysis Software. The relative expression of miR-92b was calculated by the ΔΔCt method using the U48 samples as the internal control.64,66
Tumor Implantation and Nanoparticle Administration
Animals experiments were managed according to the protocol # A8700110, approved by the Institutional Animal Care and Use Committee (IACUC) from the University of Puerto Rico, Medical Sciences Campus-following NIH Guidelines for the Care and Use of Laboratory Animals.67 The use of GL261 cells for tumor implantation was previously approved by the Research and Ethics Committee from the University of Puerto Rico, Medical Sciences Campus. GL261 cells were implanted into the brain of 12 weeks-old C57BL/6 male mice from Taconic Biosciences (Rensselaer, NY, US) following previously described methods.68–70 In brief, we first anesthetized the animals with intraperitoneal injections of ketamine cocktail. Then, mice brains were localized, and a midline scalp incision made with the Digital Just for Mouse Stereotactic Equipment from Stoelting (Wood Dale, IL). A burr hole was made to the calvaria of each mouse, 2 mm lateral, and 2 mm anterior from the bregma. About 3x105 GL261 cells (1.5x105 cells/µL in PBS 1X) were injected (2 mm ventral from drilled calvaria) at a 0.2 µL/min flow rate with the Quintessential Stereotaxic Injector (Stoelting) and a 10 µL (0.48 mm) Hamilton Syringe (Stoelting).
Two weeks after cell implantation, each nanoparticle formulation (containing 10 µg of OMIs) was administered by tail vein injection. The following groups of mice (N=4 per condition) were included in this set of experiments: PBS (non-treated), Liposome, Liposome-ApoE, Liposome-RVG, SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG. The liposomes used in this set of experiments contained the fluorescent dye 3,3ʹ-Dioctadecyl-3,3,3ʹ,3ʹ-tetramethylindocarbocyanine perchlorate (DiL, Sigma-Aldrich) [1% of DOPC (w/w)]. Six hours post-treatment, mice were anesthetized and transcardially perfused with PBS 1X, followed by 4% paraformaldehyde (PFA).71 The collected brains and livers were incubated in PFA for 24 hours, then changed to PBS 1X for 24 hours, stored in ethanol 70%, and embedded in paraffin.
Immunofluorescence and Nanoparticle Accumulation Analysis
Brain slides (10 µm) were immunostained with an antibody against the glial fibrillary acidic protein (GFAP) to localize astrocytes, reactive gliosis, and GBM foci. Briefly, brain slides were subjected to deparaffinization, followed by antigen site retrieval, quenching of endogenous peroxidase, blocking of nonspecific epitopes and serial incubation with a rabbit polyclonal anti-GFAP (1:700) (Abcam) and goat monoclonal anti-rabbit IgG (secondary antibody) labeled with Alexa-Fluor 488 (1:200) (Abcam). Liver tissue sections (5 μm) were stained with DAPI. Brain and liver slices were observed under a Nikon Eclipse Ts2R microscope. Images were taken at 4x, 20x, and 40x magnifications with the Nikon DS-Qi2 camera and subsequently analyzed with the NIS-Element Microscope Software.
Endosomal Escape Analysis of SNA-Liposome-ApoE Nanoparticles
U87 cells (3.5 X 104 cells/mL) were seeded into Lab-Tek Chamber Slides (Thermo-Fisher) and maintained under standard cell conditions. The next day, cells were treated with SNA-Liposome-ApoE nanoparticles containing Alexa-Fluor 647 labeled OMIs at a final concentration of 100 nM (OMIs) in Opti-MEM media. Cells were incubated for 2, 6, and 24 hours, followed by ethanol fixation. Then, lysosomes and late endosomes were marked with the rabbit polyclonal antibody against Lamp-1 (1:200, Abcam) followed by the goat monoclonal anti-rabbit IgG (secondary antibody) labeled with Alexa-Fluor 488 (1:200, Abcam).72–74 Cells were then counterstained with DAPI (1:5000), and slides were mounted. U87 cells were observed under a Nikon Eclipse E400 fluorescent microscope, and images were acquired at 60X magnification with the Nikon DS-Qi2 camera. The cell nucleus (DAPI), lysosomes/late endosomes (LAMP-1), and OMIs in the SNA-Liposome-ApoE nanoparticles (Alexa-Fluor 647) were identified in blue, green, and red colors, respectively.
Cell Viability of miR-92b-Targeted SNA-Liposome-ApoE
The Alamar blue dye assay (Thermo Fisher) was used to measure the cell viability of U87 cells after treatment with SNA-Liposome-ApoE targeting miR-92b.62,75,76 U87 cells (5 X 104 cells/mL) were plated into 96 well plates and incubated under normal cell conditions. The next day, cells were treated with the SNA-Liposome-ApoE nanoparticle carrying 50 nM or 25 nM of OMIs (NC-OMIs and miR92b-OMIs) in Opti-MEM media and incubated under normal cell conditions. Seventy-two hours post-treatment, the media was replaced with 95 µL of Alamar blue. Three hours later, OD values were measured, and the cell viability % analyzed as (technical replicates) relative to the non-treated control (cell viability of 100%).
Serum OMIs Stability and SNA-Liposome-ApoE Shelf-Life
For serum stability, naked-OMIs and SNA-Liposome-ApoE nanoparticles were incubated in 30% FBS at 37°C for 0, 24, 48, and 72 hours. Aliquots (containing 2 µg of RNA) were collected at each time point and separated in a 2% agarose gel electrophoresis.62 RNA bands were imaged using the ChemiDoc MP Imaging System (Bio-Rad).62 The shelf-life of SNA-Liposome-ApoE was determined by evaluating the conservation of the nanoparticle’s hydrodynamic diameter and PDI of SNA-Liposome-ApoE at room temperature (25 °C) for 0, 4, 8, and 24 hours. At each time point, the size (diameter), zeta potential, and PDI were measured by DLS with a Mobius instrument (Wyatt Technologies, Santa Barbara, CA).62
Statistical Analysis
Each experiment was performed at least in triplicates. We used GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA) for graph construction and statistical analysis. Data were analyzed with Student’s t-test for comparing two groups and one-way ANOVA for multiple group comparisons (Tukey’s Post Test). *p<0.05, **p<0.01, and ***p<0.001 was considered significant.
Results
Physicochemical Characteristics of SNAs
Figure 1A summarizes the process we used to prepare SNAs (full description in the “Materials and Methods” section). The addition of a sulfhydryl (SH) group on the 3ʹ-end of the OMIs enables the functionalization of RNA (OMIs) to AuNPs through a covalent thiol-gold bond.61 We determined the physicochemical properties of SNAs by DLS. The mean size, charge, and polydispersity index (PDI) are shown in Table 1. Figure 2A and B shows representative DLS histograms with the size distribution of AuNPs and SNAs. Our SNAs are small, neutral (−10 to +10 mV),77,78 and with a low PDI (<0.30).79 Also, the results from the loading capacity analysis show that around 1 mole of AuNPs contained 50 moles of OMIs (Table 1).
 |
Figure 2 Characterization and in vitro toxicity of SNAs in U87 GBM cells. Notes: (A) Intensity-based DLS histograms for AuNPs. (B) Intensity-based DLS histograms for SNAs. (C, D) Alamar blue dye cell viability analysis of U87 cells treated with (C) SNAs (10 nM-0.3 nM) or (D) Cisplatin (0.1 µM to 10 µM) for 72 hours. |
SNAs are Nontoxic to GBM Cells
Although AuNPs are considered inert and nontoxic to the cells, the functionalization process or drug cargo (OMIs) can result in toxic nanoparticles.25 We performed the Alamar Blue cell viability assay to determine if our SNAs carrying non-targeted negative control OMIs were toxic to U87 GBM cells (Figure 2C). Here, U87 cells were treated with serial dilutions of SNAs with AuNPs’ concentrations ranging from 0.3 nM to 10 nM of AuNP—which corresponds to 15 nM to 500 nM of OMIs (Figure 2C). As a positive control, we simultaneously incubated U87 cells with cisplatin (0.1 µM to 10 µM) for 72 hours (Figure 2D). Our results show that SNAs containing non-targeted negative control OMIs (NC-OMIs) are non-toxic to U87 cells in any of the concentrations (Figure 2C). Moreover, cisplatin treatment significantly reduced U87 cell viability at 1 µM and 10 µM by 50% (**p<0.01) and 90% (***p<0.001), respectively (Figure 2D). These results were expected since cisplatin is a chemotherapeutic agent commonly used against solid tumors.76,80
Liposome Preparation and Characterization
To synthesize brain-targeted nanoparticles, we first conjugated ApoE or RVG peptides to DSPE-PEG-Maleimide micelles (Figure 1B). We mixed these conjugated micelles with a liposomal formulation—described in the “Materials and Methods section.” These liposomes have also been described in previous studies.62,64 Table 2 shows the diameter, charge, and PDI of each nanoparticle. Figure 3 shows the DLS histograms for the nanoparticle size distribution. These DLS results (Table 2, Figure 3) show that SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG are around three times smaller than their paired OMIs-containing liposomal formulations Liposome, Liposome-ApoE, and Liposome-RVG (Table 2, Figure 3). SNA-containing liposomes ranged from 27 nm to 42 nm, compared to OMIs-containing liposomes, which ranged from 100 nm to 140 nm (Table 2, Figure 3). Curiously, the size of AuNP-PEG-Liposomes (AuNPs without OMIs) was similar to the SNA-Liposomes (Supplementary Figure 1). The zeta potential of the liposomes and SNA-Liposomes were neutral (−10 mV to +10 mV).77,78 Table 2 shows the encapsulation efficiency of OMIs (for Liposome, Liposome-ApoE, and Liposome-RVG) and SNAs (for SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG). More than 70% of OMIs and SNAs were encapsulated inside liposomes.
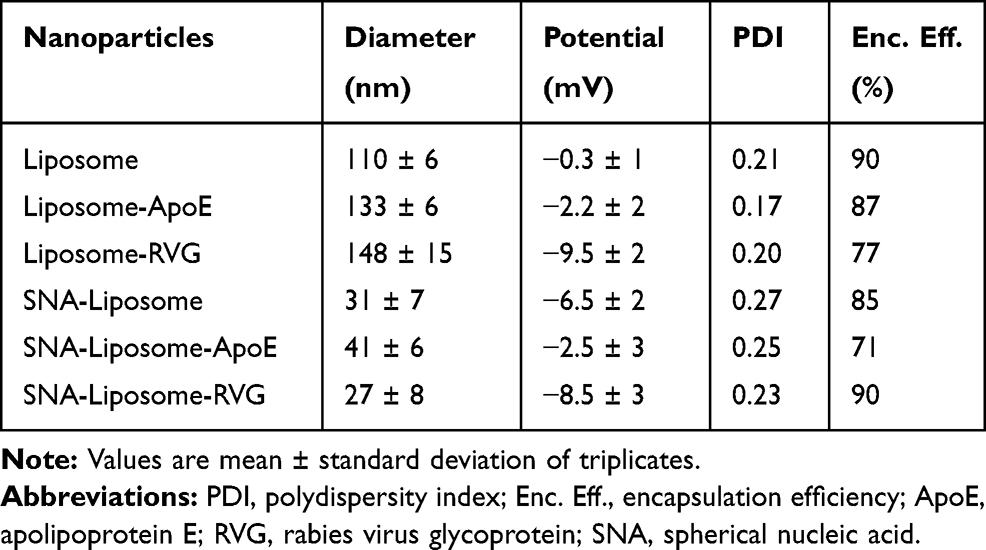 |
Table 2 Physicochemical Characteristics of OMIs-Containing Liposomes and SNA-Containing Liposomes |
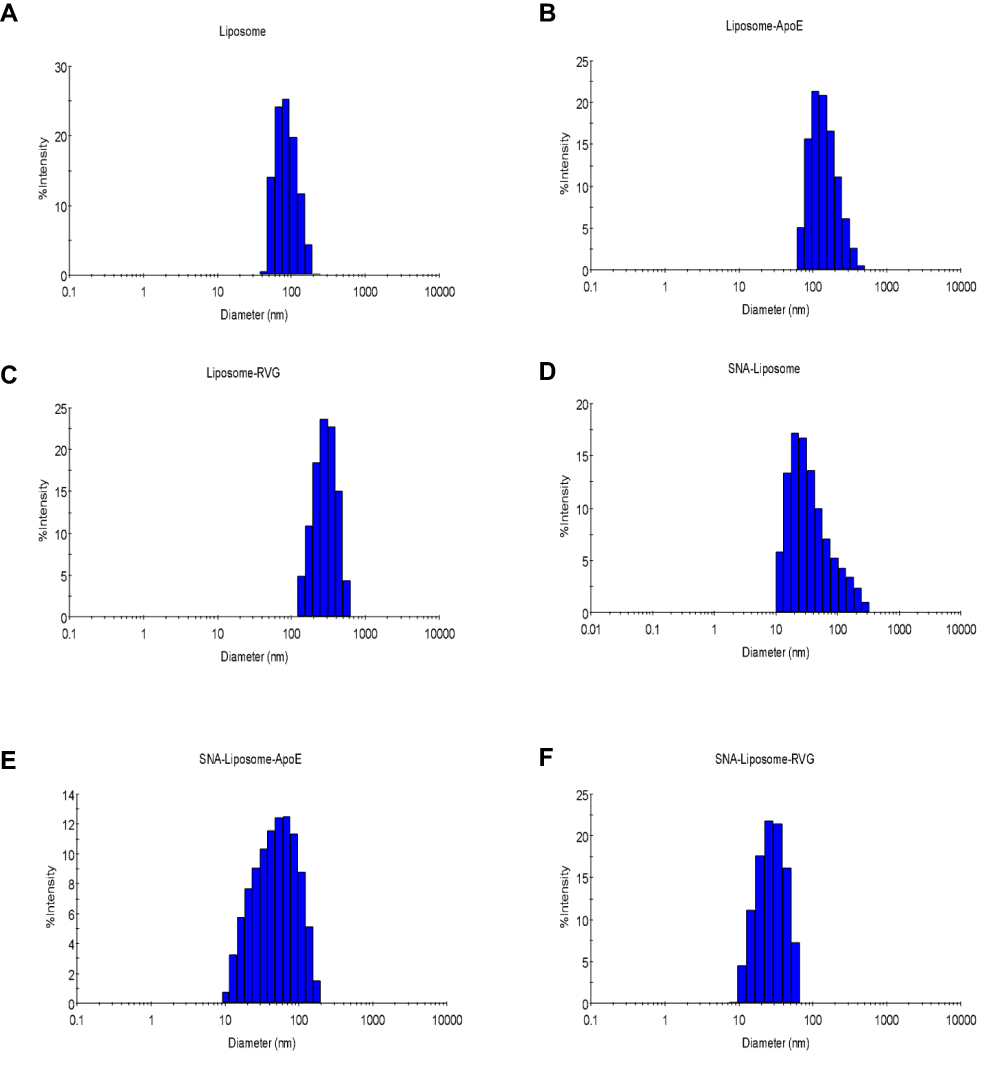 |
Figure 3 DLS histograms for (A) Liposomes, (B) Liposome-ApoE, (C) Liposome-RVG, (D) SNA-Liposome, (E) SNA-Liposome-ApoE, and (F) SNA-Liposome-RVG. |
Internalization Efficiency of Nanoparticles into GBM Cells
To evaluate in vitro cellular uptake of the nanoparticles, we treated U87 cells with 1X PBS (non-treated control), Lipofectamine (with OMIs, positive control), or each nanoparticle formulation (Liposome, Liposome-ApoE, Liposome-RVG, SNAs, SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG) containing 100 nM of Alexa-Fluor 647 labeled OMIs. Six hours after treatment, cells were fixed and stained with DAPI (nuclear fluorescent dye). We acquired microscopy images at 20x magnification and analyzed the mean fluorescent intensities using the NIS-Element Microscope Imaging Software (Figure 4). We can observe that even though all nanoparticles internalized U87 cells, SNAs, SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG had the highest fluorescence intensities (Figure 4A). Moreover, when plotting the mean fluorescent intensities, we observed a significant increase in the Lipofectamine (**P<0.01), SNAs (***P<0.001), SNA-Liposome (***P<0.001), SNA-Liposome-ApoE (***P<0.001), and SNA-Liposome-RVG (**P<0.01) treated cells when compared with the non-treated control cells (Figure 4B). Additional statistical analysis showed that SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG significantly increase OMIs internalization into U87 cells when compared to their liposomal counterparts, Liposome (***P<0.001), Liposome-ApoE (***P<0.001), and Liposome-RVG (*P<0.05) respectively (Supplementary Figure 2). SNAs and all SNA-containing liposomes had similar internalization efficiencies compared with Lipofectamine-transfected OMIs (Figure 4B).
 |
Figure 4 Fluorescence microscopy analysis of nanoparticles uptake in U87 GBM cells. Notes: (A) Images of U87 GBM cells (20X magnification). (B) Nanoparticle internalization analysis with the NIS-Elements Software. |
Targeting miR-92b in GBM Cells with Nanoparticle Formulations
The functionality of the OMIs carried by these nanoparticles was examined by Taq Man qPCR analysis. Before analysis, U87 cells were treated with 100 nM of OMIs (NC-OMIs and miR92b-OMIs contained in Lipofectamine or any of the nanoparticle formulations). Cells were analyzed after 24 hours. As expected, when cells were treated with miR92b-OMIs in combination with Lipofectamine RNAimax transfection reagent, the expression of miR-92b was reduced by 82% (***P<0.001) compared with the NC-OMIs (Figure 5). Similarly, SNAs decreased miR-92b expression by 65% (*P<0.05), SNA-Liposomes by 57% (*P<0.05), SNA-Liposome-ApoE by 78% (***P<0.001), and SNA-Liposome-RVG by 86% (***P<0.001) in U87 cells compared with their NC-OMIs counterpart (Figure 5). The Liposome-ApoE formulation (with OMIs) also decreased miR-92b expression by 50% (**P<0.01) compared with their NC-OMI formulation (Figure 5). Interestingly, SNA-Liposome-ApoE and SNA-Liposome-RVG reduced miR-92b to similar levels compared to the Lipofectamine control group, which is a transfection reagent amply used to transfect RNA oligonucleotides in cultured cells (Figure 5). Additional statistical analysis (Supplementary Figure 3) showed a significant decrease in the relative expression of miR-92b in U87 cells after treatment with SNA-Liposome-ApoE (*P<0.05) and SNA-Liposome-RVG (*P<0.05) compared to Liposome-ApoE and Liposome-RVG respectively. Together, these results indicate that ApoE and RVG peptides-targeted SNA-Liposome nanoparticles promoted cell internalization and reduced miR-92b levels in GBM cells.
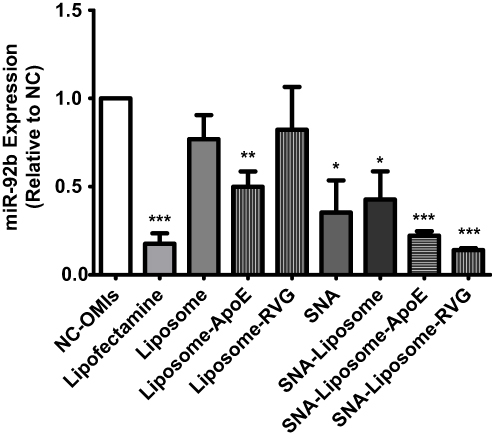 |
Figure 5 Real-time PCR-based miR-92b expression following U87 cells incubation with nanoparticles. Note: Values were expressed relative to the NC-OMI in each treatment. |
Nanoparticles Accumulation in a Syngeneic Mouse GBM Model
Then, we investigated the ability of each nanoparticle formulation to reach the tumor tissue in an intracranial GBM mouse model. Wild type C57BL/6 mice were intracranially injected with 3X105 GL261 cells, as described in the “Materials and Methods” section. We confirmed the presence of brain tumors by H&E, DAPI staining (to identify enlarged nuclei), and GFAP immunofluorescence staining (to localize astrocytoma tumor foci) (Supplementary Figure 4). Two weeks after tumor implantation, we injected mice intravenously (iv) with PBS (control non-treated group) or with each nanoparticle formulation (Liposome, Liposome-ApoE, Liposome-RVG, SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG). Liposomes were stained with DiL fluorescent lipid dye (see methods). Six hours later, mice were perfused and fixed to process the brain tissues for immunofluorescence staining and nanoparticle localization in brain tumors. To localize brain tumors and DiL (red) stained nanoparticles, we immunostained brain slices against GFAP (green) and counterstained with DAPI (blue) (Figure 6A). Our microscopy images showed prominent tumor accumulation of the following nanoparticles: SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG (Figure 6A). Of all, SNA-Liposome-ApoE and SNA-Liposome-RVG colocalized (internalize) with the tumor tissue cells (Figure 6A, merge images). Figure 6B shows the analysis for red fluorescent intensities (quantified with the NIS-Element Microscope Imaging Software) localized in brain tumors after nanoparticle administration. We observed a significant increase in fluorescent intensities for SNA-Liposome-ApoE (***P<0.001) and SNA-Liposome-RVG (**P<0.01) compared with the non-treated group (Figure 6b). Notably, the SNA-Liposome-ApoE nanoparticles showed a significant accumulation in the brain tumor area compared to SNA-Liposomes (***P<0.001) and SNA-Liposome-RVG (***P<0.001). We also analyzed the fluorescent intensities of SNA-Liposome, SNA-Liposome-ApoE, and SNA-Liposome-RVG compared to their OMIs-containing liposomal counterparts (Liposome, Liposome-ApoE, and Liposome-RVG, respectively). This analysis showed a significant increase in the fluorescent intensities of SNA-Liposome-ApoE (***P<0.001) and SNA-Liposome-RVG (*P<0.05) (Supplementary Figure 5). Also, fluorescence microscopy images taken from brain tumor-adjacent areas showed no DiL-associated fluorescence in any of the formulations tested (Supplementary Figure 6). Interestingly, the Liposome formulation showed higher accumulation in the mice liver tissue compared with the other nanoparticles (***P<0.001) (Supplementary Figure 7). Altogether, these data suggest that systemic injections of SNA-Liposome-ApoE increased the delivery of OMIs into brain tumors.
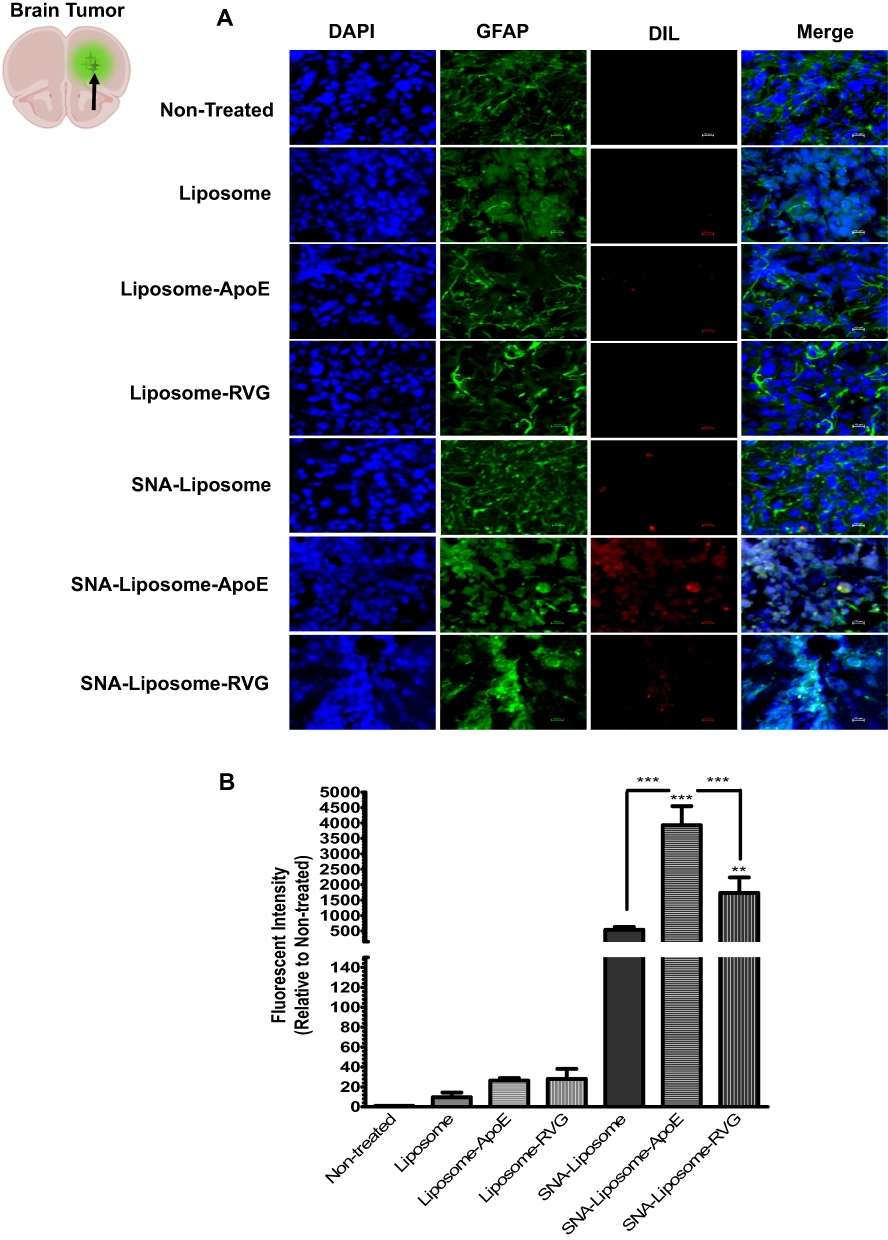 |
Figure 6 Fluorescence microscopy images showing the nanoparticle accumulation in brain tumors of GBM syngeneic mice. Notes: (A) Images of ex-vivo brain tumor tissues (40X magnification). (B) Nanoparticle distribution analysis with the NIS-Elements Software. |
SNA-Liposome-ApoE Can Escape Lysosome/Endosomes
As the SNA-Liposome-ApoE nanoparticle showed the highest accumulation in U87 cells and GBM tumors, we studied the lysosomal/late endosomal fate of these nanoparticles. We treated U87 cells with SNA-Liposome-ApoE nanoparticles for 2, 6, and 24 hours followed by cell fixation and immunostaining against the lysosome/late endosome marker, Lamp-1.29,72,73,81 Fluorescent images (60X magnification) of non-treated and SNA-Liposome-ApoE treated cells showed the internalization of Alexa-Fluor 647 labeled OMIs (red) in all of the three-time points tested (Figure 7). We detected the greatest colocalization (yellow) between SNA-Liposome-ApoE nanoparticles (red) and lysosome/late endosomes (green) at 2 hours post-treatment, although colocalization is seen at all time points (Figure 7). We identified higher non-colocalized cell regions (white arrows) as lysosome/late endosome escape at 6 hours and 24 hours (Figure 7). Our results suggest that SNA-Liposome-ApoE can notably escape lysosome/late endosomes from U87 cells after 6 hours and 24 hours of nanoparticle incubation (white arrows, Figure 7).
 |
Figure 7 Fluorescence microscopy images showing endosomal escape. Notes: Images of U87 cells (60X magnification) exposed to fluorescent oligonucleotides-containing SNA-Liposome-APOE for 2, 6, and 24 hours. |
MiR-92b-Targeted SNA-Liposome-ApoE Nanoparticles Reduced Cell Viability
As SNA-Liposome-ApoE were accumulated in tumor tissue, reduced miR-92b levels, and were able to escape from endosomes, we studied the capacity of miR-92b-targeted SNA-Liposome-ApoE to reduce cell viability in U87 cells. Our results showed that miR92b-OMIs in SNA-Liposome-ApoE significantly reduced cell viability at 50 nM (23%, ***P<0.001) and 25 nM (21%, **P<0.01) compared with NC-OMIs SNA-Liposome-ApoE (Figure 8).
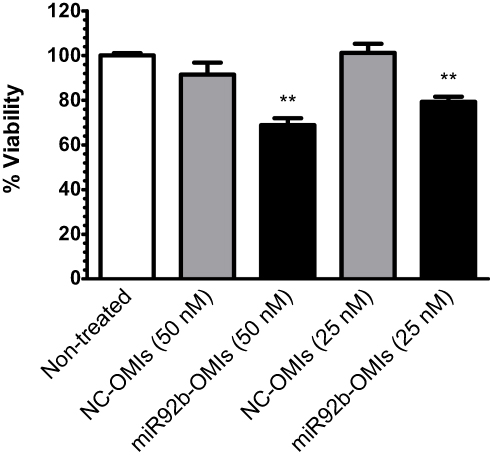 |
Figure 8 Cell viability of U87 cells incubated with miR-92b-targeted SNA-LiposomeAPoE.Note: Values were expressed relative to the untreated cells (100% cell viability). |
Serum OMIs Stability and SNA-Liposome-APOE Shelf-Life Preservation
Finally, we studied the capacity of the SNA-Liposome-APOE formulation to protect OMIs from degradation by nucleases. To in vitro imitate plasma conditions, we incubated SNA-Liposome-APoE and naked OMIs in 30% FBS at 37°C for 0, 24, 48, and 72 hours. The gel electrophoresis showed in the Supplementary Figure 8 showed that naked OMIs were degraded as soon as 24-hr incubation in FBS and were almost undetectable 72-hr after FBS incubation. Opposite, incubation of SNA-Liposome-ApoE protected OMIs from degradation at all time-points tested (Supplementary Figure 8). When we measured the shelf-life of the SNA-Liposome-ApoE, we observed that the size, charge, and PDI index were preserved at RT for 0, 4, 8, and 24 hours (Supplementary Table 2). Altogether, these studies indicate that SNA-Liposome-ApoE can protects OMI molecules from FBS degradation and maintains liposomal physical properties at RT for at least 24 hours.
Discussion
In this study, we synthesized novel brain targeted gold-liposomal nanoparticles that can effectively deliver RNAi molecules to GBM tumor cells. Specifically, SNA-Liposome-ApoE increased OMIs internalization into GBM human cells, decreased miR-92b expression, reduced GBM cell viability, and increased nanoparticles’ accumulation into intracranial GBM mouse tumors. GBM is among the deadliest types of cancers and the second most common type of primary brain tumor (National Cancer Institute). Also, GBM’s overall survival of patients (15 months) has not improved over the last two decades.1–3 Lacking effective therapies is attributed to a myriad of reasons, including the GBM’s recurrent and resistive nature and the inability of drugs to cross the BBB—which precludes more than 98% of therapies from reaching the brain.42,82-85 Therefore, the targeted gold-liposomes synthesized in this study promotes the advancement of RNAi-based therapies against GBM and other CNS disorders.
SNAs are spherical nucleic acids formed by a gold-thiol bond between SH-oligonucleotides and the AuNP core. Oligonucleotide AuNP interactions enable nucleic acids to be densely packed and radially orientated.39 The densely packed orientation of nucleic acids increases the affinity constant between complementary oligonucleotide sequences, and in turn, protects them from nuclease degradation.86 The unique 3D structure of SNAs allows them to effectively enter cells by Class A Scavenger Receptors via a caveola dependent pathway.39,87,88 Studies by Melamed, et al demonstrated that this 3D orientation led to the increased SNA’s cellular uptake in comparison to nucleic acid polyplexes.87 Additional work by Jensen et al showed that SNAs are capable of internalizing over 50 different types of cells (including U87 cells) and even crossing the BBB32,89 A clinical trial for GBM treatment using SNAs (UN-0129) carrying siRNAs against Bcl2L12 (NCT03020017) is currently underway.25,90 Despite promising results, studies reported by Wilhelm et al, show that less than 1% of SNAs and other nanoparticles reach tumor tissues while highly accumulating in the liver and spleen.91,92
The lack of knowledge about gold toxicity, as well as in aggregation and excretion in vivo, increases concerns regarding continuous treatment and long-term consequences of SNA administration in patients. Therefore, tissue-specific modifications are essential to improve the delivery of SNAs to GBM tumors rather than other extraneural tissues. To enhance the specificity of GBM cells in the brain, we encapsulated SNAs inside RVG, and ApoE peptide conjugated liposomes. The liposome formulation we used (DOPC, cholesterol, DSPE-PEG-2000) to encapsulate SNAs and OMIs has been previously characterized by our research team.62 These liposomes showed negligible toxicity both in vitro and in vivo; and no detectable immune responses.62 Also, these liposomes efficiently delivered c-MYC-targeted siRNA in a xenograft mouse model of ovarian cancer,62 and miR-143 targeted OMIs in a subcutaneous GBM mouse model.64 The dual nanoparticles we designed in this study composed of OMIs-AuNPs (SNAs), and brain targeted liposomes have two desirable characteristics: (i) highly oriented oligonucleotides that increase inhibition efficiency and (ii) tumor specificity to improve accumulation in brain tumors. These characteristics are essential to decrease peripheral nanoparticle degradation while improving their tissue specificity, as shown by the SNA-Liposome-ApoE and SNA-Liposome-RVG nanoparticles we prepared.
Unexpectedly, our SNA-Liposome nanoparticles were around 30–50 nm in diameter when their paired liposomes (containing OMIs) were twice to three times bigger (diameters between 100–150 nm). The diameter of PEG-AuNP liposomes (without OMIs) resembles SNA-Liposomes (with OMIs), suggesting that intermolecular forces between AuNPs and liposomal components may contribute to the observed size reduction. As an approximation and assuming nanoparticles as spheres, we can estimate the volume of an SNA (10 nm radium) to be 4,190 nm3. The mean size of the SNA-liposome was 20 nm (radium). Its volume is approximately 33,520 nm3, meaning that the number of entrapped SNA into liposomes could be less than eight SNAs per liposome. Previous studies have shown that AuNPs can interact with head-groups from the lipid molecules of liposomes and thus alter their physicochemical properties.93 Charged interactions between AuNPs and lipidic polar groups may occur, resulting in the reduction of nanoparticle size, as observed in our study.93 This effect on size reduction could increase their chances of crossing the BBB. Early reports showed that nanoparticles with diameters around 50 nm could accumulate with higher efficiency inside cells, escape phagocytic uptake, and deposit in tumor tissue due to the EPR effect.94–96
Our nanoparticles had neutral to slight negative charges (+10 mV to −10 mV).78 Evidence indicates that neutral nanoparticles are ideal for drug delivery due to their capacity to interact with cell membranes and to reduce immune response activation.41,97-100 Even though nanoparticles with positive charge interact easier with cell membranes,77,99 they can increase macrophage entrapment and immune response, as well as increasing the production of reactive oxygen species (ROS).100–102 On the other hand, negatively charged nanoparticles cannot interact with cell membranes decreasing in this way their internalization efficacy.100–102 Adding DSPE-PEG-2000 to our nanoparticle formulations decreases their probability of being entrapped by the mononuclear phagocyte system (MPS).39,91,100 However, the physicochemical properties could change once nanoparticles are administered systemically due to the formation of protein corona (modifications in the surface of nanoparticles), which could alter not only the charge but other nanoparticle physicochemical properties as well.37,103,104
In this study, we sought to develop a brain targeted nanoparticle that could efficiently deliver RNAi molecules into GBM tumors. We chose ApoE and RVG peptides because of their promising results delivering biomolecules to the brain parenchyma.43,47,55 ApoE and RVG peptide labeled nanoparticles have enhanced the delivery of antifungal treatments, siRNAs, and proteins across the BBB and into the CNS.43,47,55 ApoE is a small peptide that targets LDL receptors, commonly present in brain endothelium and GBM cells.43,49,105 On the other hand, RVG is a neurotropic virus peptide that binds to the nicotinic acetylcholine receptors and facilitates nanoparticle delivery to the brain and brain tumors.47,53-55,60 When evaluating nanoparticles’ capacity to internalize GBM cells and inhibit the expression of miR-92b in vitro, we observed that SNAs, SNA-Liposomes, SNA-Liposome-ApoE, and SNA-Liposome-RVG had similar delivery efficiencies than the lipofectamine used as a positive control. Our in vivo results confirmed our hypothesis that ApoE and RVG peptides would enable RNAi systemic delivery into brain tumor cells. Specifically, SNA-Liposome-ApoE significantly improved tumor accumulation compared to each of the other nanoformulations created. Also, SNA-Liposome-ApoE was able to achieve endosomal escape from U87 GBM cells. Endosomal escape serves as an indicator of OMIs release into the cytoplasm, hence demonstrating the nanoparticle’s effective delivery. Furthermore, the OMIs contained in SNA-Liposome-ApoE were protected from FBS degradation suggesting a higher payload of OMIs into the brain tumors when exposed to high protein and nuclease concentration present in plasma.
At present, there are no available explanations of how SNA-Liposomes accumulate into brain tumor tissue. SNA-Liposomes could be reaching brain tumors by EPR due to the incomplete vascularization of brain endothelial cells during tumorigenesis.106 However, additional studies are necessary to fully understand the mechanism by which SNA-Liposomes can cross the BBB and reach brain tumor cells. Further studies should also address the mechanism by which the SNA-liposome formulations scape of liver uptake we observed here.
Our results propose novel nanoparticle conjugates for the delivery of RNAi-based therapies and other drugs to brain tumors and other brain-related diseases. Future studies should assess the therapeutic effects of targeting specific deregulated miRNAs (ie, miR-92b) or other aberrantly abundant genes (with siRNA) using SNA-Liposome-ApoE or SNA-Liposome-RVG in GBM mouse models.
Conclusion
Despite the therapeutic potential of RNAi-based therapies, their poor stability and delivery efficiency delays their translation to the clinic. The nanoparticles we developed and characterized in this study are composed of SNAs encapsulated inside ApoE or RVG peptide-conjugated liposomes. They were able to effectively be internalized by GBM cells and target a highly abundant miRNA (miR-92b) in these cells. Furthermore, these nanoparticles were able to cross the BBB and reach the tumor tissue in a GBM mouse model. These nanoparticles could be, therefore, optimal vehicles for future RNAi-based treatments against GBM and other CNS malignancies.
Acknowledgments
This manuscript is a partial fulfillment of the doctoral dissertation of Nilmary Grafals-Ruiz. This project was supported in part by the RCMI grant U54 MD007600 (National Institute on Minority Health and Health Disparities) (PEVM), the Puerto Rico Science, Technology & Research Trust (PEVM), the NIGMS-RISE (National Institute of General Medical Sciences-Research Training Initiative for Student Enhancement) Program Grant Number R25-GM061838 (NGR, BIQD, GSS, and RN), and institutional seed funds from the University of Puerto Rico Comprehensive Cancer Center (PEVM). The authors acknowledge Dr. Kimberleve Rolón-Roldán and Dr. Lilia Y Kucheryavykh for their technical support with the glioblastoma intracranial tumor implantation. The authors also want to thank Drs. Gabriel Barletta for the critical revision of the manuscript.
Author Contributions
PEVM conceived and supervised the overall study. NGR contributed to the nanoparticle design and preparation, experimental design, conducted the experiments, prepared the figures, and wrote the manuscript. BQD, GMZ, and RNR performed nanoparticle characterization experiments. ELD, GMZ, GSS, and FV did in vivo experiments. CRV and FV performed immunostaining experiments. YSR performed qPCR experiments, edited figures, and contributed to the graphical design of Figure 1. All authors contributed to data analysis, drafting and revising the article with the supervision of PEVM, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Alcedo-Guardia R, Labat E, Blas-Boria D, Vivas-Mejia PE. Diagnosis and new treatment modalities for glioblastoma: do they improve patient survival? Curr Mol Med. 2016;16(5):447–464. doi:10.2174/1566524016666160429120150
2. Tamimi AF, Juweid M. Epidemiology and Outcome of Glioblastoma. Codon Publications; 2017. doi:10.15586/CODON.GLIOBLASTOMA.2017.CH8
3. Ostrom QT, Cioffi G, Gittleman H, et al. CBTRUS Statistical Report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21(Supplement_5):v1–v100. doi:10.1093/neuonc/noz150
4. Friedman RC, Farh KKH, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105. doi:10.1101/gr.082701.108
5. Grün D, Wang Y-L, Langenberger D, Gunsalus KC, Rajewsky N. microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comput Biol. 2005;1(1):e13. doi:10.1371/journal.pcbi.0010013
6. Wang H, Xu T, Jiang Y, Yan Y, Qin R, Chen J. MicroRNAs in human glioblastoma: from bench to beside. Front Biosci (Landmark Ed.). 2015;20:105–118. doi:10.2741/4300
7. Lebrun DG, Min L. MicroRNAs in glioblastoma multiforme: profiling studies and therapeutic impacts. Mol Cell Pharmacol. 2011;3(3):93–105. doi:10.4255/mcpharmacol.11.14
8. Zhang Y, Dutta A, Abounader R. The role of microRNAs in glioma initiation and progression. Front Biosci (Landmark Ed). 2012;17:700–712. doi:10.2741/3952
9. Wen KC, Sung PL, Yen MS, Chuang CM, Liou WS, Wang PH. MicroRNAs regulate several functions of normal tissues and malignancies. Taiwan J Obstet Gynecol. 2013;52(4):465–469. doi:10.1016/j.tjog.2013.10.002
10. Tan SJ, Kiatwuthinon P, Roh YH, Kahn JS, Luo D. Engineering nanocarriers for siRNA delivery. Small. 2011;7(7):841–856. doi:10.1002/smll.201001389
11. Gao S, Dagnaes-Hansen F, Nielsen EJB, et al. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther. 2009;17(7):1225–1233. doi:10.1038/mt.2009.91
12. Huang Y, Hong J, Zheng S, et al. Elimination pathways of systemically delivered siRNA. Mol Ther. 2011;19(2):381–385. doi:10.1038/mt.2010.266
13. Hickerson RP, A V V, Wang Q, et al. Stability study of unmodified siRNA and relevance to clinical use. Oligonucleotides. 2008;18(4):345–354. doi:10.1089/oli.2008.0149
14. Díaz MR, Vivas-Mejia PE. Nanoparticles as drug delivery systems in cancer medicine: emphasis on RNAi-containing nanoliposomes. Pharmaceuticals. 2013;6(11):1361–1380. doi:10.3390/ph6111361
15. McNeil SE. unique benefits of nanotechnology to drug delivery and diagnostics. Methods Mol Biol. 2011;3–8. doi:10.1007/978-1-60327-198-1_1
16. Lozada-Delgado EL, Grafals-Ruiz N, Vivas-Mejía PE. RNA interference for glioblastoma therapy: innovation ladder from the bench to clinical trials. Life Sci. 2018;188:26–36. doi:10.1016/j.lfs.2017.08.027
17. Crucho CIC, Anselmo AC, Mitragotri S. Nanoparticles in the Clinic. ChemMedChem. 2015;55(1):24–38. doi:10.1002/cmdc.201402290
18. Mc Carthy DJ, Malhotra M, O’Mahony AM, Cryan JF, O’Driscoll CM. Nanoparticles and the blood-brain barrier: advancing from in-vitro models towards therapeutic significance. Pharm Res. 2015;32(4):1161–1185. doi:10.1007/s11095-014-1545-6
19. Dewhirst MW, Secomb TW. Transport of drugs from blood vessels to tumour tissue. Nat Rev Cancer. 2017;17(12):738–750. doi:10.1038/nrc.2017.93
20. Li J, Liang H, Liu J, Poly WZ. (amidoamine) (PAMAM) dendrimer mediated delivery of drug and pDNA/siRNA for cancer therapy. Int J Pharm. 2018;546(1–2):215–225. doi:10.1016/j.ijpharm.2018.05.045
21. Thomas M, Lange-Grünweller K, Dayyoub E, et al. PEI-complexed LNA antiseeds as miRNA inhibitors. RNA Biol. 2012;9(8):1088–1098. doi:10.4161/rna.21165
22. Cao N, Cheng D, Zou S, Ai H, Gao J, Shuai X. The synergistic effect of hierarchical assemblies of siRNA and chemotherapeutic drugs co-delivered into hepatic cancer cells. Biomaterials. 2011;32(8):2222–2232. doi:10.1016/j.biomaterials.2010.11.061
23. Puri A, Loomis K, Smith B, et al. Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit Rev Ther Drug Carrier Syst. 2009;26(6):523–580. doi:10.1615/critrevtherdrugcarriersyst.v26.i6.10
24. Lujan H, Griffin WC, Taube JH, Sayes CM. Synthesis and characterization of nanometer-sized liposomes for encapsulation and microrna transfer to breast cancer cells. Int J Nanomedicine. 2019;14:5159–5173. doi:10.2147/IJN.S203330
25. Jensen SA, Day ES, Ko CH, et al. Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med. 2013;5(209):209ra152. doi:10.1126/scitranslmed.3006839
26. Grumezescu AM. Advances in nanobiomaterials for oncology medicine. In: Nanobiomaterials in Cancer Therapy: Applications of Nanobiomaterials. Vol. 7. Elsevier; 2016.
27. Figueiredo S, Cabral R, Luís D, Fernandes AR, Baptista PV. Conjugation of Gold nanoparticles and liposomes for combined vehicles of drug delivery in cancer. Nanomedicine. 2014;48–82.
28. Deng L, Zhang Y, Ma L, et al. Comparison of anti-EGFR-Fab’ conjugated immunoliposomes modified with two different conjugation linkers for siRNA delivery in SMMC-7721 cells. Int J Nanomedicine. 2013;8:3271–3283. doi:10.2147/IJN.S47597
29. Liu Y, He M, Niu M, et al. Delivery of vincristine sulfate-conjugated gold nanoparticles using liposomes: a light-responsive nanocarrier with enhanced antitumor efficiency. Int J Nanomedicine. 2015;10:3081–3095. doi:10.2147/IJN.S79550
30. Marqués-Gallego P, De Kroon AIPM. Ligation strategies for targeting liposomal nanocarriers. Biomed Res Int. 2014;2014. doi:10.1155/2014/129458.
31. Silverman JA, Deitcher SR. Marqibo (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother Pharmacol. 2013;71(3):555–564. doi:10.1007/s00280-012-2042-4
32. Giljohann DA, Seferos DS, Daniel WL, Massich MD, Patel PC, Mirkin CA. Gold nanoparticles for biology and medicine. Angew Chem Int Ed Engl. 2010;49(19):3280–3294. doi:10.1002/anie.200904359
33. Panyala NR, Peña-Méndez EM, Havel J. Gold and nano-gold in medicine: overview, toxicology and perspectives. J Appl Biomed. 2009;7(2):75–91. doi:10.32725/jab.2009.008
34. Ding Y, Jiang Z, Saha K, et al. Gold nanoparticles for nucleic acid delivery. Mol Ther. 2014;22(6):1075–1083. doi:10.1038/mt.2014.30
35. McCully M, Hernandez Y, Conde J, et al. Significance of the balance between intracellular glutathione and polyethylene glycol for successful release of small interfering RNA from gold nanoparticles. Nano Res. 2015;8(10):3281–3292. doi:10.1007/s12274-015-0828-5
36. Levy R, Shaheen U, Cesbron Y, See V, Lévy R, Sée V. Gold nanoparticles delivery in mammalian live cells: a critical review. Nano Rev. 2010;1:1–18. doi:10.3402/nano.v1i0.4889
37. Singh P, Pandit S, Mokkapati VRSS, Garg A, Ravikumar V, Mijakovic I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int J Mol Sci. 2018;19:7. doi:10.3390/ijms19071979
38. Prades R, Guerrero S, Araya E, et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials. 2012;33(29):7194–7205. doi:10.1016/j.biomaterials.2012.06.063
39. Kapadia CH, Melamed JR, Day ES. Spherical nucleic acid nanoparticles: therapeutic potential. BioDrugs. 2018;32(4):297–309. doi:10.1007/s40259-018-0290-5
40. Taton TA. Preparation of gold nanoparticle-DNA conjugates. In: Beaucage SL, et al, editor, Current Protocols in Nucleic Acid Chemistry; 2002. doi:10.1002/0471142700.nc1202s09
41. Choi CHJ, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. Proc Natl Acad Sci. 2013;110(19):7625–7630. doi:10.1073/pnas.1305804110
42. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx. 2005;2(1):3–14. doi:10.1602/neurorx.2.1.3
43. Bockenhoff A, Cramer S, Wolte P, et al. Comparison of five peptide vectors for improved brain delivery of the lysosomal enzyme arylsulfatase A. J Neurosci. 2014;34(9):3122–3129. doi:10.1523/JNEUROSCI.4785-13.2014
44. Neves AR, Queiroz JF, Weksler B, Romero IA, Couraud P-O RS. Solid lipid nanoparticles as a vehicle for brain-targeted drug delivery: two new strategies of functionalization with apolipoprotein E. Nanotechnology. 2015;26(49):495103. doi:10.1088/0957-4484/26/49/495103
45. Nikanjam M, Blakely EA, Bjornstad KA, Shu X, Budinger TF, Forte TM. Synthetic nano-low density lipoprotein as targeted drug delivery vehicle for glioblastoma multiforme. Int J Pharm. 2007;328(1SPEC. ISS.):86–94. doi:10.1016/j.ijpharm.2006.07.046
46. Son S, Hwang DW, Singha K, et al. RVG peptide tethered bioreducible polyethylenimine for gene delivery to brain. J Control Release. 2011;155(1):18–25. doi:10.1016/j.jconrel.2010.08.011
47. Kumar P, Wu H, McBride JL, et al. Transvascular delivery of small interfering RNA to the central nervous system. Nature. 2007;448(7149):39–43. doi:10.1038/nature05901
48. Elshourbagy NA, Liao WS, Mahley RW, Taylor JM. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc Natl Acad Sci U S A. 1985;82(1):203. doi:10.1073/PNAS.82.1.203
49. Murakami M, Ushio Y, Morino Y, Ohta T, Matsukado Y. Immunohistochemical localization of Apolipoprotein E in human glial neoplasms. J Clin Invest. 1989;82(1):177–188. doi:10.1172/JCI113568
50. Béduneau A, Saulnier P, Benoit JP. Active targeting of brain tumors using nanocarriers. Biomaterials. 2007;28(33):4947–4967. doi:10.1016/j.biomaterials.2007.06.011
51. Youn P, Chen Y, Furgeson DY. A myristoylated cell-penetrating peptide bearing a transferrin receptor-targeting sequence for neuro-targeted siRNA delivery. Mol Pharm. 2014;11(2):486–495. doi:10.1021/mp400446v
52. Wiley DT, Webster P, Gale A, Davis ME. Transcytosis and brain uptake of transferrin-containing nanoparticles by tuning avidity to transferrin receptor. Proc Natl Acad Sci. 2013;110(21):8662–8667. doi:10.1073/pnas.1307152110
53. Oswald M, Geissler S, Goepferich A. Targeting the ventral nervous system (CNS): a review of rabies virus-targeting strategies. Mol Pharm. 2017;14(7):2177–2196. doi:10.1021/acs.molpharmaceut.7b00158
54. Gooding M, Malhotra M, McCarthy DJ, et al. Synthesis and characterization of rabies virus glycoprotein-tagged amphiphilic cyclodextrins for siRNA delivery in human glioblastoma cells: in vitro analysis. Eur J Pharm Sci. 2015;71:80–92. doi:10.1016/j.ejps.2015.02.007
55. Chen W, Zhan C, Gu B, et al. Targeted brain delivery of itraconazole via RVG29 anchored nanoparticles. J Drug Target. 2011;19(3):228–234. doi:10.3109/1061186X.2010.492523
56. Qin H, Jiang Y, Zhang J, Deng C, Zhong Z. Oncoprotein inhibitor Rigosertib loaded in ApoE-Targeted smart polymersomes reveals high safety and potency against human glioblastoma in mice. Mol Pharm. 2019;16(8):3711–3719. doi:10.1021/acs.molpharmaceut.9b00691
57. Pang HH, Huang CY, Chou YW, et al. Bioengineering fluorescent virus-like particle/RNAi nanocomplexes act synergistically with temozolomide to eradicate brain tumors. Nanoscale. 2019;11(17):8102–8109. doi:10.1039/c9nr01247h
58. Li Y, Wu M, Zhang N, et al. Mechanisms of enhanced antiglioma efficacy of polysorbate 80-modified paclitaxel-loaded PLGA nanoparticles by focused ultrasound. J Cell Mol Med. 2018;22(9):4171–4182. doi:10.1111/jcmm.13695
59. Hwang DW, Son S, Jang J, et al. A brain-targeted rabies virus glycoprotein-disulfide linked PEI nanocarrier for delivery of neurogenic microRNA. Biomaterials. 2011;32(21):4968–4975. doi:10.1016/j.biomaterials.2011.03.047
60. Zou L, Tao Y, Payne G, et al. Targeted delivery of nano-PTX to the brain tumor-associated macrophages. Oncotarget. 2017;8(4):6564–6578. doi:10.18632/oncotarget.14169
61. Li J, Zhu B, Yao X, et al. Synergetic approach for simple and rapid conjugation of gold nanoparticles with oligonucleotides. ACS Appl Mater Interfaces. 2014;6(19):16800–16807. doi:10.1021/am504139d
62. Reyes-Gonzalez JM, Armaiz-Pena GN, Mangala LS, et al. Targeting c-MYC in platinum-resistant ovarian cancer. Mol Cancer Ther. 2015;14(10):2260–2269. doi:10.1158/1535-7163.MCT-14-0801
63. Patel MM, Goyal BR, S V B, Bhatt JS, Amin AF. Getting into the brain: approaches to enhance brain drug delivery. CNS Drugs. 2009;23(1):35–58. doi:10.2165/0023210-200923010-00003
64. Lozada-Delgado EL, Grafals-Ruiz N, Miranda-Román MA, et al. Targeting MicroRNA-143 leads to inhibition of glioblastoma tumor progression. Cancers (Basel). 2018;10:10. doi:10.3390/cancers10100382
65. Rivera-Díaz M, Miranda-Román MA, Soto D, et al. MicroRNA-27a distinguishes glioblastoma multiforme from diffuse and anaplastic astrocytomas and has prognostic value. Am J Cancer Res. 2015;5(1):201–218.
66. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25(4):402–408. doi:10.1006/METH.2001.1262
67. National Research Council. Guide for the Care and Use of Laboratory Animals.The. Washington, DC: National Academic Press; 2011.
68. Rolón-Reyes K, Y V K, Cubano LA, et al. Microglia activate migration of glioma cells through a Pyk2 intracellular pathway. PLoS One. 2015;10(6):e0131059. doi:10.1371/journal.pone.0131059
69. Kucheryavykh LY, Y V K, Rolón-Reyes K, et al. Visualization of implanted GL261 glioma cells in living mouse brain slices using fluorescent 4-(4-(dimethylamino)-styryl)-N-methylpyridinium iodide (ASP +). Biotechniques. 2012;53(5):305–309. doi:10.2144/000113940
70. Kucheryavykh LY, Rolón-Reyes K, Kucheryavykh YV, et al. Glioblastoma Development in Mouse Brain: General Reduction of OCTs and Mislocalization of OCT3 Transporter and Subsequent Uptake of ASP + Substrate to the Nuclei. Vol. 3. 2014. doi:10.1166/jnsne.2014.1091
71. Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012;65:e3564. doi:10.3791/3564
72. Dunster K, Toh BH, Sentry JW. Early endosomes, late endosomes, and lysosomes display distinct partitioning strategies of inheritance with similarities to Golgi-derived membranes. Eur J Cell Biol. 2002;81(3):117–124. doi:10.1078/0171-9335-00232
73. Wu XA, Choi CHJ, Zhang C, Hao L, Mirkin CA. Intracellular fate of spherical nucleic acid nanoparticle conjugates. J Am Chem Soc. 2014;136(21):7726–7733. doi:10.1021/ja503010a
74. Oh N, Park JH. Endocytosis and exocytosis of gold nanoparticles in mammalian cells. Int J Nano. 2014;9(Supplement 1):51–63. doi:10.2147/IJN.S26592
75. Báez-Vega PM, Vargas IME, Valiyeva F, et al. Targeting miR-21-3p inhibits proliferation and invasion of ovarian cancer cells. Oncotarget. 2016;7:24. doi:10.18632/oncotarget.9216
76. Echevarría-Vargas IM, Valiyeva F, Vivas-Mejía PE. Upregulation of miR-21 in cisplatin resistant ovarian cancer via JNK-1/c-Jun pathway. Navarro A, ed. PLoS One. 2014;9(5):e97094. doi:10.1371/journal.pone.0097094
77. Clogston JD, Patri AK. zeta potential measurement. In: Methods in Molecular Biology (Clifton, N.J.). Vol. 697. 2011;63–70. doi: 10.1007/978-1-60327-198-1_6
78. Bhattacharjee S. DLS and zeta potential - What they are and what they are not? J Control Release. 2016;235:337–351. doi:10.1016/j.jconrel.2016.06.017
79. Garnaes J, Nielsen L, Metrology DF, et al. Requirements to Measurements of Nanomaterials and Nanoproducts. The Danish Environmental Protection Agency; 2015. ISBN: 978-87-93352-95-7 Available from: http://www2.mst.dk/Udgiv/publications/2016/12/978-87-93352-95-7.pdf.
80. Chavez JD, Hoopmann MR, Weisbrod CR, Takara K, Bruce JE. Quantitative proteomic and interaction network analysis of cisplatin resistance in HeLa cells. Uversky VN, ed. PLoS One. 2011;6(5):e19892. doi:10.1371/journal.pone.0019892
81. Varkouhi AK, Scholte M, Storm G, Haisma HJ. Endosomal escape pathways for delivery of biologicals. J Control Release. 2011;151(3):220–228. doi:10.1016/j.jconrel.2010.11.004
82. Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi:10.1016/j.ccr.2009.12.020
83. Rao SAM, Santosh V, Somasundaram K. Genome-wide expression profiling identifies deregulated miRNAs in malignant astrocytoma. Mod Pathol. 2010;23(10):1404–1417. doi:10.1038/modpathol.2010.135
84. Abdouh M, Facchino S, Chatoo W, Balasingam V, Ferreira J, Bernier G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29(28):8884–8896. doi:10.1523/JNEUROSCI.0968-09.2009
85. Huang Z, Cheng L, Guryanova OA, Wu Q, Bao S. Cancer stem cells in glioblastoma-molecular signaling and therapeutic targeting. Protein Cell. 2010;1(7):638–655. doi:10.1007/s13238-010-0078-y
86. Seferos DS, Prigodich AE, Giljohann DA, Patel PC, Mirkin CA. Polyvalent DNA nanoparticle conjugates stabilize nucleic acids. Nano Lett. 2009;9(1):308–311. doi:10.1021/nl802958f
87. Melamed JR, Kreuzberger NL, Goyal R, Day ES. Spherical nucleic acid architecture can improve the efficacy of polycation-mediated siRNA delivery. Mol Ther Nucleic Acids. 2018;12:207–219. doi:10.1016/J.OMTN.2018.05.008
88. Rosi NL, Giljohann DA, Thaxton CS, AKR L-J, Han MS, Mirkin CA. Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science. 2006;312(5776):1027–1030. doi:10.1126/science.1125559
89. Patel PC, Giljohann DA, Daniel WL, Zheng D, Prigodich AE, Mirkin CA. Scavenger receptors mediate cellular uptake of polyvalent oligonucleotide-functionalized gold nanoparticles. Bioconjug Chem. 2010;21(12):2250–2256. doi:10.1021/bc1002423
90. Kumthekar P NU-0129 in treating patients with recurrent glioblastoma or gliosarcoma undergoing surgery - Full text view - clinicaltrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT03020017.
91. Wilhelm S, Tavares AJ, Dai Q, et al. Analysis of nanoparticle delivery to tumours. Nat Rev Mater. 2016;1(5):1–12. doi:10.1038/natrevmats.2016.14
92. Kouri FM, Hurley LA, Daniel WL, et al. miR-182 integrates apoptosis, growth, and differentiation programs in glioblastoma. Genes Dev. 2015;29(7):732–745. doi:10.1101/gad.257394.114
93. Mhashal AR, Roy S. Effect of gold nanoparticle on structure and fluidity of lipid membrane. PLoS One. 2014;9(12):e114152. doi:10.1371/journal.pone.0114152
94. Sonavane G, Tomoda K, Makino K. Biodistribution of colloidal gold nanoparticles after intravenous administration: effect of particle size. Colloids Surf B. 2008;66(2):274–280. doi:10.1016/j.colsurfb.2008.07.004
95. Tohidkia MR, Barar J, Asadi F, Omidi Y. Molecular considerations for development of phage antibody libraries. J Drug Target. 2012;20(3):195–208. doi:10.3109/1061186X.2011.611517
96. Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11(17–18):812–818. doi:10.1016/j.drudis.2006.07.005
97. Rahme K, Guo J, Holmes JD, O’Driscoll CM. Evaluation of the physicochemical properties and the biocompatibility of polyethylene glycol-conjugated gold nanoparticles: a formulation strategy for siRNA delivery. Colloids Surf B. 2015;135:604–612. doi:10.1016/j.colsurfb.2015.08.032
98. Arvizo RR, Miranda OR, Thompson MA, et al. Effect of nanoparticle surface charge at the plasma membrane and beyond. Nano Lett. 2010;10(7):2543–2548. doi:10.1021/nl101140t
99. Gratton SEA, Ropp PA, Pohlhaus PD, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci U S A. 2008;105(33):11613–11618. doi:10.1073/pnas.0801763105
100. Sharma A, S V M, Robertson GP. Toxicological considerations when creating nanoparticle-based drugs and drug delivery systems. Expert Opin Drug Metab Toxicol. 2012;8(1):47–69. doi:10.1517/17425255.2012.637916
101. Hang C, Choi J, Hao L, Narayan SP, Auyeung E, Mirkin CA. Mechanism for the endocytosis of spherical nucleic acid nanoparticle conjugates. PNAS. 2013;110(19):. doi:10.1073/pnas.1305804110
102. Nasser F, Davis A, Valsami-Jones E, Lynch I. Shape and charge of gold nanomaterials influence survivorship, oxidative stress and moulting of Daphnia magna. Nanomaterials. 2016;6(12). doi:10.3390/NANO6120222
103. Albanese A, Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW. Secreted biomolecules alter the biological identity and cellular interactions of nanoparticles. ACS Nano. 2014;8(6):5515–5526. doi:10.1021/nn4061012
104. Vogt C, Pernemalm M, Kohonen P, et al. Proteomics analysis reveals distinct corona composition on magnetic nanoparticles with different surface coatings: implications for interactions with primary human macrophages. PLoS One. 2015;10(10):e0129008. doi:10.1371/journal.pone.0129008
105. Roheim PS, Carey M, Forte T, Vega GL. Apolipoproteins in human cerebrospinal fluid. Proc Natl Acad Sci U S A. 1979;76(9):4646–4649. doi:10.1073/pnas.76.9.4646
106. Cheng Y, Dai Q, Morshed RA, et al. Blood-brain barrier permeable gold nanoparticles: an efficient delivery platform for enhanced malignant glioma therapy and imaging. Small. 2014;10(24):5137–5150. doi:10.1002/smll.201400654
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.