Back to Journals » Drug Design, Development and Therapy » Volume 20
Brain Endothelial Glycocalyx as a Blood-Facing Translational Interface in Alzheimer’s Disease: Beyond “Leaky” Barriers Toward Repair-First Stratification
Authors Zhang C ![]() , Fang D, Zhang L
, Fang D, Zhang L
Received 25 April 2026
Accepted for publication 18 June 2026
Published 26 June 2026 Volume 2026:20 619901
DOI https://doi.org/10.2147/DDDT.S619901
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Chungang Zhang,1,* Deyu Fang,2,* Lin Zhang3
1College of Pharmacy, Liaoning University of Traditional Chinese Medicine, Dalian, People’s Republic of China; 2College of Artificial Intelligence and Information Engineering, Liaoning University of Traditional Chinese Medicine, Shenyang, People’s Republic of China; 3College of Basic Medical Sciences, Liaoning University of Traditional Chinese Medicine, Shenyang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chungang Zhang, College of Pharmacy, Liaoning University of Traditional Chinese Medicine, No. 77, Life One Road, DD Port, Dalian, 116600, People’s Republic of China, Email [email protected] Lin Zhang, College of Basic Medical Sciences, Liaoning University of Traditional Chinese Medicine, No. 79, Chongshan East Road, Shenyang, 110847, People’s Republic of China, Email [email protected]
Abstract: Alzheimer’s disease (AD) pathogenesis is increasingly recognized as involving blood-brain barrier (BBB) and neurovascular unit (NVU) destabilization. The brain endothelial glycocalyx—a blood-facing glycan-rich interface—represents a critical but under-characterized determinant of BBB dysfunction in AD. In this review, we systematically reappraised glycocalyx abnormality mechanisms, distinguishing robust causal evidence from murine models from limited human observational data. We propose a four-dimensional interface-state framework (structural, glycosylation, transport, inflammatory) to stratify patients beyond binary “leaky versus intact” classifications. Glycocalyx deterioration in AD reflects compartmentalized remodeling (early mucin-domain depletion) rather than uniform shedding. A reversible therapeutic window exists in APOE4 carriers and mild cognitive impairment, but collapses with concurrent cerebral amyloid angiopathy (CAA) or amyloid-related imaging abnormalities (ARIA). Pathological glycocalyx disruption amplifies nonproductive vascular retention rather than parenchymal penetration. We advocate a hierarchical “repair-first, transport-engineering-second, exploitation-last” strategy. This repositions the glycocalyx as a decisive arbiter governing BBB-targeted interventions in AD, not merely a structural appendage.
Keywords: Alzheimer’s disease, blood-brain barrier, endothelial glycocalyx, mucin-type O-glycosylation, therapeutic stratification
Introduction
The pathological framework of Alzheimer’s disease (AD) is transitioning from a mono-centric model focused on neuronal damage and amyloid-beta/tau pathological aggregation toward an interface-pathology model incorporating blood-brain barrier (BBB) and neurovascular unit (NVU) destabilization. Current evidence indicates that increased BBB permeability may not represent a mere passive accompaniment to AD but rather actively participates in pathological cascades involving neuroinflammation and microcirculatory disturbances by facilitating the entry of blood-derived proteins and inflammatory mediators into brain parenchyma.1–7
Population-based imaging studies further reveal hippocampal BBB leakage at early cognitive impairment stages, partially independent of amyloid-beta/tau biomarker abnormalities.1–5,7 This suggests vascular interface alterations may progress in parallel with, rather than merely downstream of, classical proteinopathies.
Currently, AD vascular pathology discussions remain focused on structural abnormalities within the endothelial cell layer itself: tight junction disruption, pericyte loss, basement membrane remodeling, and transcytosis imbalance.1,3,6,7 The glycocalyx, by contrast, remains systemically under-characterized.
In vivo two-photon imaging has directly visualized the cerebral endothelial glycocalyx in murine cerebral vessels;8 ultrastructural studies further demonstrate that brain capillary glycocalyx exhibits greater density compared to continuous capillaries in the heart and lungs;9 while in vivo imaging and transport analyses indicate that the glycocalyx, endothelial layer, and perivascular compartment collectively determine the passive barrier properties of the BBB.10 These findings establish that the brain endothelial glycocalyx constitutes an integral component of the blood-facing structural organization of the BBB.8–10
Functionally, the glycocalyx forms a negatively charged surface layer through its mesh-like structure enriched in glycosaminoglycans (GAGs)—including heparan sulfate (HS) and hyaluronic acid (HA)—and glycoproteins.11–13 It participates in mechanotransduction, shear sensing, and molecular sieving, and it regulates leukocyte adhesion while maintaining permeability.
In BBB research, fluid shear stress can induce upregulation of extracellular matrix (ECM) and glycocalyx-associated genes and pathways, enhancing negative surface charge in brain-like endothelial cells, thereby suggesting a functional association between glycocalyx status and BBB phenotype maintenance.14 Concurrently, studies examining brain endothelial surface charge and adsorptive-mediated transcytosis suggest that blood-facing surface chemical properties may influence initial interaction modalities between cationic drugs, protein therapeutics, nanocarriers, and the BBB.15,16 From the perspectives of pharmaceutical design and delivery, the brain endothelial glycocalyx potentially constitutes a blood-facing functional interface connecting hemodynamics, inflammatory adhesion, microcirculatory homeostasis, BBB integrity, and interface behaviors of delivery systems, yet its potential as a “translational interface” requires further validation.
This conceptualization is beginning to acquire more direct evidentiary support in AD research. A 2024 review on AD proposed that cerebrovascular glycocalyx dysfunction may participate in pathological events underlying neurovascular imbalance and neurodegenerative progression.17 More critically, recent studies demonstrate extensive dysregulation of brain endothelial glycocalyx in aging and neurodegenerative disease contexts, with significant downregulation of mucin-domain glycoproteins and their associated mucin-type O-glycosylation pathways. In aged murine models, these alterations correlate with BBB functional impairment and may induce cerebral hemorrhage phenotypes. Analogous molecular signatures have been observed in brain microvasculature from human AD patients.18
Furthermore, preclinical studies employing endothelial/glycocalyx-targeted nanoparticles indicate that interventions targeting the blood-facing interface can reversibly modulate BBB permeability and enhance cerebral accumulation in animal models, suggesting that the glycocalyx may serve not merely as a disease severity biomarker but also as a candidate intervention target, though its targetability and safety in human AD remain uncharacterized.19 Combined with recent BBB repair/rejuvenation frameworks, blood-facing interface mechanisms are transitioning from pathological phenomena to potential intervention entry points. However, robust mechanistic and interventional evidence currently derives primarily from murine aging and disease-associated models; direct demonstration that specific glycocalyx defects drive BBB collapse in human AD remains incomplete.20
Building upon the evidence outlined above, this review advances three interrelated conceptual propositions that constitute its core novelty (Figure 1): (i) The brain endothelial glycocalyx should be repositioned as a blood-facing translational interface in AD—not merely a structural accessory, but a functional frontier governing mechanotransduction, inflammatory adhesion, and transcytosis. (ii) We introduce a four-dimensional interface-state framework (structural, glycosylation, transport, inflammatory) that operationalizes patient stratification beyond binary “leaky versus intact” classifications. (iii) We advocate a hierarchical “repair-first, transport-engineering-second, exploitation-last” strategic sequence, explicitly distinguishing robust murine causal evidence from limited human observational data. This framework is particularly timely given the expanding clinical use of anti-amyloid immunotherapies, in which amyloid-related imaging abnormalities (ARIA) have underscored the fragility of the cerebrovascular substrate and the urgent need for blood-facing interface stratification to guide safe therapeutic delivery.
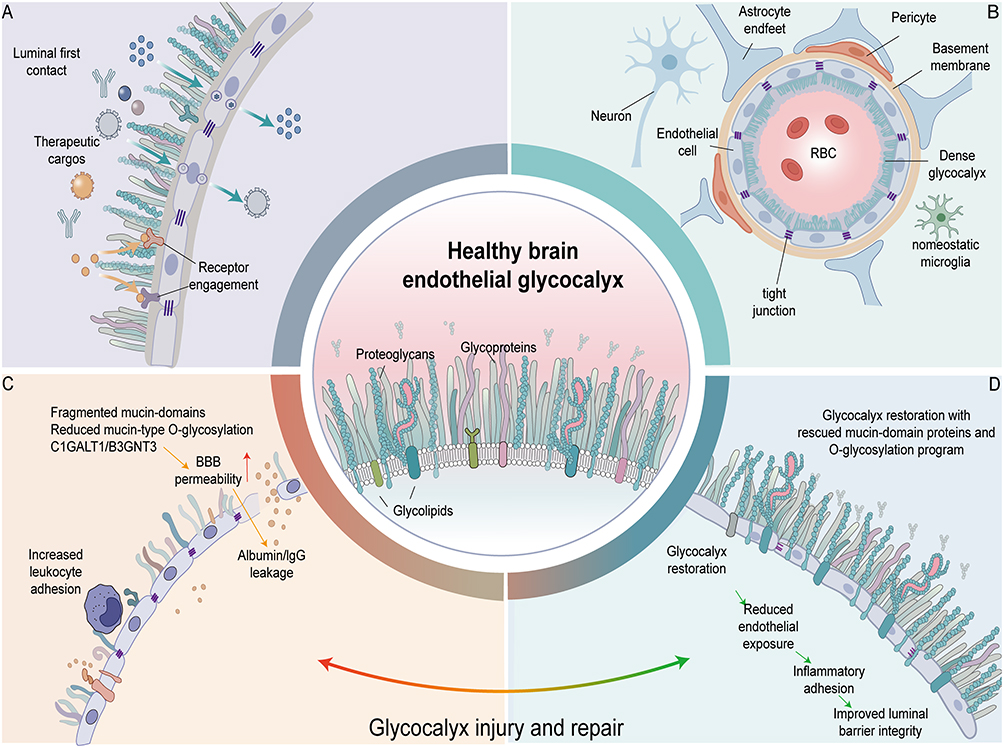 |
Figure 1 Impairment and Repair of the Cerebral Endothelial Glycocalyx in Alzheimer’s Disease and Its Implications for Drug Delivery. (A) The cerebral endothelial glycocalyx as the primary blood-facing interface of the blood-brain barrier. The cerebral endothelial glycocalyx coats the luminal surface of cerebral microvascular endothelial cells, serving as the first physical and biochemical interface encountered by circulating therapeutic cargoes—including antibodies, protein therapeutics, and nano-delivery systems. Blue arrows indicate the approach of therapeutic cargoes toward the endothelial luminal surface and their initial contact with the cerebral endothelial glycocalyx; yellow arrows indicate the subsequent binding of certain cargoes to luminal surface receptors; green arrows and intracellular vesicles denote receptor-mediated endocytosis, intracellular vesicular trafficking, and basolateral release, i.e., regulated transcytosis. This pathway does not imply that cargoes directly penetrate endothelial cells or cross intact tight junctions to enter brain tissue. (B) Structural composition of the healthy cerebral endothelial glycocalyx and its barrier microenvironment. Under healthy conditions, the cerebral endothelial glycocalyx is composed primarily of proteoglycans, glycoproteins, and glycolipids, forming a dense coating over the luminal surface of cerebral microvascular endothelial cells. Cerebral endothelial cells, tight junctions, the basement membrane, pericytes, and astrocytic endfeet collectively constitute the neurovascular unit and maintain the structural and functional integrity of the blood-brain barrier. RBC denotes red blood cells; microglia are depicted in a homeostatic state. (C) Glycocalyx injury and blood-brain barrier dysfunction. Under pathological conditions associated with aging and Alzheimer’s disease, the cerebral endothelial glycocalyx undergoes structural disruption and glycosylation abnormalities, manifested as mucin-domain fragmentation, reduced mucin-type O-glycosylation, and downregulation of relevant glycosylation regulators including C1GALT1 and B3GNT3. Glycocalyx injury increases luminal surface exposure and leukocyte adhesion, accompanied by elevated blood-brain barrier permeability and extravasation of blood-derived components such as albumin and IgG into the brain, thereby promoting cerebrovascular interface dysfunction. Red arrows indicate increased BBB permeability; yellow arrows indicate albumin and IgG extravasation. (D) Glycocalyx repair and improvement of vascular luminal barrier function. Restoration of glycocalyx-associated mucin-domain proteins and O-glycosylation programs promotes reconstruction of the cerebral endothelial glycocalyx. Glycocalyx repair helps reduce abnormal endothelial surface exposure and inflammatory cell adhesion, and improves the continuity and integrity of the vascular luminal barrier. Green arrows indicate the sequential process from reduced endothelial exposure and decreased inflammatory adhesion to improved luminal barrier function following glycocalyx reconstruction. Central magnified schematic: Summarizes the major constituents of the healthy cerebral endothelial glycocalyx, including proteoglycans, glycoproteins, and glycolipids. The bottom arc transitioning from red to green represents the dynamic shift between glycocalyx injury and repair: from pathological states characterized by glycocalyx disruption, elevated BBB permeability, and plasma protein extravasation, toward glycocalyx restoration and improved vascular luminal barrier function. Abbreviations: BBB, blood-brain barrier; IgG, immunoglobulin G; RBC, red blood cell. Notes: This is an author-drawn mechanistic schematic illustrating the composition, pathological injury, and repair process of the cerebral endothelial glycocalyx, and their relationship to trans-BBB drug delivery. |
Brain Endothelial Glycocalyx in AD: From Blood-Facing Frontline to Translational Interface
Brain Endothelial Glycocalyx as the Blood-Facing Frontline Interface of the BBB
Traditionally, the blood-brain barrier (BBB) has been defined primarily through the endothelial cell layer, emphasizing tight junctions, low-level nonspecific transcytosis, and efflux transport systems.21,22 However, from the blood-facing perspective, circulating components first encounter the endothelial glycocalyx covering the luminal surface of brain endothelium. As illustrated in Figure 2, the brain endothelial glycocalyx resides at the foremost frontier where circulating components contact cerebral vascular endothelium, constituting the blood-facing frontline interface rather than an accessory layer external to classical barrier structures. Existing imaging and diffusion analyses suggest that the BBB can be conceptualized as a multi-layered resistance system comprising the glycocalyx, endothelial layer, and abluminal compartments, wherein the glycocalyx provides the primary diffusion resistance.10,21
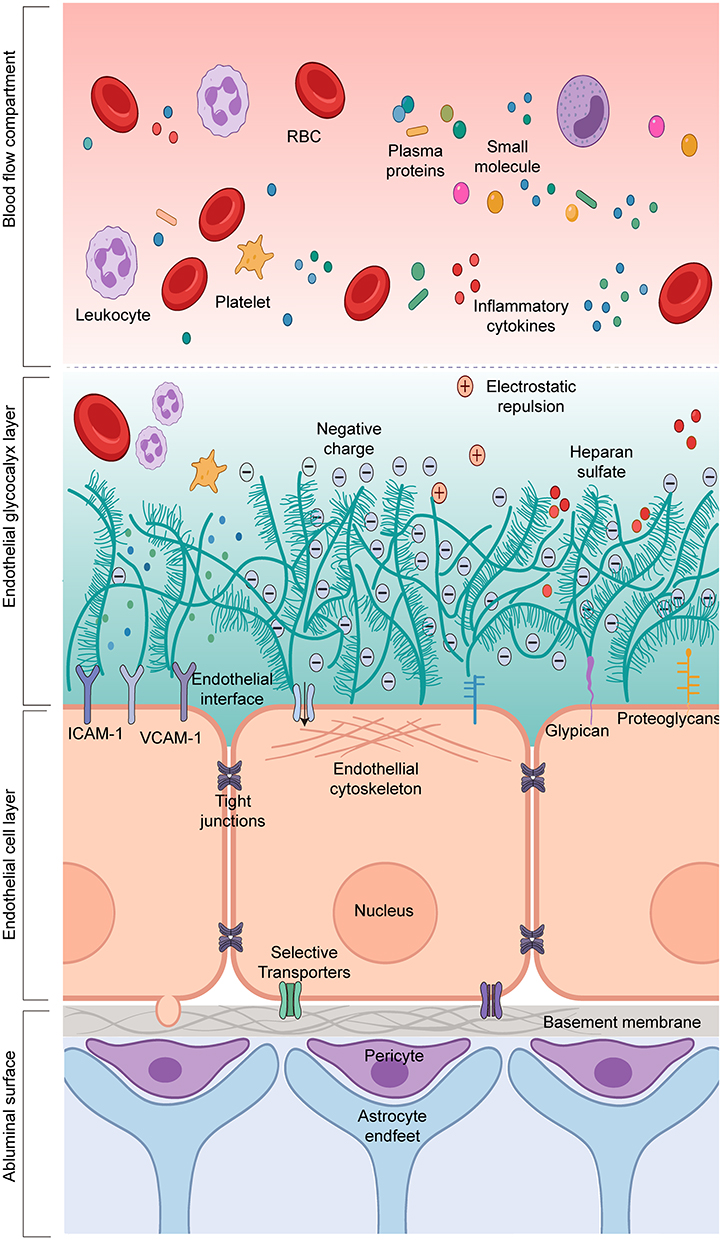 |
Figure 2 Layered Architecture of the Normal Blood-Brain Barrier. This figure illustrates the layered architecture of the normal blood-brain barrier from the vascular lumen to the brain parenchyma, emphasizing that the endothelial luminal glycocalyx is the frontline interface facing circulating blood, rather than an accessory layer external to classical barrier structures. The glycocalyx, together with the endothelial layer and abluminal compartments, constitutes a continuous multi-layered resistance system and collectively participates in blood-brain barrier homeostasis. Abbreviations: RBC, red blood cell; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular cell adhesion molecule-1. Notes: This schematic was hand-drawn by the authors using Adobe Illustrator; no AI image generation tools were used. |
The specificity of brain endothelial glycocalyx lies in its not being a simple replication of general microvascular glycocalyx within the brain. Ultrastructural studies demonstrate that brain capillary glycocalyx exhibits superior coverage and length compared to cardiac and pulmonary capillaries, retaining more residual structure following lipopolysaccharide (LPS) injury.9 Subsequent reviews further indicate that brain endothelial glycocalyx is generally thicker, denser, and more negatively charged—characteristics consistent with the highly selective barrier function of the BBB.23–25 Recent studies suggest that brain endothelial glycocalyx integrity may be associated with mucin-domain glycoproteins and mucin-type O-glycosylation; under aging and disease-associated states, dysregulation of this program may accompany glycocalyx thinning, elevated BBB permeability, and impaired tight junction formation.18 These characteristics suggest adaptive specialization between brain endothelial glycocalyx and BBB physiological demands.
The “frontline” nature of the glycocalyx manifests not merely spatially through preferential exposure, but functionally through pre-positioned regulation. As an initial sieving layer, the glycocalyx influences local distribution of molecules before they reach the endothelial membrane, with differential accessibility for molecules of varying sizes within its volume.10 As a blood-facing charge interface, its negative charge influences initial interactions between macromolecules, particles, and drugs with the BBB. As a frontline regulatory layer for inflammatory and mechanical signaling, the glycocalyx participates in shear stress transduction and, under inflammatory conditions, alters contact conditions between blood cells, cytokines, and endothelial adhesion molecules through structural degradation.14,26,27 Therefore, while the glycocalyx does not singularly determine BBB integrity or selectivity, it can execute early contact, sieving, and gating of material, mechanical, and inflammatory signals prior to their action on endothelial programs. Importantly, positioning the glycocalyx at the blood-facing frontline of the BBB does not imply its substitutability for endothelial cell layers, tight junctions, low-background transcytosis, basement membranes, or pericyte/astrocyte-associated structures. Accordingly, the BBB can be viewed as a continuous multi-layered functional system extending from the blood-facing side toward the brain parenchymal side, with the glycocalyx defining the interface engaging in early interactions with circulating components (Figure 2).10,21,22,28
BBB/NVU Destabilization in AD: Re-Focusing Attention on Blood-Facing Glycocalyx
The glycocalyx merits renewed attention in AD because the pathological framework of AD has progressively shifted from a purely neuron-centric narrative toward an integrated model encompassing cerebrovascular pathology, the neurovascular unit (NVU), and BBB destabilization. Previous reviews have proposed that BBB dysfunction, cerebral blood flow reduction, hypoxia, and entry of blood-derived toxic molecules into the brain may precede, occur in parallel with, or interact reciprocally with cognitive decline, amyloid-beta deposition, and brain atrophy in certain individuals, forming mutually amplifying pathological loops with amyloid-beta clearance impairment, neuroinflammation, and neurodegenerative changes.4,7 Thus, the BBB/NVU perspective does not negate amyloid-beta/tau mechanisms but rather indicates that early AD pathogenesis and individual heterogeneity cannot be adequately explained by classical proteinopathy-centric linear models alone. Human studies provide crucial support for this conceptualization. Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) studies demonstrate that hippocampal BBB disruption increases with age, is more pronounced in mild cognitive impairment (MCI) individuals, and exhibits brain-region selectivity.29 Subsequent studies suggest that early AD may be accompanied by reduced cerebral blood flow or regional blood volume and increased BBB leakage, indicating that cerebrovascular destabilization may simultaneously manifest as perfusion dysregulation and barrier functional abnormalities, rather than singular structural loosening.30 Moreover, early cognitive decline individuals may exhibit capillary injury and BBB disruption in the hippocampus and parahippocampal gyrus, and in certain studies, these changes can occur independently of amyloid-beta and tau biomarker status.2 Apolipoprotein E ε4 (APOE4) carriers may exhibit BBB dysfunction in the hippocampus and medial temporal lobe even at pre-symptomatic cognitive stages, correlating with subsequent cognitive decline.31 Elevated cerebrospinal fluid (CSF) angiopoietin-2 (ANGPT-2) in association with BBB leakage, soluble platelet-derived growth factor receptor-β (sPDGFRβ), and neuroinjury-associated markers further supports the presence of vascular activation or destabilization states in early AD.32 Building upon these foundations, multi-omics studies further indicate that BBB abnormalities in AD should not be oversimplified as mere “leakage.” Human brain vascular atlas studies reveal significant expression of AD risk genes in cerebrovascular cells, with enrichment in endothelial protein transport, immune response, and ECM-related pathways.33 Single-nuclear transcriptomic studies further demonstrate that AD cerebrovascular abnormalities involve cell type-specific state reprogramming, including alterations in endothelial transport programs, pericyte homeostasis, and vascular-associated cellular interactions.34–36 Therefore, within the AD context, BBB impairment is more appropriately understood as cerebrovascular interface reprogramming rather than merely elevated static permeability.
Nevertheless, existing evidence remains insufficient to characterize AD as a unified “early BBB leakage disease.” On one hand, not all cohorts support BBB changes consistently preceding amyloid-beta/tau pathology; on the other hand, different technologies define “BBB abnormalities” disparately, potentially corresponding to distinct dimensions including permeability, transcytosis, vascular activation, pericyte injury, or regional perfusion coupling.2,29–32,37,38 Consequently, a more circumspect conclusion holds that cerebrovascular/NVU/BBB destabilization constitutes an important pathological axis in AD that may be exposed relatively early in certain individuals, yet its dominant forms, temporal relationships, and magnitudes are inconsistent; existing evidence better supports its progression prior to or in parallel with classical amyloid/tau pathologies across different populations, rather than universally constituting a singular upstream event. Against this backdrop, discussion priorities have expanded beyond whether the BBB ultimately leaks to encompass where vascular interface destabilization may initiate and how it progressively expands into endothelial activation, inflammatory amplification, and transport abnormalities. Following this logic, the brain endothelial glycocalyx at the foremost frontier ceases to be merely a structural nomenclature, becoming instead a critical blood-facing interface meriting separate discussion in AD.
Brain Endothelial Glycocalyx as a Blood-Facing Translational Interface in AD
Based on the foregoing discussion, brain endothelial glycocalyx in AD is more appropriately situated within an analytical framework of “blood-facing translational interface” rather than merely as an accessory component in BBB structural inventories. Within this framework, brain endothelial glycocalyx in AD may be better defined as a blood-facing functional interface connecting pathological drivers, therapeutic plasticity, and delivery boundaries. Located at the foremost frontier before peripheral blood-derived information enters the cerebrovascular wall, the glycocalyx may pre-positionally influence how the BBB perceives hemodynamic forces, circulating inflammatory mediators, blood cells, and exogenous therapeutic inputs. Consequently, the glycocalyx may represent not merely a concomitant phenotype following BBB damage, but an important preceding state variable influencing how blood-derived perturbations are translated into endothelial activation, inflammatory amplification, and barrier destabilization.
It must be noted that currently robust mechanistic and interventional evidence derives primarily from animal and aging/disease-associated models rather than human AD per se. Recent studies demonstrate that intervening upon brain endothelial mucin-type O-glycosylation can induce or ameliorate glycocalyx thinning, BBB functional impairment, neuroinflammation, and behavioral phenotypic changes in mice. These findings enhance the credibility that “glycocalyx abnormalities may reside upstream of BBB destabilization,” yet remain insufficient to support the conclusion that “specific glycocalyx defects have been directly demonstrated to drive BBB collapse in human AD.”18,39 Therefore, at current evidence levels, brain endothelial glycocalyx has qualified as a candidate interface for BBB repair, though its causal status in human AD-specific pathology remains unproven. All intervention evidence cited below derives primarily from murine aging or disease-associated models; direct human AD validation is currently lacking.18,39
This interface positioning carries methodological significance for drug development, though at this stage it is more appropriate to consider brain endothelial glycocalyx as an interface of conceptual importance yet experimentally under-explored in aging/AD brain drug delivery. Existing evidence suggests that glycocalyx status may alter initial contact conditions on the blood-facing side of the BBB, thereby influencing subsequent interactions between therapeutic inputs and the barrier. This understanding currently rests more upon BBB surface biology and general nanomedicine principles rather than abundant AD-specific delivery data.14,17,24,25 In this sense, brain endothelial glycocalyx represents not merely an important component of BBB biology, but a candidate frontline interface connecting pathological mechanisms, barrier repair, and brain delivery strategies. Accordingly, Table 1 demonstrates that current evidence from human imaging, fluid biomarkers, multi-omics, and model studies sufficiently supports incorporating brain endothelial glycocalyx into AD BBB/NVU discussion frameworks and considering it as a blood-facing frontline interface meriting separate attention; however, this support currently manifests primarily as framework-level and translational relevance rather than completed causal closure at the human level.
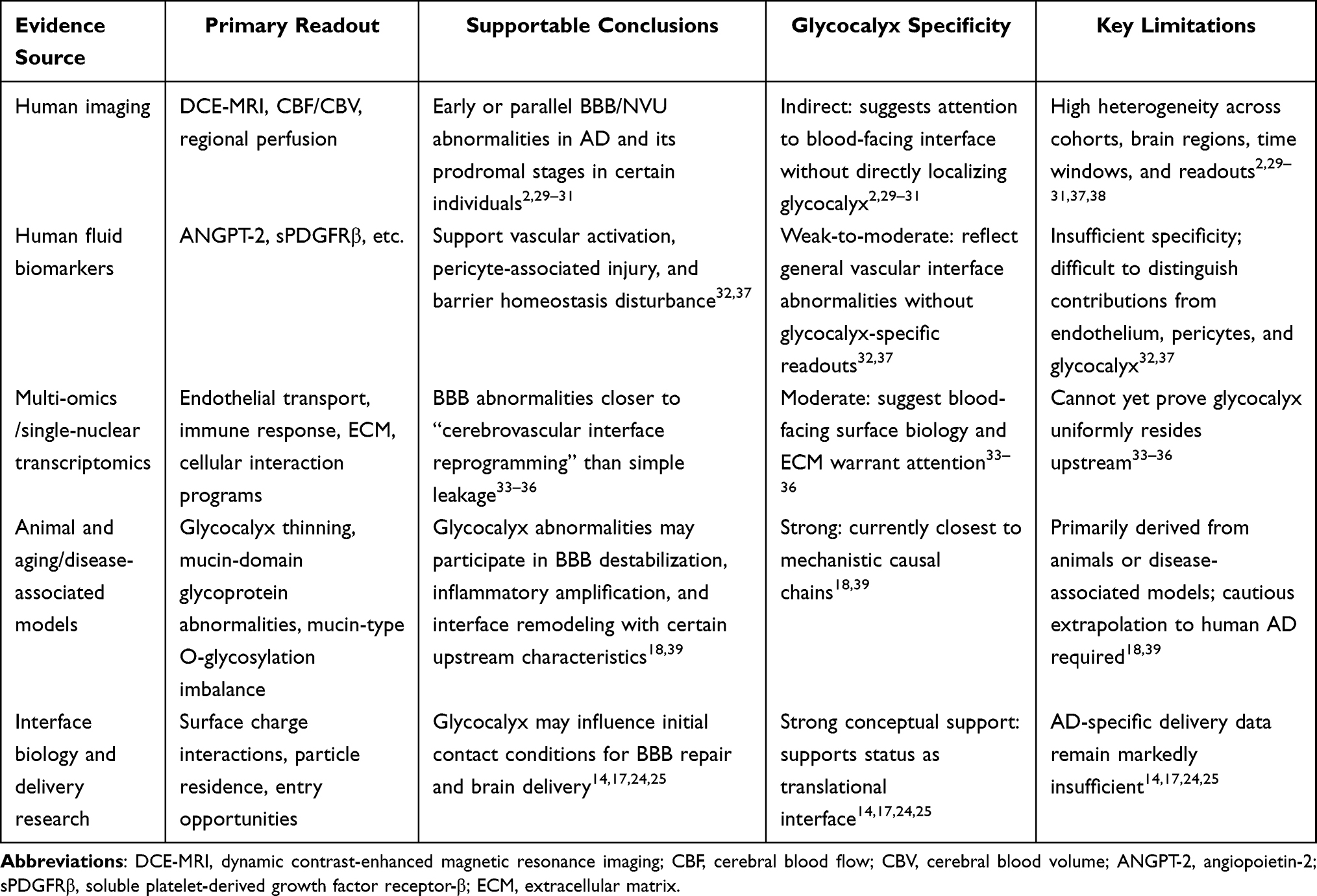 |
Table 1 Evidence Hierarchy, Inferential Boundaries, and Critical Gaps Regarding BBB/NVU Destabilization and Translational Significance of Brain Endothelial Glycocalyx in AD |
Evidence Hierarchy of Brain Endothelial Glycocalyx Abnormalities in Alzheimer’s Disease BBB Injury
Distinct from Brain Endothelial Glycocalyx in AD: From Blood-Facing Frontline to Translational Interface, which primarily elucidates why brain endothelial glycocalyx should be incorporated into AD BBB/NVU discussion frameworks from an evidence-source perspective, this section further focuses on glycocalyx abnormalities themselves, evaluating evidence strength and inferential boundaries at structural, molecular, functional, and causal levels. In the AD/aging context, brain endothelial glycocalyx abnormalities should not be defined merely as glycocalyx shedding or thickness reduction, but more appropriately understood as stratified remodeling occurring at the BBB luminal interface. This concept encompasses at least three dimensions: structural-level thinning, coverage reduction, or continuity impairment; molecular-level alterations in component composition, spatial distribution, and glycosylation programs; and BBB functional abnormalities including elevated permeability, decreased tight junction continuity, enhanced inflammatory adhesion, and impaired shear stress responsiveness. Only when both perturbation and rescue evidence exist can such alterations further support their possession of pathological driver significance.
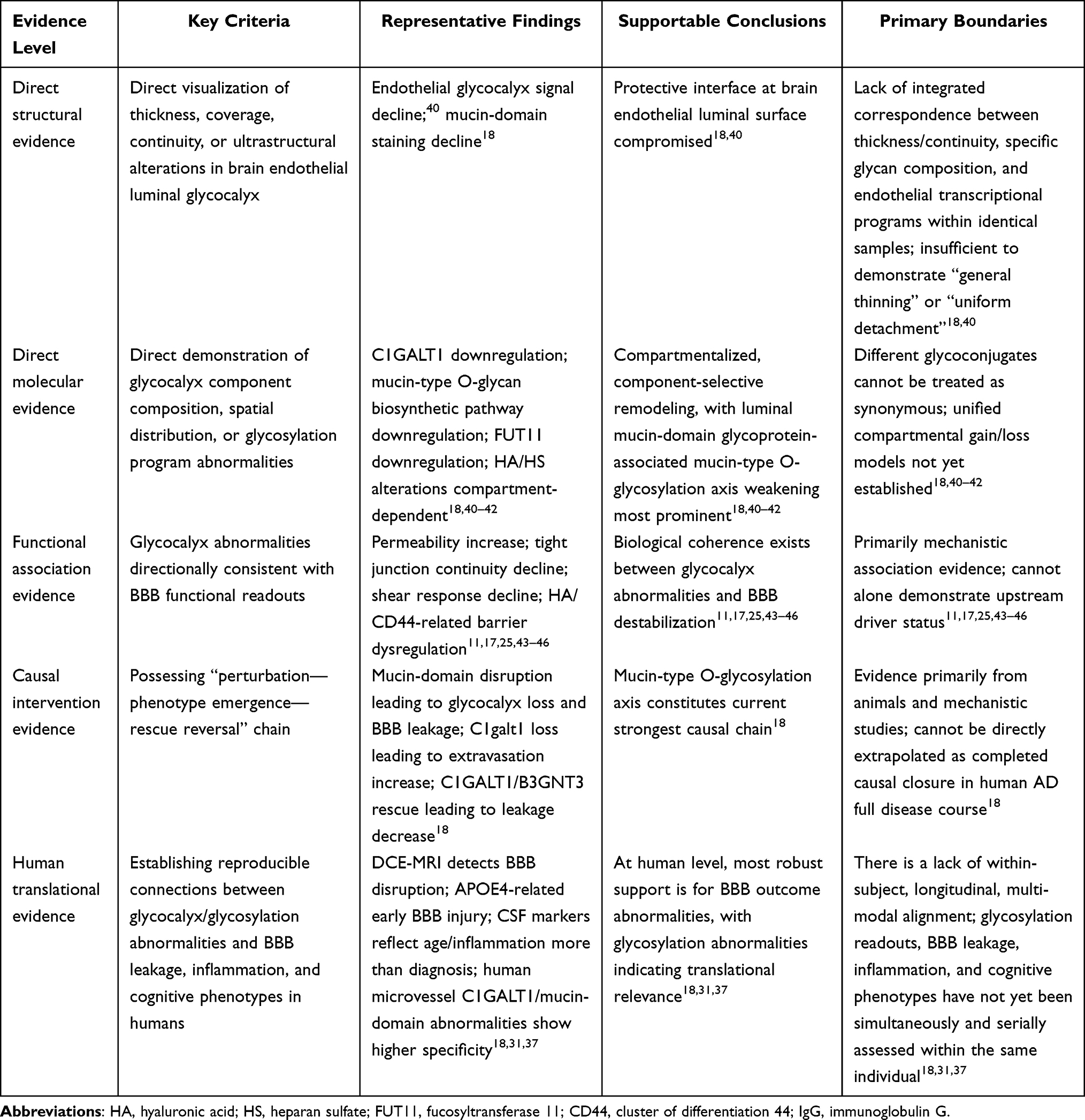 |
Table 2 Evidence Stratification of AD-Associated Brain Endothelial Glycocalyx Abnormalities and BBB Injury |
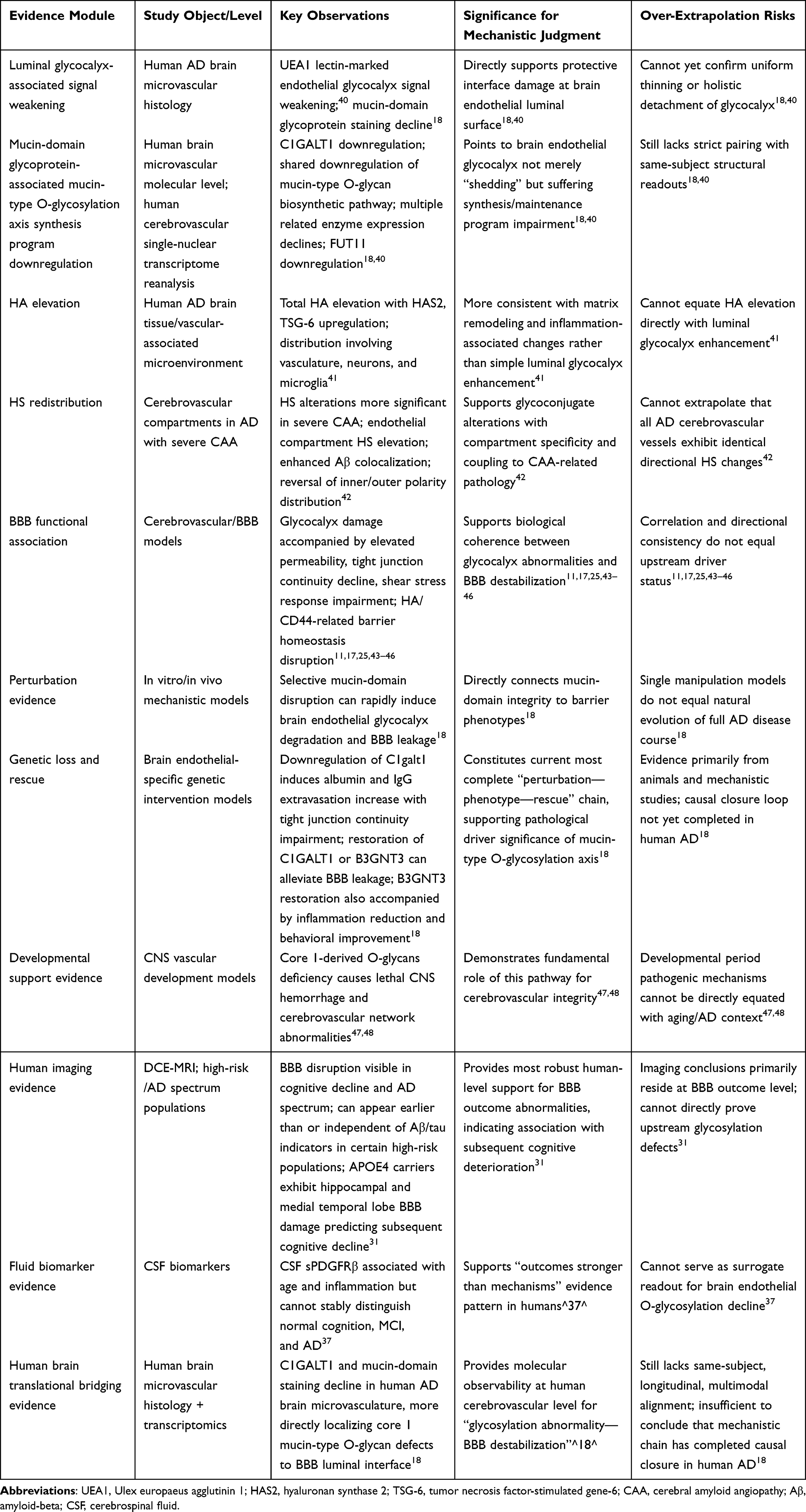 |
Table 3 Extended Evidence Matrix of AD-Associated Brain Endothelial Glycocalyx Abnormalities and BBB Injury |
Under this premise, Table 2 stratifies by “direct structural evidence—direct molecular evidence—functional association evidence—causal intervention evidence—human translational evidence” to delineate evidence strength and inferential boundaries for different findings. To facilitate holistic comprehension of this section’s evidence hierarchy framework and logical relationships between levels, Figure 3 provides schematic summarization of this stratification approach. Table 3 further illustrates the actual distribution of each evidence level across specific glycocalyx components, compartments, and research scenarios, demonstrating that current evidence strength concentrates most heavily on damage to the luminal mucin-domain glycoprotein-associated mucin-type O-glycosylation axis. In contrast, characterizing AD-associated brain endothelial glycocalyx abnormalities as uniform, holistic detachment currently rests primarily on inference, lacking direct evidentiary support.
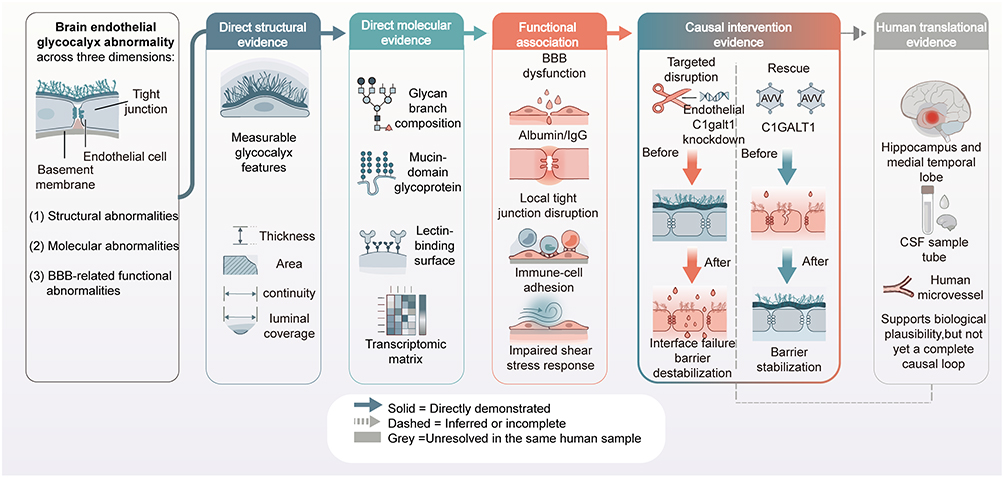 |
Figure 3 Evidence Hierarchy of Brain Endothelial Glycocalyx Abnormalities in Aging and Alzheimer’s Disease. This schematic illustrates the evidence hierarchy for Alzheimer’s disease-associated brain endothelial glycocalyx abnormalities and blood-brain barrier injury. The left panel summarizes major categories of glycocalyx abnormalities; subsequent columns present direct structural evidence, direct molecular evidence, functional association evidence, causal intervention evidence, and human translational evidence. Arrows indicate logical or causal links between evidence tiers; solid lines indicate directly supported links, dashed lines indicate inferred or incomplete links, and gray shading indicates associations that have not yet been mechanistically closed within the same human subjects. Colored boxes indicate different evidence sources: blue for structural/molecular evidence, red for functional/causal intervention evidence, and gray for human translational evidence. Abbreviations: AD, Alzheimer’s disease; BBB, blood-brain barrier; CSF, cerebrospinal fluid; IgG, immunoglobulin G; AAV, adeno-associated viral vector; C1GALT1, core 1 β1,3-galactosyltransferase. Notes: This schematic was hand-drawn by the authors using Adobe Illustrator; no AI image generation tools were used. |
Structural and Compositional Evidence: Brain Endothelial Glycocalyx Abnormalities Indicate Compartmentalized Remodeling Rather Than Uniform Loss
AD-associated brain endothelial glycocalyx structural and compositional alterations are more appropriately characterized as compartmentalized, component-selective remodeling rather than holistic detachment as conceptualized in peripheral vascular contexts. Currently, more direct evidence primarily points to impaired luminal protective mucin-domain glycoprotein-associated mucin-type O-glycosylation networks (Figure 4A and B).9,18,40 Shi et al confirmed in AD patient-derived brain microvasculature that both C1GALT1 and mucin-domain glycoprotein staining were reduced; reanalysis of existing human cerebrovascular single-nuclear transcriptome data further revealed that the mucin-type O-glycan biosynthetic pathway represents a shared downregulated pathway in AD brain endothelium, with decreased expression of multiple mucin-type O-glycan biosynthetic enzymes.18 Smyth et al also observed in AD patient middle temporal gyrus vasculature that Ulex europaeus agglutinin 1 (UEA1)-marked endothelial glycocalyx signals were markedly weakened, persisting after correction for vascular density differences using collagen IV as a total vascular marker; concurrently, downregulated fucosyltransferase 11 (FUT11) expression in AD cerebrovascular endothelial cells further supports impaired glycocalyx generation and maintenance programs. However, direct structural evidence regarding AD luminal glycocalyx damage remains currently limited, particularly lacking studies integrating ultrastructural thickness changes, specific glycan composition, and endothelial transcriptional programs within identical human brain samples. Therefore, definitive judgments regarding “luminal glycocalyx thinning” remain premature at this stage.
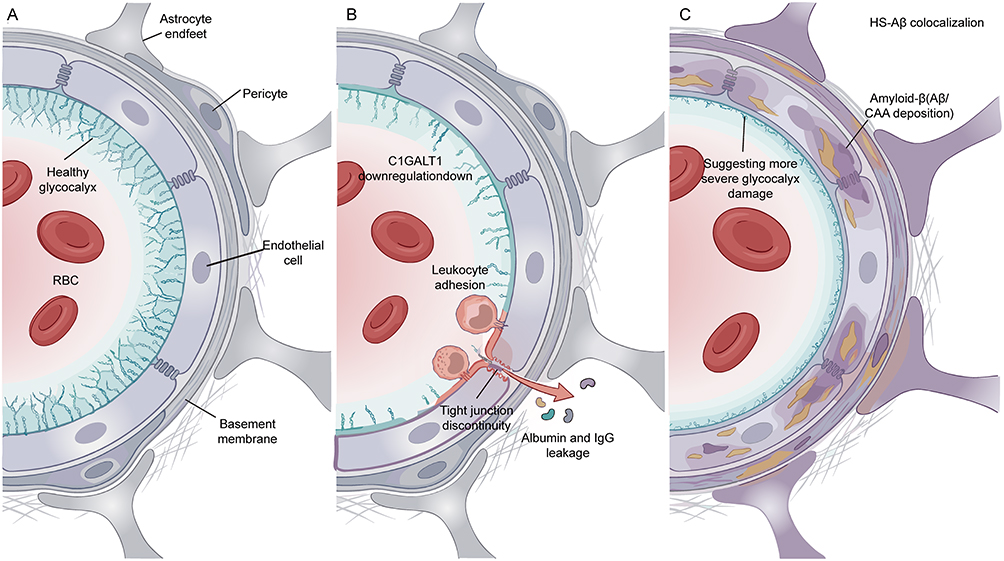 |
Figure 4 Compartmentalized Remodeling of the Brain Endothelial Luminal Glycocalyx. (A) Brain microvascular interface under glycocalyx homeostasis. Under normal conditions, the brain endothelial glycocalyx covers the endothelial luminal surface, maintains endothelial homeostasis and blood-brain barrier integrity, and participates in neurovascular unit homeostasis together with tight junctions, basement membrane, pericytes, and astrocytic end-feet. (B) Vascular wall compartment remodeling in aging or Alzheimer’s disease. In aging and Alzheimer’s disease-associated states, luminal protective mucin-domain glycoproteins and their mucin-type O-glycosylation networks are weakened, accompanied by C1GALT1 downregulation, increased endothelial exposure, enhanced leukocyte adhesion, focal tight junction abnormalities, and increased albumin and IgG extravasation. Red arrows indicate directions of inflammatory cell adhesion and plasma protein extravasation. (C) Vascular wall compartment remodeling in Alzheimer’s disease with severe cerebral amyloid angiopathy. In Alzheimer’s disease with severe cerebral amyloid angiopathy, enhanced HS-Aβ colocalization and altered polarity distribution are observed, suggesting further glycocalyx injury or spatial remodeling. Yellow/brown deposit-like signals indicate Aβ/CAA-related vascular wall deposition; luminal cyan brush-like structures indicate the brain endothelial glycocalyx. Abbreviations: AD, Alzheimer’s disease; Aβ, amyloid-beta; CAA, cerebral amyloid angiopathy; C1GALT1, core 1 β1,3-galactosyltransferase 1; HS, heparan sulfate; IgG, immunoglobulin G; RBC, red blood cell. Notes: This schematic was hand-drawn by the authors using Adobe Illustrator; no AI image generation tools were used. |
Concurrently, evidence for increased glycoconjugates in AD should not be directly interpreted as “luminal glycocalyx enhancement.” Reed et al reported elevated total hyaluronic acid (HA) in brain tissue with high AD neuropathological burden, accompanied by upregulation of hyaluronan synthase 2 (HAS2) and tumor necrosis factor-stimulated gene-6 (TSG-6); however, relevant molecules distribute across vasculature, neuronal nuclei (NeuN)-positive neurons, and ionized calcium-binding adapter molecule 1 (Iba1)-positive microglia, suggesting these more likely reflect matrix remodeling and inflammation-associated changes in AD brain tissue and vascular-associated microenvironments rather than simple luminal glycocalyx enhancement.41 Similarly, McMillan et al found that cerebrovascular heparan sulfate (HS) alterations do not demonstrate uniform enhancement in overall AD versus control comparisons, but are more significant in AD with severe cerebral amyloid angiopathy (CAA), manifesting as endothelial compartment HS elevation, enhanced HS-Aβ colocalization, and reversal of inner/outer polarity distribution (Figure 4C).42 Therefore, the currently more appropriate generalization is that AD cerebrovascular glycoconjugate alterations exhibit clear compartment specificity, with luminal protective mucin-domain glycoprotein-associated mucin-type O-glycosylation network weakening demonstrating the greatest consistency, while certain HA/HS alterations more reflect vascular-associated, vascular wall, or CAA-related compartment remodeling.
This evidence pattern suggests that AD-associated brain endothelial glycocalyx abnormalities should be understood as remodeling processes occurring across different vascular compartments and glycoconjugate levels rather than uniform, unidirectional holistic detachment; conceptual schematics are illustrated in Figure 4.
Functional and Interventional Evidence: The Mucin-Type O-Glycosylation Axis Constitutes the Current Strongest Causal Chain
Cerebrovascular and BBB model studies demonstrate that glycocalyx abnormalities can influence BBB function through elevated permeability, impaired shear stress responsiveness, and HA/CD44-related barrier homeostasis disruption.11,17,25,43–46 Building upon this foundation, abnormality types with relatively complete evidentiary chains currently concentrate primarily on the brain endothelial mucin-domain glycoprotein-associated mucin-type O-glycosylation axis. Selective mucin-domain disruption can rapidly induce brain endothelial glycocalyx degradation and BBB leakage; brain endothelial-specific downregulation of C1galt1 not only reduces luminal mucin-domain glycoprotein markers but also induces albumin and immunoglobulin G (IgG) extravasation with impaired tight junction continuity; conversely, restoration of C1GALT1 or B3GNT3 expression in aged mice significantly alleviates BBB leakage, with B3GNT3 restoration additionally accompanied by reduced inflammation indicators and behavioral improvements.18 Earlier developmental studies further demonstrated that core 1-derived O-glycan deficiency causes lethal central nervous system hemorrhage and cerebrovascular network abnormalities, indicating fundamental roles for this pathway in cerebrovascular integrity.47,48
Synthesizing existing evidence, brain endothelial mucin-type O-glycosylation downregulation should no longer be viewed merely as an accompanying alteration, but more likely represents a relatively upstream component in the currently most complete causal chain (Figure 5).18,47,48 However, it must also be recognized that this judgment remains primarily established upon animal experiments and mechanistic studies, insufficient for direct extrapolation as completed causal closure in the full course of human AD.17,18 The chain of “glycosylation program downregulation—luminal mucin-domain depletion—interface destabilization—BBB leakage—brain consequences” constitutes the currently relatively complete mechanistic evidence; however, obvious gaps remain between this and human translational evidence (Figure 5).
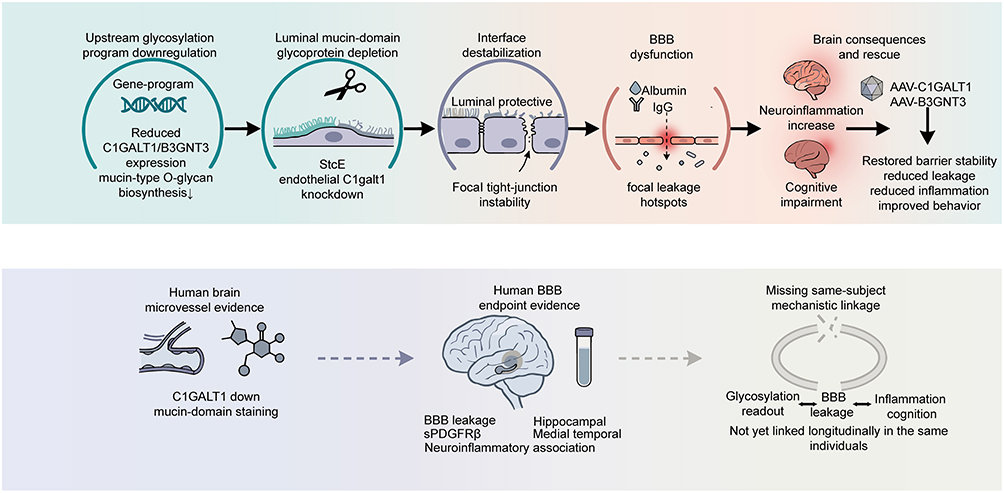 |
Figure 5 Causal Chain from Mucin-Type O-Glycosylation Dysregulation to BBB Dysfunction and Its Translational Gap. This figure summarizes the mechanistic chain linking cerebral endothelial mucin-type O-glycosylation dysregulation to blood-brain barrier dysfunction, and the translational gap at the human level. Upper panel: Experimental causal chain. Downregulation of glycosylation programs leads to luminal mucin-domain glycoprotein depletion, interface destabilization, and BBB leakage, whereas restoration of C1GALT1 or B3GNT3 expression partially reverses these abnormalities. The DNA icon is a conceptual schematic representing upstream downregulation of glycosylation-related gene programs; downward arrows indicate reduced expression of C1GALT1/B3GNT3 and downregulation of mucin-type O-glycosylation programs. Circular borders are used to demarcate distinct mechanistic modules or pathological stages; black arrows denote dynamic regulatory or pathological progression nodes. Lower panel: Cerebral microvascular abnormalities and BBB endpoint evidence at the human level, highlighting the current lack of direct mechanistic evidence longitudinally linking glycosylation abnormalities, BBB leakage, and inflammatory or cognitive phenotypes within the same subjects. Solid arrows indicate causality directly validated by experimental evidence; dashed arrows indicate inferred or incomplete connections. Abbreviations: BBB, blood-brain barrier; IgG, immunoglobulin G; AAV-C1GALT1, adeno-associated viral vector expressing C1GALT1; AAV-B3GNT3, adeno-associated viral vector expressing B3GNT3; C1GALT1, core 1 β1,3-galactosyltransferase 1; B3GNT3, β1,3-N-acetylglucosaminyltransferase 3; SPDGFRβ, stromal platelet-derived growth factor receptor β (vascular wall marker). |
Translational Evidence: Human Studies Support BBB Abnormalities But Have Not Yet Formed Complete Mechanistic Closure
At the human evidence level, the most robust support currently remains for the outcome level of BBB functional abnormalities rather than upstream brain endothelial glycosylation mechanisms themselves. DCE-MRI and related studies suggest that BBB disruption is visible in cognitive decline and AD spectrum populations, and can appear earlier than or independent of typical amyloid-beta/tau indicators in certain high-risk populations.31 This demonstrates that blood-brain barrier destabilization represents a relatively robust observational endpoint in human AD-associated vascular pathology, though it cannot directly prove brain endothelial glycocalyx abnormalities, particularly not definitively establishing whether these derive from O-glycosylation defects. Fluid biomarkers also support this “outcomes stronger than mechanisms” evidence pattern: for example, CSF sPDGFRβ correlates with age and inflammation-related indicators but cannot stably distinguish normal cognition, MCI, and AD, and is insufficient as a surrogate readout for brain endothelial O-glycosylation decline.37
In contrast, direct human evidence pointing to cerebrovascular glycosylation abnormalities, though less abundant, exhibits higher specificity. Histological and transcriptomic results at the brain microvascular level suggest that aging and AD are accompanied not merely by vague vascular glycocalyx damage, but more prominently by endothelial glycocalyx abnormalities rich in mucin-type O-glycosylation; in human AD brain microvasculature, both C1GALT1 and mucin-domain staining exhibit decline, thereby more directly localizing core 1 mucin-type O-glycan defects to the BBB endothelial luminal interface.18 Consistently, Yang et al’s established human cerebrovascular single-nuclear atlas reveals systematic transcriptional remodeling in cerebrovascular and perivascular cells under AD backgrounds; previous reanalysis upon this foundation further suggests possible overall downregulation of mucin-type O-glycan biosynthetic pathways.18 The significance of these studies lies in their transformation of “glycosylation abnormalities” from speculation to molecular observability at the human cerebrovascular level.
However, from the perspective of human translational evidence completeness, the current true boundary lies not in whether relevant phenomena exist, but in the lack of same-subject, longitudinal, multimodal mechanistic alignment. Currently, evidence capable of connecting BBB leakage to clinical phenotypes in vivo derives primarily from imaging and fluid biomarkers rather than synchronous correspondence with glycosylation readout indicators. Montagne et al discovered that APOE4 carriers exhibit BBB disruption in the hippocampus and medial temporal lobe, with baseline BBB injury indicators predicting subsequent cognitive decline independent of amyloid-beta and tau pathology.31 Preis et al further demonstrated in cross-sectional studies that hippocampal BBB leakage is primarily observed in AD dementia stages rather than MCI stages, with CSF sPDGFRβ increasing with age and correlating with chitinase-3-like protein 1 (YKL-40) and other neuroinflammation indicators, but not consistently with overall AD biomarker positivity status.37
Therefore, existing human studies more strongly support biological coherence among “vascular surface glycosylation abnormalities—BBB destabilization—inflammation/cognitive deterioration,” yet lack direct evidence longitudinally aligning glycosylation readout indicators, BBB leakage, and inflammatory or cognitive phenotypes within identical subjects, remaining insufficient to conclude that this mechanistic chain has completed causal closure in human AD. Overall, brain endothelial glycocalyx abnormalities in AD better fit the characterization of interface imbalance centered on luminal mucin-domain glycoprotein-associated mucin-type O-glycosylation axis weakening; causal evidence derives primarily from animal and mechanistic studies, while human AD evidence currently provides primarily pathological correlation and translational feasibility support.
Theoretical Basis and Translational Value of Brain Endothelial Glycocalyx Repair as an Intervention Strategy in Alzheimer’s Disease
If brain endothelial glycocalyx abnormalities in Alzheimer’s disease (AD) represent not merely concomitant phenomena but rather frontline interface abnormalities associated with BBB destabilization, neuroinflammatory amplification, and deterioration of the brain microenvironment, then the question requiring further answer shifts from merely “is the glycocalyx important” to “can the glycocalyx become an actionable intervention target.” Regarding existing research, interventions targeting brain endothelial glycocalyx do not manifest as single pathways but rather divide into three main categories: one focusing on preventing continued destabilization of the blood-facing interface, another aiming to restore endogenous glycosylation programs and promote glycocalyx reconstruction, and a third attempting surface remodeling through exogenous interface write-back. Concurrently, “utilizing existing BBB gaps to enhance drug delivery,” while strategically intuitive, follows a translational logic distinct from the aforementioned repair pathways. Based upon these distinctions, Table 4 provides parallel summarization of core positioning, primary potential benefits, and translational judgments for different strategic pathways.
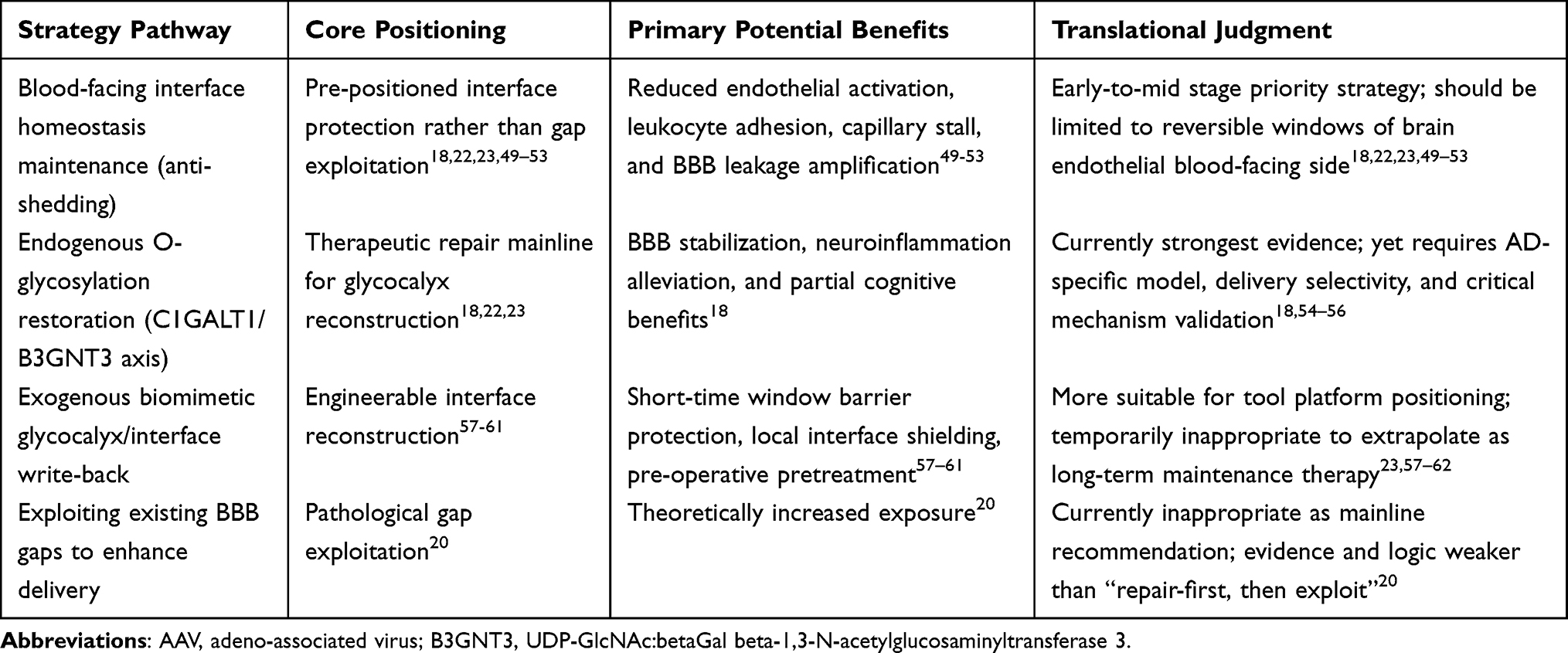 |
Table 4 Translational Positioning and Application Boundaries of Brain Endothelial Glycocalyx Intervention Strategies in Alzheimer’s Disease |
Preventing Blood-Facing Interface Destabilization: Glycocalyx Protection Should Precede Gap Exploitation
AD brain endothelial glycocalyx should no longer be characterized as a dispensable “surface decorative layer” beyond the BBB, but rather defined as the first interface that first withstands pathological impact and first determines endothelial homeostasis maintenance at the blood-facing side of the BBB (Figure 6A and B). Important support for this understanding derives from 2025 research demonstrating abnormalities in mucin-domain glycoproteins and core 1 mucin-type O-glycan axes in AD-associated brain endothelium and human AD brain microvasculature, with restoration of this glycosylation program improving BBB function, alleviating neuroinflammation, and partially improving relevant behavioral phenotypes, suggesting that glycocalyx abnormalities represent not merely concomitant phenomena following BBB destruction, but constituent links in BBB destabilization chains.18 Combined with recent BBB/glycocalyx reviews, it becomes further apparent that AD cerebrovascular pathology may not primarily manifest as terminal tight junction loosening, but more likely first occurs as thinning, disruption, and disorder of the low-adhesion, low-inflammation, low-permeability interface at the blood-facing side (Figure 6B).9,22,23 Therefore, the primary goal of AD-associated glycocalyx intervention should not be “exploiting already-formed gaps,” but must be preventing continued collapse of this initial interface.
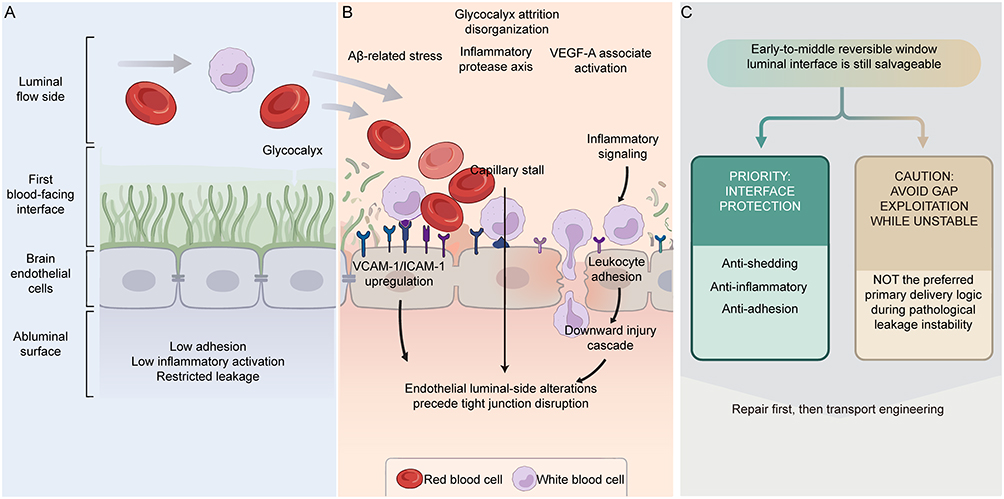 |
Figure 6 Pathological Destabilization of the Blood-Facing Endothelial Interface in Alzheimer’s Disease and Strategic Implications. (A) Protective blood-facing interface under homeostasis. The glycocalyx covers the endothelial luminal surface, maintaining low adhesion, low inflammatory activation, and restricted leakage. Gray arrows indicate blood flow direction and movement paths of cells or circulating components at the blood-facing interface. (B) Pathological interface destabilization and leakage amplification. Aβ-associated stress, inflammatory-protease axes, and VEGF-A activation can drive glycocalyx depletion, accompanied by VCAM-1/ICAM-1 upregulation, enhanced leukocyte adhesion, and capillary stalling. Black arrows indicate causal pathological flow directions. (C) Strategic options within early-to-mid-stage reversible windows. Intervention strategies may prioritize interface protection (anti-shedding, anti-inflammatory, anti-adhesion) to reduce sustained destabilization. Branching arrows indicate strategic choice directions; dashed arrows indicate inferred mechanisms. Abbreviations: AD, Alzheimer’s disease; Aβ, amyloid-beta; BBB, blood-brain barrier; ICAM-1, intercellular adhesion molecule-1; VEGF-A, vascular endothelial growth factor A; VCAM-1, vascular cell adhesion molecule-1. Notes: This schematic was hand-drawn by the authors using Adobe Illustrator; no AI image generation tools were used. |
Furthermore, in AD, glycocalyx dilution and endothelial activation are not two parallel events, but continuous links in the same pathological circuit. Clinical studies demonstrate that cerebrospinal fluid vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) are already significantly elevated in early AD patients, correlating with tau pathology, cortical thinning, and subsequent cognitive deterioration, indicating that cerebrovascular endothelial activation has already entered early disease stages.49,50 At the mechanistic level, amyloid-beta 42 can directly induce BBB endothelial VCAM-1 upregulation, promoting transition of the blood-facing surface toward more adherent activated states.51 At the pathological level, enhanced leukocyte-cerebrovascular interactions are visible in both AD patients and model animals, with glycocalyx reduction likely constituting one of the interface bases enabling such abnormal adhesion.52,53 More critically, this blood-facing activated interface represents not a static marker, but a reversible pathological surface with clear functional consequences: in AD mice, whether relieving neutrophil adhesion or inhibiting blood-facing vascular endothelial growth factor A (VEGF-A)-related pro-adhesion, pro-permeability signals, both can reduce capillary stall, enhance cerebral blood flow, and improve BBB integrity within short timeframes.52,53 Correspondingly, elevated peripheral VCAM-1/activated leukocyte cell adhesion molecule (ALCAM) also accompany AD cognitive decline, inflammatory burden, and brain atrophy phenotypes.50 This indicates that what truly requires priority cessation in AD is not merely “barrier gaps” in the simple sense, but the pathological endothelial surface that is being continuously rewritten toward high-adhesion, high-permeability, and inflammation-amplification susceptibility at the blood-facing interface (Figure 6C). Although amyloid-beta-RAGE, DR6/Wnt imbalance, and blood-facing VEGF-A abnormalities all suggest brain endothelial activation reversibility, existing evidence also indicates obvious compartment and stage dependencies for relevant pathways; therefore, the anti-shedding proposition establishable in AD should be strictly limited within reversible windows of brain endothelial blood-facing side, disease early-to-mid stages: priority suppression of inflammation-protease axes continuously driving shedding, blocking amplification chains of leukocyte adhesion, microcirculatory stagnation, and BBB leakage, and preserving stable starting surfaces for subsequent glycocalyx reconstruction. This logic can be further generalized as: within early-to-mid stage reversible windows of AD, what truly requires priority cessation is not exploitation of existing gaps, but continued destabilization of the blood-facing luminal interface; pathological expansion and strategic implications are illustrated in Figure 6.
Driving Glycocalyx Reconstruction Through O-Glycosylation Restoration: Brain Functional Benefits and Translational Limitations
Alterations in brain endothelial glycocalyx represent not isolated morphological phenomena, but structural imbalances anchorable at the molecular level. Therefore, glycocalyx reconstruction should be understood as repair of plastic structural units at the blood-facing side of the BBB, rather than simple transposition of peripheral experiences into the brain.9,23 Previous glycocalyx injury research concentrated primarily on heparan sulfate (HS)/heparan sulfate proteoglycans (HSPGs), syndecans, hyaluronic acid (HA)/CD44, and shedding, emphasizing inflammation, adhesion, and elevated permeability.23,44,45,63 Recent key advances further localize aged brain endothelial glycocalyx abnormalities to mucin-domain glycoproteins and their dependent core 1 mucin-type O-glycosylation axis. Aged cerebrovasculature exhibits selective weakening of mucin-domain signals, not primarily due to universal glycoprotein scaffold loss, but more consistent with O-glycosylation program attenuation: downregulation of mucin-type O-glycan biosynthesis-related genes, with Galnt10, B3gnt3, Galnt2, and C1galt1 most prominent; concurrently, C1GALT1 and B3GNT3 protein levels decline, correlating positively with mucin-domain marker intensity.18 This signifies that the focus of brain endothelial glycocalyx reconstruction is not exogenous supplementation of specific components, but restoration of endogenous O-glycosylation capacity supporting mucin-like interfaces.
Further causal evidence demonstrates that brain endothelial-specific downregulation of C1galt1 can directly induce albumin and IgG extravasation, indicating that core 1 O-glycosylation decline itself is sufficient to induce BBB leakage.18 Acute cleavage of blood-facing mucin-domain glycoproteins with mucinase (StcE mucinase) similarly increases BBB permeability, with sustained injury even producing meningeal and intraventricular hemorrhage. This process accompanies claudin-5 (CLDN5) decline and oxidative stress enhancement, consistent with previous findings of glycocalyx destruction-induced permeability increases through caveolin-1 (CAV1)-dependent transcytosis.18,44 Earlier studies also demonstrated that core 1-derived O-glycan deficiency can cause embryonic cerebral hemorrhage and vascular structural abnormalities.47,48 These results indicate that restoring brain endothelial O-glycosylation may not merely correct molecular markers, but may directly translate into therapeutic repair effects of BBB stabilization.
Concurrently, O-glycosylation restoration currently forms relatively complete in vivo rescue evidence. In 17-month-old mice, brain endothelial-specific overexpression of C1GALT1 or B3GNT3 can enhance mucin-domain glycoprotein markers, significantly reduce whole-brain sulfo-NHS-biotin extravasation, and bring BBB status closer to young controls.18 Notably, B3GNT3 effects further extend to brain functional levels: improving Y-maze spontaneous alternation and contextual fear conditioning, suggesting spatial working memory and hippocampus-dependent learning and memory benefits, while reducing CD68-positive microglial activation and shifting excitatory neurons, oligodendrocytes, and glial inflammation-associated transcriptional states toward younger directions.18 This indicates that brain endothelial glycocalyx reconstruction is not limited to reducing vascular leakage, but can translate into brain functional improvements through BBB stabilization, neuroinflammation mitigation, and optimization of the brain microenvironment.18
At this stage, this pathway more closely approaches therapeutic repair rather than pathological exploitation of damaged barriers. Compared with “enhancing delivery through glycocalyx alterations,” O-glycosylation reconstruction merits priority advancement precisely because it already possesses continuous evidentiary chains of “targeted intervention—BBB restoration—inflammation alleviation—partial cognitive improvement.”18,22 However, current strongest evidence derives primarily from aging models, with human AD evidence still dominated by post-mortem samples and transcriptomic associations.18,23 Employed adeno-associated virus (AAV) delivery systems also retain cross-species applicability and brain endothelial specificity limitations.54–56 Furthermore, truly efficacy-critical substrates and downstream pathways remain incompletely characterized.18 Therefore, brain endothelial O-glycosylation reconstruction satisfies proof-of-concept standards, yet three translational gaps constrain immediate clinical applicability. First, murine cerebrovascular glycosylation programs differ quantitatively from human brain microvascular transcriptomes in baseline enzyme expression and substrate specificity. Second, adeno-associated virus (AAV) vectors employed in preclinical rescue studies face unresolved challenges in brain endothelial tropism, immunogenicity, and scalable manufacturing for human CNS delivery. Third, the precise glycoprotein substrates whose O-glycan restoration is both necessary and sufficient for BBB stabilization in human AD remain incompletely mapped. Until these gaps are closed, O-glycosylation reconstruction should be regarded as a high-priority mechanistic target rather than an imminent clinical intervention.
Brain Endothelial Glycocalyx Interface Engineering: From Mechanistic Inspiration to Application Boundaries
The core of glycocalyx engineering in AD lies not in vague “thickening” or “preserving remaining glycocalyx,” but in treating brain endothelial glycocalyx as a programmable molecular interface at the blood-facing side of the BBB, rewriting specific glycosylation modules to reshape its functions governing adhesion restriction, mechanosensation, and inflammatory regulation.10,22,52 Molecular modules associated with mucin-type O-glycosylation can be viewed as “programmable units” not merely because they are passively damaged during disease progression, but because their upstream expression can be directionally manipulated while their downstream effects can link to barrier stability and inflammatory status—such glycosylation units are simultaneously interface structural components and intervention nodes determining interface functional outputs.18 Therefore, glycocalyx engineering significance in AD extends beyond merely “preserving remaining glycocalyx” to reshaping functional rules of the BBB blood-facing interface through rewriting specific glycosylation modules. However, existing evidence simultaneously indicates that engineering priorities do not involve abstract “glycocalyx thickening.” Although both C1GALT1 and B3GNT3 can improve BBB integrity, cognitive behavior and neuroglial inflammatory program improvements concentrate more prominently in the B3GNT3 axis, indicating that critical determinants of therapeutic effects may be which glycan classes are restored, assembled onto which mucin-domain carrier classes, and how these structures participate in adhesion restriction, shear stress sensing, inflammatory regulation, and barrier homeostasis coupling.18 Glycocalyx functions should not be simplified as singular physical permeability barriers, but understood as composite interfaces integrating mechanosensation, leukocyte adhesion, endothelial activation thresholds, and local inflammatory amplification/attenuation.22,52
If brain endothelial glycocalyx is regarded as a remodelable BBB blood-facing interface, then one of the most direct intervention pathways involves “writing back” exogenous biomimetic interfaces onto cell surfaces. Distinct from prioritizing endogenous glycan synthesis restoration, this strategy attempts to reconstruct interface spatial exclusion, surface electronegativity, and molecular crowding through glycopolymer shielding, synthetic mucin display, or glycosaminoglycan (GAG) mimetic coating. Relevant methods have been incorporated into broader glycoengineering frameworks, including metabolic glycoengineering, bioorthogonal functionalization, synthetic glycopolymers, mucin-mimetic materials, and enzymatic glycan editing.23,53 However, in AD and BBB research, such exogenous “physical layer reconstruction” should currently be characterized as mechanistic inspiration and technical reserve rather than mature therapeutic pathways.22,23,53 The most compelling proof-of-concept for this direction derives from transplantation fields rather than BBB research. Siren et al reported that immunosuppressive glycopolymers can be enzymatically connected in situ to vascular graft endothelial surfaces under cold ischemia conditions, thereby protecting glycocalyx and alleviating acute and chronic rejection in the absence of systemic immunosuppression.57 Notably, this surface engineering not only improved endothelial morphology but altered functional outcomes, including reduced ischemia-reperfusion injury, decreased immune cell adhesion and immune-mediated cytotoxicity, and ultimately improved graft fate.57 Its significance for the BBB field lies in first explicitly demonstrating that endothelial glycocalyx itself can serve as an engineerable therapeutic target surface, with in situ re-coating capable of reshaping inflammation and injury processes.57 Correspondingly, synthetic mucin research further provides platform-level support. Wardzala et al demonstrated that synthetic mucins with cholesterol amide anchor terminals can effectively insert into diverse cell membranes, enabling quantitative assessment of surface density, residence time, half-life, and cellular tolerance.58 These artificial mucins maintain detectable signals on cell surfaces for several days, with half-lives of approximately 20–62 hours, without significant cytotoxicity.58 Although such results cannot be directly extrapolated to BBB therapeutics, they at least demonstrate that key physical properties of glycocalyx including spatial volume, molecular crowding, and surface exclusion can be directly conferred onto cell surfaces through exogenous materials.58 In other words, glycocalyx represents not merely an endogenously synthesized product, but an engineerable interface structure.
However, boundaries for this strategy within the BBB are equally clear. First, whether exogenous biomimetic layers can stably reside in brain microvascular high-shear, continuous perfusion environments remains without direct evidence. Second, BBB glycocalyx exhibits significant organ specificity, while its precise composition and microdomain distribution remain incompletely characterized; with native templates yet unclear, any biomimetic write-back can only approximate reconstruction rather than precise replication. Third, glycocalyx is not merely a protective layer—it simultaneously determines membrane receptor exposure states. Studies have indicated that cell surface glycocalyx can significantly influence drug delivery system accessibility to membrane receptors.59 Therefore, glycocalyx reconstruction may on one hand enhance BBB stability and suppress endothelial activation, while on the other hand may weaken receptor-mediated transport or targeted nanocarrier binding due to spatial occlusion.62 In the BBB, this more closely resembles interface functional reallocation rather than unidirectional gain. Accordingly, exogenous biomimetic glycocalyx in AD is more appropriately positioned as mechanism and tool platform rather than long-term chronic disease treatment. Its practical value primarily lies in demonstrating that brain endothelial surfaces can be re-coated, re-shielded, and re-orchestrated, providing methodological foundations for more refined local interface editing, such as short-time window barrier protection, acute or peri-operative BBB stabilization, and blood-facing pretreatment before local delivery.23,53,58 In contrast, direct extrapolation as long-term maintenance therapy remains obviously limited by residence time, repeated dosing, receptor occlusion, material immunocompatibility, and chronic safety factors.53,60–62 Therefore, at least at current stages, exogenous biomimetic glycocalyx will more likely first serve short-time window interface protection and local intervention rather than long-term maintenance therapy for AD.
Synthesizing existing evidence, brain endothelial glycocalyx in AD is more appropriately positioned as a pathogenic interface for priority repair. Centered on mucin-domain glycoproteins and core 1 mucin-type O-glycosylation axis, preliminary continuous evidentiary chains have formed from structural imbalance, BBB destabilization, to in vivo rescue and partial brain functional improvement. In comparison, exploiting existing interface gaps to enhance delivery or long-term exogenous biomimetic coating implementation currently remain primarily at stages of mechanistic rationality but translational pending verification.
Remodeling of BBB Delivery Rules by Brain Endothelial Glycocalyx Alterations in AD
Changing the Starting Boundary: How Glycocalyx Redefines BBB Luminal Contact Conditions
In AD-associated BBB research, brain endothelial glycocalyx alterations may be misread as “the vasculature is leakier, therefore more favorable for drug delivery.” However, from a delivery perspective, consequences of brain endothelial glycocalyx abnormalities in AD may not be a simply “leakier” BBB, but rather a systematic rewriting of the entire delivery logic from luminal contact, target recognition, trans-endothelial pathways, to brain distribution allocation. Specifically, glycocalyx remodeling may first influence initial contact conditions between blood-facing cargo and endothelial membranes rather than directly altering terminal flux; subsequently, it may further influence receptor recognition, trans-endothelial route selection, brain distribution, and corresponding toxicity costs.64 Therefore, the significance of glycocalyx abnormalities in AD may be inappropriately simplified as “more entry,” but should be understood as front-positioned alterations in BBB recognition and shunting rules for therapeutic inputs (Figure 7).
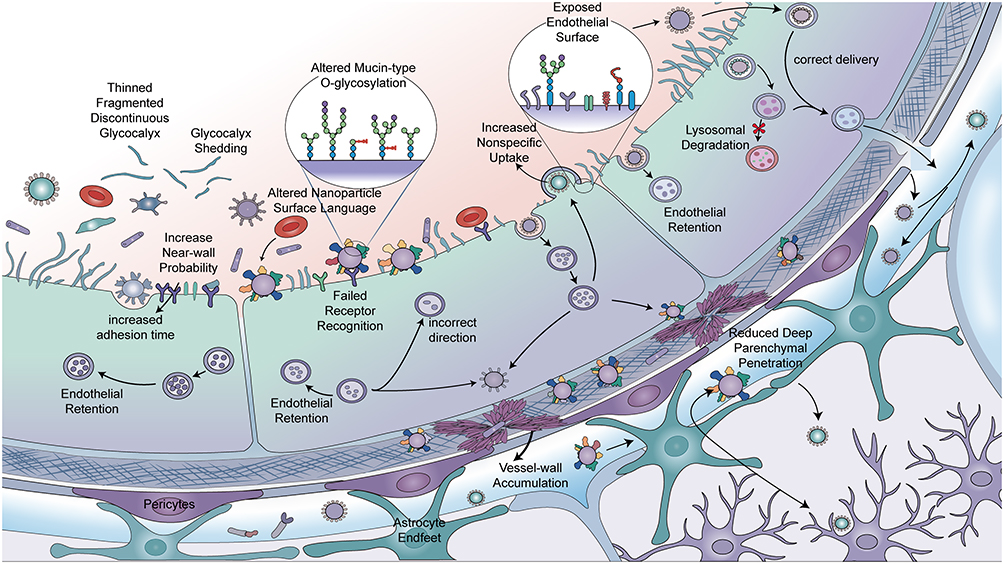 |
Figure 7 Hypothetical Model of How Brain Endothelial Glycocalyx Remodeling Rewires the BBB Blood-Facing Delivery Interface. This hypothetical working model illustrates how brain endothelial glycocalyx abnormalities alter the blood-brain barrier blood-facing delivery interface and influence the trans-barrier behavior of delivery particles. Glycocalyx thinning, fracture, shedding, and mucin-type O-glycosylation changes may expose abnormal endothelial surfaces and alter particle interactions with the blood-facing interface. These changes may lead to increased near-wall probability, prolonged adhesion, altered surface Corona language, failed receptor recognition, erroneous directionality, increased nonspecific uptake, and enhanced endothelial intracellular retention. Subsequent outcomes may include lysosomal degradation, vascular wall accumulation, decreased blood-brain barrier delivery efficiency, and reduced deep brain parenchymal penetration. Conversely, under conditions of relatively normal interface recognition and transport pathways, delivery particles can cross the vascular wall in the correct direction and enter the brain parenchyma. Black arrows indicate movement directions or potential transport pathways of delivery particles; arrows pointing into endothelial cells indicate particle uptake or endothelial retention; arrows pointing to lysosome-like structures indicate possible degradation pathways; arrows crossing the vascular wall toward the brain parenchyma side indicate relatively effective blood-brain barrier delivery; arrows within or around the vascular wall indicate possible vascular wall accumulation or off-target deviation. Red asterisks indicate lysosomal degradation or ineffective delivery outcomes. Particles, cells, and glycocalyx structures are schematic and not to scale. Abbreviation: BBB, blood-brain barrier. Notes: This schematic was hand-drawn by the authors using Adobe Illustrator; no AI image generation tools were used. |
For any antibody, protein therapeutic, or nanocarrier, BBB delivery first confronts not a bare endothelial membrane, but the near-wall luminal environment defined by the glycocalyx. Existing studies indicate that brain endothelial glycocalyx resides at the foremost frontier between blood and cerebrovascular vessels, participating in blood-facing processes including signal transduction, adhesion, transport, and membrane morphology.18 This suggests that therapeutic inputs must first experience near-wall contact, local residence, and spatial sieving established by the glycocalyx before effective interaction with membrane receptors can occur. This point carries particular importance for drug delivery. Imaging and diffusion studies suggest that the glycocalyx itself serves as a macromolecular pre-sieving layer at the BBB luminal surface, with differential accessibility for different-sized molecules into its effective volume.10 Simultaneously, restrictive properties of healthy BBB manifest not merely as tight junction closure, but include exceptionally low levels of nonspecific fluid-phase/vesicular transcytosis.64
Therefore, when glycocalyx thinning, continuity impairment, or mucin-type O-glycosylation abnormalities occur in AD, what may be rewritten first are “near-wall probabilities” for cargo reaching membrane surfaces, effective collision frequencies with receptors, and background conditions for shunting toward nonspecific channels, without necessarily waiting for obvious tight junction destruction to manifest delivery differences.18 Glycocalyx abnormalities bring not a simply “more open” BBB, but a BBB with adjusted luminal contact rules. For certain cargo, such changes may increase membrane surface exposure and initial adhesion opportunities; for others, they may increase nonspecific adsorption, abnormal retention, erroneous shunting, or vascular wall deposition risks. Therefore, discussing BBB delivery boundaries in AD contexts, evaluation priorities should appropriately balance “whether brain entry is easier” with “how cargo first approaches endothelium, in what manner it is recognized, and at what cost it traverses this starting interface” (Figure 7).
Overall, AD-associated brain endothelial glycocalyx remodeling alters not single permeability indicators, but the entire delivery rule system from starting contact, trans-endothelial route selection, to brain distribution allocation. For comparison convenience, these adjusted levels and their impacts on delivery design and evaluation are summarized in Table 5.
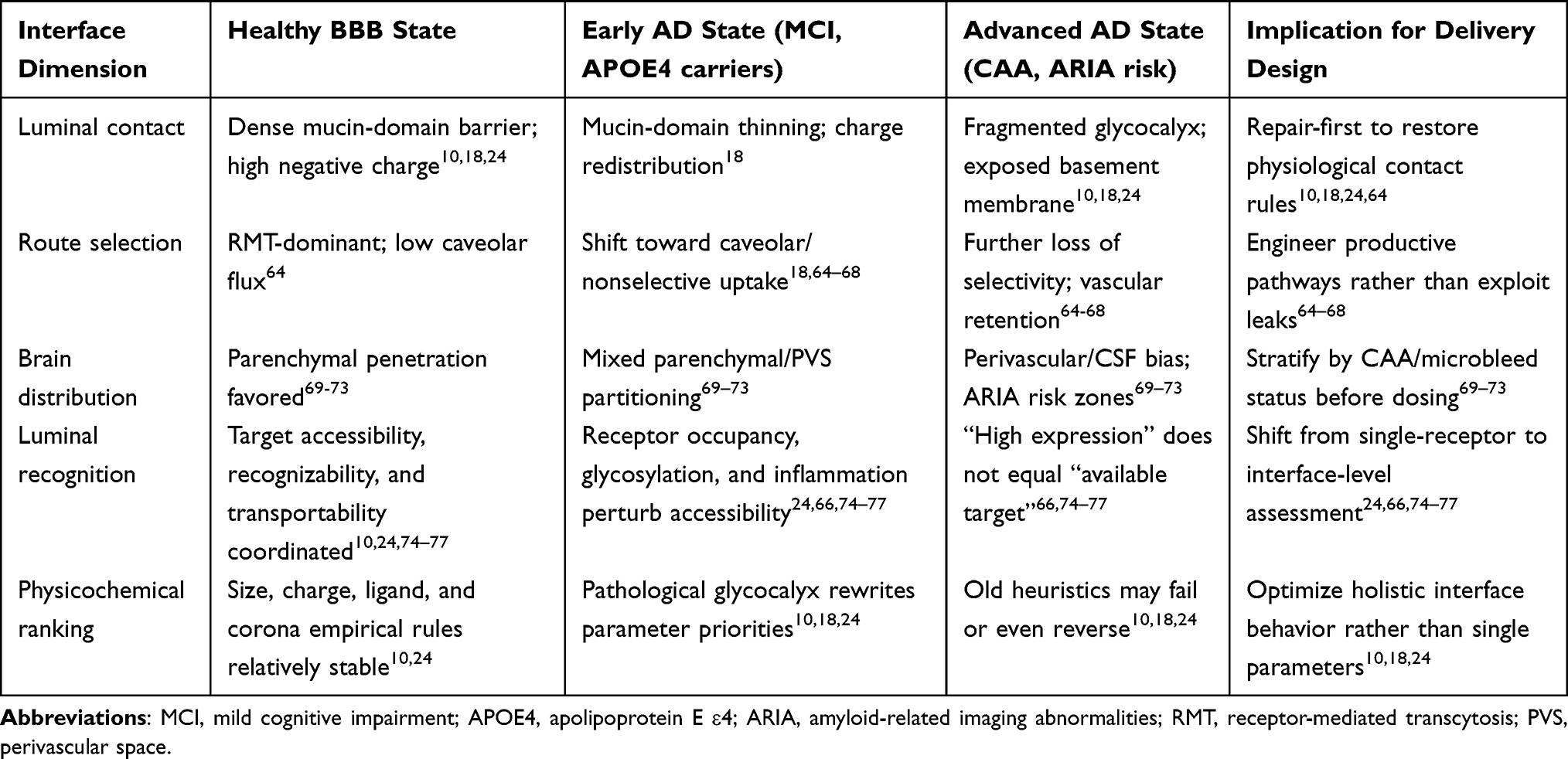 |
Table 5 How Brain Endothelial Glycocalyx Remodeling in AD Rewrites BBB Delivery Rules |
Remodeling of the Luminal Recognition Interface: Coupled Changes in Target Accessibility, Recognition Efficiency, and Transport Outcomes
Active targeting across the BBB rests on three assumptions. First, target molecules must remain accessible on the vascular luminal surface. Second, exogenous ligands must bind these targets stably despite systemic circulation complexity. Third, binding must trigger productive endocytosis or transcytosis rather than surface trapping or lysosomal degradation. However, this assumption does not necessarily hold in AD contexts. Whether active delivery can be achieved may depend upon at least three prerequisites: first, targets must be continuously exposed on the vascular luminal side, maintaining accessibility for circulating delivery systems; second, exogenous antibodies or ligands must maintain sufficient recognition efficiency and binding capacity in complex physiological environments; third, target-binding-formed complexes must enter effective trans-endothelial transport programs rather than remaining on cell surfaces, stagnating in endosomal systems, or diverting into lysosomal degradation pathways. Existing studies have indicated that BBB physiological barrier properties and disease-associated alterations can significantly influence precise delivery of therapeutics following systemic administration.74 Therefore, when evaluating whether a BBB target possesses delivery value in AD, judgments should not be based solely upon expression levels, but rather upon whether it remains in a functional state that is accessible, recognizable, and transportable for blood-facing delivery systems. BBB target “availability” represents not merely an expression issue, but an interface issue with obvious state dependence.
As the first interface between blood and cerebrovascular vessels, brain endothelial glycocalyx is encountered before receptors. Previous studies suggest that brain endothelial glycocalyx and its negative surface charge constitute important BBB defense system components, capable of influencing interactions between macromolecules, nanoparticles, and drugs with endothelial surfaces.10,24 When AD-associated glycocalyx remodeling occurs, what is altered is not merely “whether pathways exist,” but “what is first encountered,” “at what distance and angle contact occurs,” and “whether charge and hydration layers cause repulsion or capture.” Receptor exposure is not a static fact nakedly exposed to blood flow, but a result continuously defined and sieved by the glycocalyx. Therefore, glycocalyx and receptor extracellular domains should not be viewed in isolation. The glycocalyx determines “what is first encountered”; receptor extracellular domains determine “whether recognition occurs after contact.” But these two steps are not independent. Drugs truly face not an isolated naked receptor, but a continuous interface jointly constituted by glycocalyx thickness and composition, receptor extracellular domain integrity, glycosylation status, inflammatory environment, and endogenous ligand competition. Notably, the glycocalyx may physically shield or modulate the accessibility of luminal receptors such as LRP1. By governing the near-wall molecular crowding and charge environment, an intact glycocalyx can limit or direct ligand access to the LRP1 ectodomain; conversely, glycocalyx shedding or thinning may expose cryptic epitopes or accelerate ectodomain shedding, thereby altering the balance between productive transcytosis and decoy receptor competition. Finally, this review does not systematically address.
BBB targeting confronts not merely a specific receptor, but a “luminal recognition interface.” Failure of traditional BBB targeting strategies often does not mean receptors have completely disappeared, but more commonly indicates luminal recognition interface remodeling. Taking transferrin receptor (TfR) as an example: circulating transferrin can continuously occupy receptors, influencing effective binding of exogenous ligands or antibodies; simultaneously, different epitopes and affinities alter intracellular sorting, causing decreased transcytosis efficiency.75 Furthermore, low-density lipoprotein receptor-related protein 1 (LRP1) and other receptors, following ectodomain shedding, can generate soluble fragments acting as “decoy receptors” competitively binding ligands, reducing membrane surface available target density while weakening transport function.66,76 Under inflammatory or disease states, glycocalyx shedding, glycosylation abnormalities, and epitope conformational changes further perturb initial contact and subsequent recognition, even causing “false targets” or functional target loss.25,77 This explains why some traditional targeting strategies fail—not because receptors have completely vanished, but because the external “recognition environment” has changed. Therefore, BBB drug design must expand from “single receptor nomenclature” to holistic assessment of luminal recognition interfaces.
How Glycocalyx Damage Reorganizes Trans-Endothelial Pathways
A key consequence of AD-associated glycocalyx abnormalities is not that all cargo more easily traverses the BBB, but that endothelial route choice for different cargoes is rewritten: originally highly selective, controlled trans-endothelial transport weakens, while low-selectivity uptake, erroneous endocytosis, and abnormal shunting proportionally rise.64 Consistently, brain endothelial proteomics research demonstrates that vesicle-mediated transport globally declines during aging, accompanied by systematic imbalances across multiple vesicular pathways including endocytosis, receptor recycling, macropinocytosis, and lysosomal degradation.65 Human brain single-nuclear transcriptomics and NVU studies also suggest that AD vascular pathology involves not merely barrier homeostasis impairment, but concurrent rearrangement of endothelial transport, pericyte support, angiogenic responses, and gliovascular signaling.26,34,36 Therefore, AD-associated BBB abnormalities are more appropriately understood as “transport pathway disorder” rather than a natural delivery window arbitrarily exploitable by any cargo.26,34,36,64,65
This point is particularly clear in LRP1-related research. Lee et al demonstrated that brain endothelial ANKS1A (ankyrin repeat and SAM domain-containing protein 1A) deficiency reduces cell surface LRP1, impairs amyloid-beta clearance across the BBB, and exacerbates amyloid-beta pathology and cognitive impairment in AD mice.66 Conversely, Chen et al, utilizing LRP1-targeting polymersomes with different multivalencies, discovered that cargo size and multivalency can determine sorting fate even when targeting identical receptors; by biasing LRP1 pathways toward productive transcytosis, researchers enhanced amyloid-beta efflux and improved cognitive performance.67 This suggests that what is truly damaged in AD is not merely “barrier tightness,” but whether cargo can be directed into correct, transport-task-completing pathways. Similarly, Moyer et al further demonstrated that alkaline phosphatase (ALPL) can serve as a cerebrovascular receptor for engineered AAV vectors crossing the BBB.68 Therefore, the same BBB may exhibit certain endogenous clearance route impairments under pathological states, yet provide new route handles for specific cargoes in engineering designs; keys determining delivery success lie not in whether the BBB is “leakier,” but which pathway cargo is directed toward, where it ultimately arrives, and whether that pathway remains productive.67,68
Therefore, AD-associated glycocalyx damage functions more as a “pathway reorganizer” than a simple “permeability amplifier.” Its results are not uniform increases for all inputs, but gradual attenuation of high-fidelity receptor-dependent transport and amyloid-beta clearance networks, with rising proportions of low-selectivity uptake, erroneous endocytosis, and abnormal shunting, subsequently amplified by endothelial activation and NVU abnormalities. From a drug design perspective, this implies that delivery strategies should not default to “entering through leaks,” but rather focus on how to restore or reconstruct productive pathways, or engineer cargoes according to existing pathway logic.18,67,68 However, it must be cautiously noted that current research still lacks studies directly completing closed-loop verification of “glycocalyx damage—decreased RMT/increased pathological caveolar flux—specific cargo flux rewriting” within standardized AD systems using identical cargoes. Yet pathway selection changes are not endpoints. Different pathways possess therapeutic implications not merely because they determine whether crossing occurs, but because they further shape cargo deposition locations within the brain.
Brain Distribution Reallocation: How Glycocalyx Remodeling Influences Parenchymal versus Vascular-Associated Exposure
In BBB delivery research, crossing the endothelium is not an endpoint but a starting point for drug redistribution within the brain. For macromolecular therapeutics, “brain entry” involves two distinct processes: completing trans-endothelial transport, and determining final localization after crossing. The critical question is not whether drugs entered the brain, but where they ultimately deposit—vascular walls, perivascular spaces (PVS), cerebrospinal fluid (CSF)-related regions, or deeper brain parenchyma approaching pathological targets. For Alzheimer’s disease (AD), this distinction carries particular importance, because elevated total brain exposure does not necessarily equate to elevated effective parenchymal exposure; conversely, it may imply increased vascular-associated or perivascular exposure, which more directly associates with cerebral amyloid angiopathy (CAA), vascular inflammation, and amyloid-related imaging abnormalities (ARIA). Therefore, in AD BBB delivery evaluation, spatial distribution often carries greater explanatory power than simple total brain entry quantity.69–73
This conceptual shift—from “how much entered” to “where it settled after entering”—has received direct support from recent BBB shuttle protein research. Pizzo et al’s 2025 Science study demonstrated that transferrin receptor (TfR)-mediated anti-amyloid-beta constructs not only increased brain antibody levels approximately 5-8-fold, but more critically, shifted brain distribution from peri-arterial/vascular-associated enrichment characteristic of traditional anti-amyloid-beta antibodies toward broader parenchymal distribution, accompanied by marked reductions in vascular inflammation and ARIA-like pathology.69 A concurrent Science review further noted that safety improvements likely derived not merely from “reduced effective dosimetric burden,” but rather from altered brain entry pathways reducing spatial encounters between antibodies and vascular wall amyloid-beta/CAA while increasing contact with parenchymal targets.70 Khoury et al’s study provided powerful comparison: even under matched total brain exposure conditions, BBB transport vehicles demonstrated higher parenchymal distribution, while non-BBB-targeted IgG primarily localized to meninges, choroid plexus, and perivascular/CSF-related regions, barely entering deep parenchymal cells.71 Similarly, TfR1 shuttle fused with Aducanumab not only enhanced brain delivery but improved distribution uniformity and reduced peri-arterial bias, suggesting that altering entry mechanisms carries greater importance than simply increasing dosage.72 Wells et al further demonstrated that receptor types and affinities simultaneously influence brain concentration, exposure kinetics, cellular association, and intracellular processing, indicating that BBB entry functions not as a simple “open or closed” gate, but rather as an active program shaping brain distribution trajectories.73
Within this context, the importance of brain endothelial glycocalyx becomes pronounced—not merely as a BBB structural component, but as a functional regulatory unit. Recent studies demonstrate that AD-associated pathological backgrounds induce significant glycocalyx dysregulation, thereby influencing BBB function and neuroinflammatory processes.18 Earlier ultrastructural studies also indicated relatively prominent glycocalyx structures on brain capillary endothelium, suggesting a special interface with charge-sieving and spatial organization functions on the BBB blood-facing surface.9 Furthermore, accumulating research suggests glycocalyx functions not merely as a “mucus layer” passively coating endothelial surfaces, but as an active structural factor regulating how circulating drug carriers approach endothelial receptors and enter endocytic/transcytotic pathways.59 Therefore, in AD, if brain endothelial glycocalyx undergoes remodeling, its most likely consequence is not simply uniform elevation or reduction in total brain entry, but first rewriting the “distribution allocation rules” for drugs after brain entry.
Mechanistically, glycocalyx alterations can likely rewrite at least four categories of brain “distribution allocation rules.”
First, it may alter the balance between blood-facing surface residence and effective transcytosis. Glycocalyx directly participates in initial contact, charge sieving, and nanoscale receptor accessibility regulation for drugs at the blood-facing side. Therefore, when glycocalyx thinning or negative charge barrier weakening occurs, drug residence time on endothelial luminal surfaces and nonproductive adhesion may both increase, causing more exposure to remain on vascular walls or endothelial surfaces rather than effective transport into brain parenchyma.
Second, it may alter route selection between capillary-brain parenchyma pathways and plasma-CSF/perivascular pathways. Existing BBB shuttle protein research clearly demonstrates that different brain entry pathways correspond to distinctly different spatial outcomes: nontargeted antibodies more easily distribute to brain surfaces, CSF, or perivascular regions, while optimized BBB carriers more favor parenchymal delivery.69–73 If glycocalyx remodeling alters receptor accessibility, endocytic preferences, or surface microenvironments, the same cargo may be redirected from originally capillary-dominated parenchymal delivery pathways toward more CSF- or perivascular-biased pathways.
Third, it may alter regional distribution preferences, particularly the balance between peri-arterial/vascular-associated enrichment and more uniform brain parenchymal distribution. This carries particular importance in AD because vascular wall amyloid-beta and CAA themselves constitute high-risk “anatomical traps” for anti-amyloid-beta drugs. If glycocalyx abnormalities cause drugs to more easily deposit on vascular walls or peri-arterial regions, then even without significant total brain exposure changes, vascular inflammation, microhemorrhage tendencies, and ARIA risks may be amplified; conversely, if drug distribution becomes more uniform with greater deposition in brain parenchyma, target utilization efficiency may improve and safety windows may narrow.
Fourth, it may also influence post-entry fates, including intracellular transport, catabolism, residence time, and cell homing. Studies have demonstrated that different receptor platforms and affinity combinations alter whether drugs exhibit “rapid entry but faster decomposition” or “slower entry but more persistent brain residence.”73 Upon this foundation, if glycocalyx further alters receptor presentation modes, membrane surface organization states, or post-endocytic sorting processes, it will likely continue influencing whether drugs remain at vascular interfaces, stagnate in PVS, or ultimately enter deeper brain parenchyma approaching true cellular and pathological targets.
From a translational research perspective, these “distribution allocation rules” collectively point to a core judgment: in AD therapeutics, what is truly critical is not abstract “brain exposure,” but the ratio between brain parenchymal exposure and vascular wall/PVS exposure. Amyloid-beta exists both in brain parenchymal plaques and deposited on cerebrovascular walls forming CAA, while ARIA risk is highly associated with vascular fragility, inflammatory activation, and spatial contact with CAA. Therefore, even if a drug “enters the brain,” if it is primarily directed toward vascular-associated or peri-arterial regions, therapeutic efficacy may remain limited or even toxicity may rise; conversely, even if total brain entry increase magnitudes are modest, as long as distribution is redirected toward brain parenchyma and away from vascular walls, more efficient target engagement and improved safety may result.69–72 This also suggests that future AD BBB delivery evaluation should no longer remain at single brain uptake indicators, but should shift toward distribution frameworks simultaneously examining parenchymal delivery, vascular/PVS burden, and corresponding inflammation or hemorrhage-associated readouts. Table 6 further summarizes upstream determinants of “distribution allocation.” Overall, the key lies not merely in different transport pathways corresponding to different intracellular destinations, but in whether cargo can be effectively contacted, specifically recognized, and entered into productive transcytosis programs on the blood-facing side. Therefore, if brain spatial distribution differences are further traced upstream, their root still points toward alterations in luminal recognition interfaces. Based upon this, “increased total brain entry” cannot substitute for judgments regarding parenchymal exposure and vascular-associated burden.
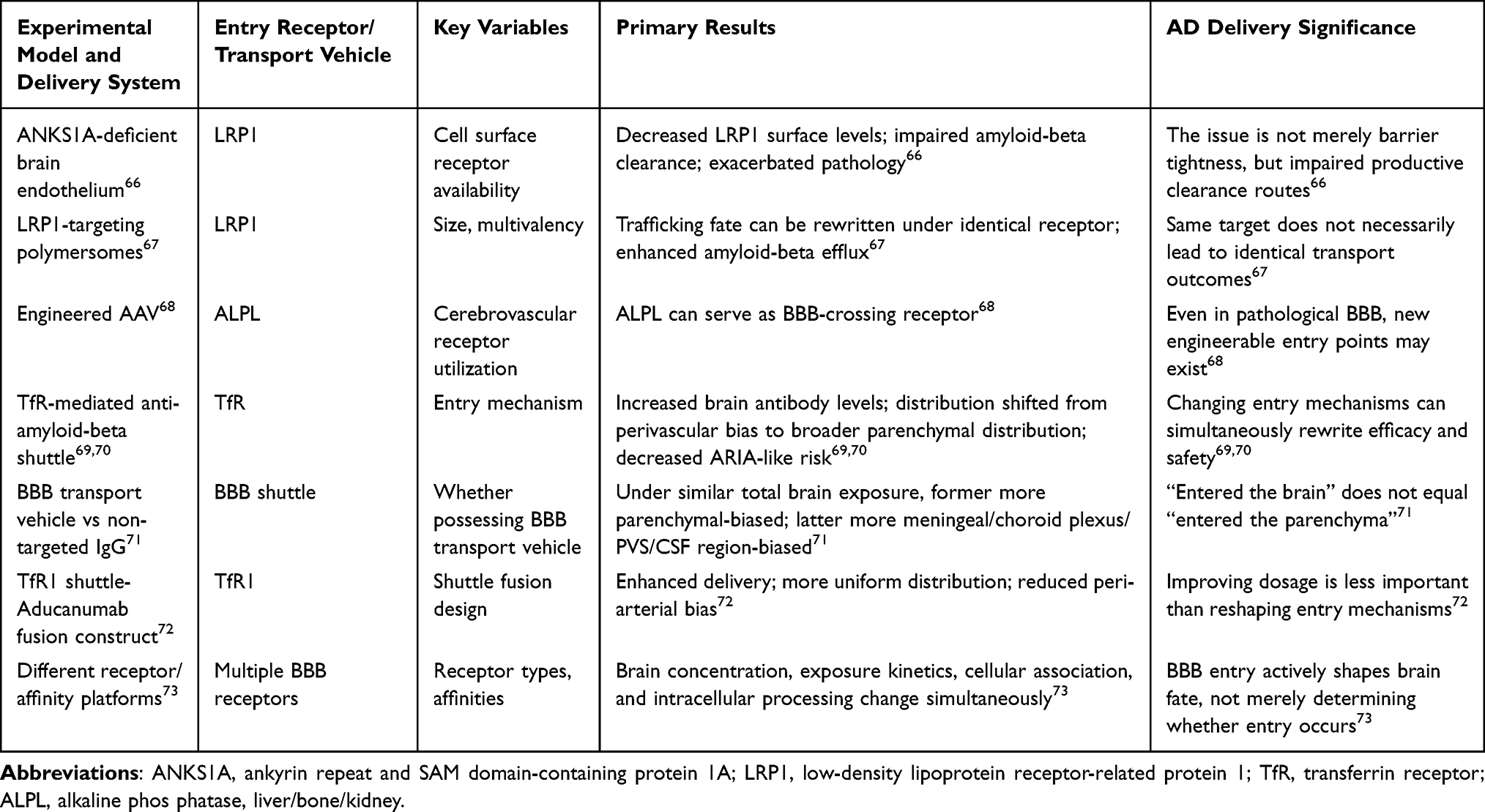 |
Table 6 Representative Studies Supporting “Pathway Reorganization and Spatial Redistribution” Rather Than “Simple Leakage Increase” |
Physicochemical Parameter Re-Ranking Under Pathological Glycocalyx: Size, Charge, Ligands, and Protein Corona
The brain endothelial glycocalyx contains glycosaminoglycans (GAGs), proteoglycans, sialylated glycans, and their hydration layers. It is electronegative and spatially sieving. Consequently, cargo properties—hydrated particle size, surface charge, and ligand presentation—are re-stratified at the luminal surface before the cargo reaches membrane receptors.10,24 If glycocalyx thickness, negative charge density, and mucin-type O-glycosylation undergo rearrangement in AD, many empirical rules summarized on healthy BBB may fail—not merely “efficiency reduction.”10,18,24
Regarding size, in vivo two-photon imaging suggests that glycocalyx itself serves as a pre-positioned sieving layer at the blood-facing side, with small molecules more easily entering while larger molecules are more restricted.10 This implies that what determines whether nanocarriers have opportunities to approach membrane surfaces involves not merely nominal particle size, but also hydrated size, outer layer flexibility, and early adsorption layers.
Regarding charge, classical BBB delivery often posits that positively charged systems more easily engage in adsorptive interactions with endothelium, but this heuristic assumes stable glycocalyx electronegativity; glycocalyx is precisely an important source of BBB negative surface charge, so when sialic acid or heparan sulfate (HS) and other anionic glycans change, nanosystems face no longer the same “electric field.”24 Existing evidence also suggests charge effects are not linear. In vitro endothelial studies demonstrate that mature glycocalyx can reduce cytotoxicity of certain cationic nanosystems, while some anionic systems display more obvious interactions only after glycan degradation.78 Brain endothelial-related research also indicates that removing surface sialic acid can alter cellular uptake of mildly negatively charged nanosystems.24 Therefore, “cations are more easily taken up” or “anions are safer” are not universal propositions; their validity depends upon glycocalyx maturity and composition.
Ligand modifications are similarly not exemption certificates. Porous silicon nanoparticle studies demonstrate that BBB crossing efficiency does not increase unidirectionally with ligand density, but is optimal under smaller particle sizes and moderate ligand density; excessive ligand density may instead limit crossing.79 The cARLA human BBB model demonstrates that BBB closer to in vivo phenotypes simultaneously possess higher glycocalyx density, stronger efflux activity, and lower endocytosis levels, therefore providing more reliable predictions for brain penetration of drugs and targeted nanoparticles.80 This implies that in models with more mature or in vivo-like glycocalyx states, many “high uptake” designs do not necessarily correspond to truly effective trans-BBB delivery.
Protein coronas are also not static backgrounds. ACS Nano research has demonstrated that nanoparticles undergo significant corona remodeling during BBB crossing, with blood-facing initial coronas not directly predictive of brain-side fates.81 If glycocalyx is further incorporated, issues become more complex: glycocalyx influences not merely particle near-wall residence and contact geometry, but may also alter corona exchange, adsorption, and desorption temporal sequences.10,24,81 Against this background, traditional design variables including size, charge, ligand density, protein corona, and intracellular fate should no longer be treated as independent parameters, but understood as a set of coupled factors re-ranked on pathological glycocalyx interfaces. Corresponding potential failure points and recommended evaluation readouts are summarized in Table 7. This signifies that AD-associated glycocalyx abnormalities alter not isolated steps, but the entire delivery logic chain from first contact to brain landing. Therefore, future BBB delivery design under AD conditions should not target exploiting “more leaky” barriers as objectives, but should focus on parsing and reconstructing recognition logic and transport programs of luminal interfaces following pathological glycocalyx remodeling.
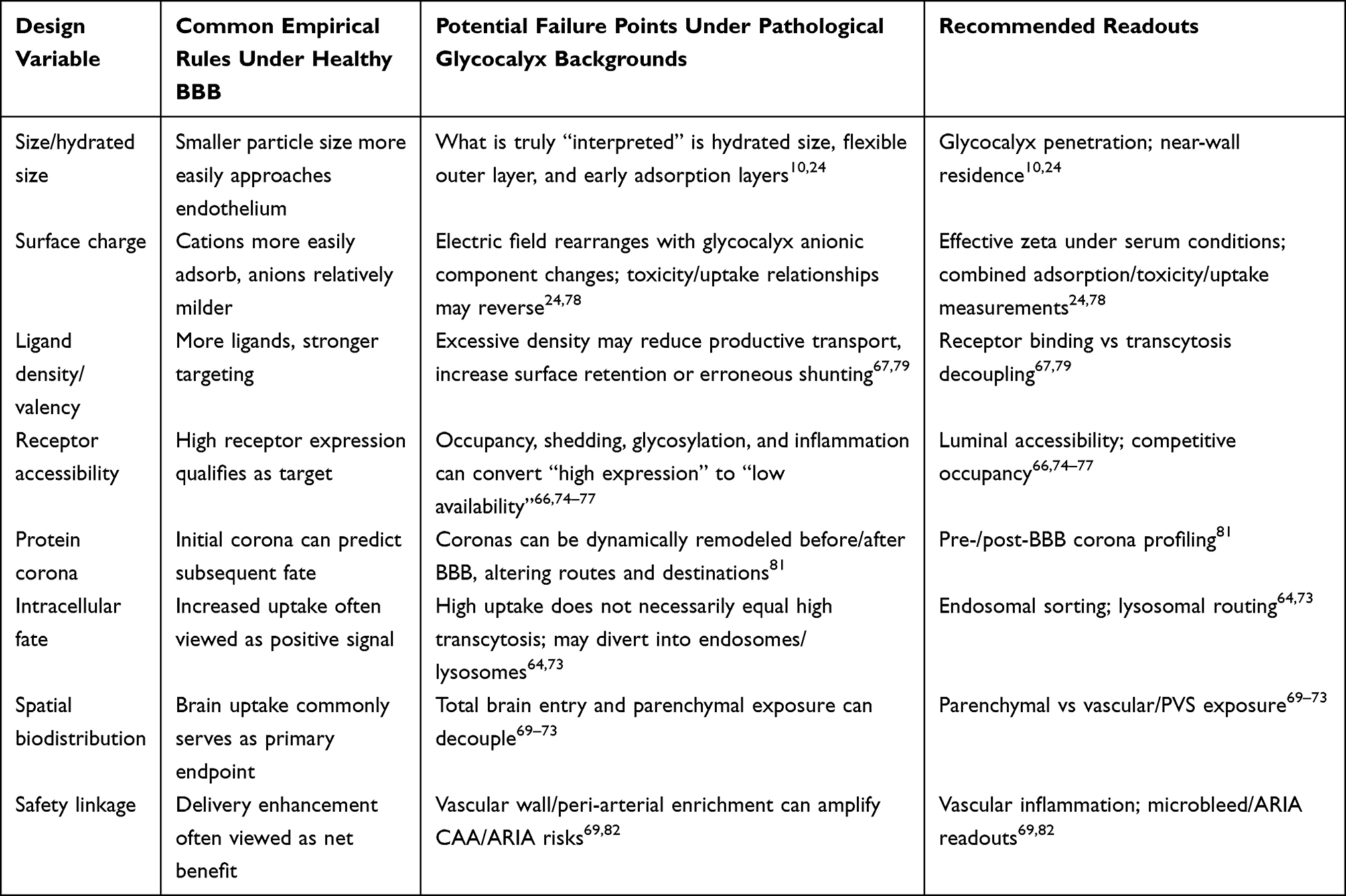 |
Table 7 Design Variable Re-Ranking for BBB Targeting and Nanodelivery Under Pathological Glycocalyx Backgrounds |
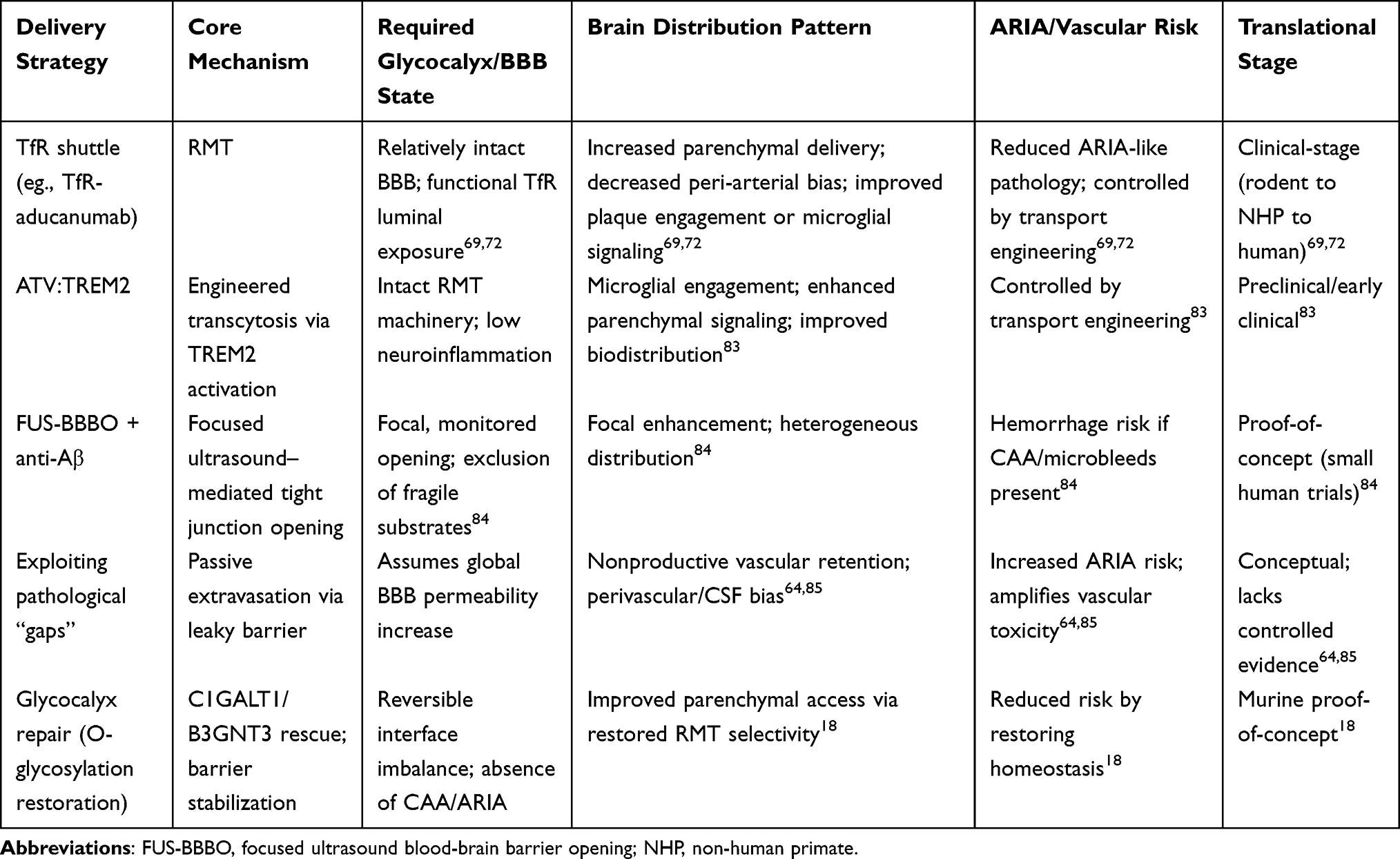 |
Table 8 Comparative Summary of BBB Delivery Strategies Under AD-Associated Glycocalyx Remodeling |
The foregoing analysis indicates that AD-associated glycocalyx remodeling does not create a uniform delivery window, but instead differentially rewrites the feasibility, distribution, and risk profiles of distinct BBB-crossing strategies. Table 8 provides a comparative summary of representative delivery modalities under glycocalyx-remodeled conditions, integrating their core mechanisms, required BBB states, brain distribution patterns, ARIA/vascular risk implications, and current translational stages. This comparison underscores why a one-size-fits-all exploitation of “leaky” barriers is untenable, and sets the stage for the risk-stratified, sequencing-based framework discussed in the following section.
Interface State Interpretation, Risk Stratification, and Translational Thresholds: Conversion Boundaries of Brain Endothelial Glycocalyx Abnormalities in AD
No Unified “Glycocalyx Delivery Window” Exists in Alzheimer’s Disease
Understanding damaged glycocalyx in AD as a unified, statically existing, brain-wide delivery window does not conform to existing evidence. This is because glycocalyx does not function in isolation, but is embedded within a dynamic interface shaped by endothelial subtypes, local fluid mechanics, brain region differences, disease stage, and neurovascular unit status. First, AD cerebrovascular endothelial cells themselves exhibit significant heterogeneity. Single-nuclear transcriptomics and multi-brain region vascular atlas studies demonstrate that endothelium, pericytes, and other vascular-associated cells present different inflammation, transport, aging, and angiogenic imbalance patterns across different brain regions, vascular segments, and genetic backgrounds.34,36 Therefore, glycocalyx is not the same coat covering “identical endothelium,” and its delivery consequences are inherently region- and vascular segment-specific.
Second, mechanical microenvironments further rewrite BBB and glycocalyx phenotypes. Shear stress, pulsatile strain, and substrate stiffness can all influence BBB integrity; flow closer to physiological conditions often enhances barrier properties, induces glycocalyx-related genes and more negative surface charge, while abnormally high shear or pulsatile flow, elevated substrate stiffness may impair barriers and promote inflammatory phenotypes.14,86–88 In AD, cerebral blood flow reduction, capillary constriction, immune cell stalling, and hypoxia further amplify this local mechanical heterogeneity.89 This implies that identical delivery systems face different glycocalyx operating points across different microcirculatory regions.
Third, brain region and vascular segment differences determine which luminal faces delivery systems first encounter. Human cerebrovascular single-cell atlases suggest extensive molecular program and endothelial-perivascular cell interaction differences across different vascular segments and brain regions.90 Imaging and pathology studies regarding AD also demonstrate that hippocampus, precuneus, parietal white matter and other regions exhibit inconsistent relationships between BBB injury, hypoperfusion, pericyte marker loss, and amyloid-beta burden.2,37,91 These results collectively negate simple narratives of “unified BBB/glycocalyx window existing in AD.”
Finally, disease progression is not linearly “later equals more open.” Early stages may already exhibit local BBB functional abnormalities and pericyte injury signals, while later stages more likely manifest as imageable leakage, inflammatory amplification, and elevated vascular fragility.2,37 Therefore, what is continuously switched in AD is not the size of the same window, but local rules regarding “which molecular classes more easily approach—adhere—cross—retain.” Based upon this inference, what is more meaningful is not “whether AD has a glycocalyx window,” but rather: which brain region classes, which vascular segments, which disease stages, and which vascular cell state/molecular subtype glycocalyx alterations will produce reproducible, directionally clear, and predictable impacts upon specific delivery systems.
Brain Endothelial Glycocalyx Abnormalities Should Be Interpreted Through Multidimensional Interface States
Incorporating brain endothelial glycocalyx abnormalities in AD into blood-brain barrier discussions occurs not because once damaged it leaves a simple gap for drug delivery “drilling,” but because it resides at the frontline of BBB-blood contact, simultaneously influencing physical/charge barriers, inflammatory adhesion thresholds, membrane morphology, trans-endothelial transport behaviors, and fluid mechanical perception.18,21,43,86 Recent studies have defined brain endothelial glycocalyx as the first interface between blood and cerebrovascular vessels,18 noting that within BBB hierarchical structures, endothelial glycocalyx resides before tight junctions.21 Concurrently, reviews regarding BBB mechanobiology further emphasize that shear stress, cyclic strain, tissue stiffness, and ECM remodeling actively rewrite BBB states, with glycocalyx representing one of the foremost blood-facing mechanosensory layers.43,86 Therefore, if “whether BBB leaks” remains the core question, it becomes difficult to accommodate new evidence suggesting active remodeling of BBB frontline interfaces.
Previous extensive BBB literature indeed tended to approximate barrier abnormalities as tight junction destruction, or to employ gadolinium, sucrose, or albumin extravasation as primary readouts.2,18,92 However, for brain endothelial glycocalyx, elevated permeability should be viewed as one outcome of interface abnormalities rather than the interface state itself. When the glycocalyx thins, sheds, or undergoes glycosylation program rewriting, the earliest changes may not be large-scale extravasation. Instead, the interface rules change: which molecules can approach the endothelium; which transport pathways are mobilized; which inflammatory cells adhere more easily; and which mechanical stimuli are amplified into endothelial signals.18,21,43,86 In other words, from an evaluative perspective, “leakage” is merely a downstream readout rather than the sole standard for adjudicating blood-facing interface states.18,21,43,86 This also aids in re-understanding long-standing opposing conclusions in the AD-BBB field. Some human studies suggest BBB changes can appear at preclinical or mild cognitive impairment (MCI) stages, partially independent of classical amyloid-beta and tau biomarkers, with DCE-MRI studies reporting hippocampal and medial temporal lobe-predominant BBB disruption or leakage burden increases in normal aging, MCI, early AD, and APOE4 carriers.2,29,31,92 However, other cohorts do not support “AD necessarily accompanied by extensive, brain-wide, passive leakage”: some studies only observed hippocampal Ktrans elevation in AD dementia stages rather than MCI stages, with CSF sPDGFRβ more associated with age and neuroinflammation rather than directly corresponding to cognitive groupings.37,93 In Tg2576 models, stable isotope-labeled [13C12]sucrose also showed no significantly enhanced small molecule passive paracellular flux in whole brain or hippocampus, cortex, or cerebellum regions.85 These results suggest that rather than mechanically questioning “whether AD BBB leaks or not,” it is better to further question which layer, stage, and pathological substrate of BBB destabilization different studies actually measured.
The above divergences first arise because measured objects are not identical. Ktrans in DCE-MRI reflects contrast agent slow extravasation kinetics in specific regions.2,92 sPDGFRβ more closely approximates fluid biomarkers for BBB-associated pericyte injury rather than simple equivalents of barrier permeability at arbitrary molecular scales.37,92,93 CSF/serum albumin ratio (QAlb) biases toward larger molecule-level barrier abnormalities, inherently insensitive to low-grade, subtle paracellular leakage.64 [13C12]sucrose primarily tests small hydrophilic molecule passive trans-barrier flux.37,85 Therefore, equating “pericyte injury,” “regional gadolinium extravasation,” “large molecule plasma protein leakage,” and “small molecule passive paracellular flux changes” as synonymous expressions is not rigorous.2,37,64 Second, spatial scales also alter answers. Existing human evidence more commonly suggests regional, low-grade, chronic BBB destabilization rather than brain-wide synchronous, dramatic, passive “permeabilization.”2,29,31,92 Conversely, Tg2576 research primarily negates “significantly enhanced passive flux for sucrose in whole brain range or plaque-adjacent regions,” rather than all vascular pathological changes.85 Therefore, “focal hotspot destabilization” and “global small molecule passive integrity maintenance” can simultaneously hold.29,85 Third, AD itself is not a vascular-level homogeneous disease label. Age, APOE4, cerebral amyloid angiopathy (CAA), inflammatory background, metabolic comorbidities, and disease stage may all shape different BBB dysfunction phenotypes, so different cohorts or models likely are not converging toward identical BBB endpoints. Finally, glycocalyx “invisibility” itself is a bias source. Brain endothelial glycocalyx is extremely fragile and often difficult to preserve faithfully under conventional fixation and staining conditions; studies have noted that under conditions lacking lanthanum nitrate staining, brain capillary luminal glycocalyx is nearly invisible, while modified perfusion fixation combined with lanthanum staining renders its position and morphology clearly visible.9 Therefore, at the glycocalyx level, “abnormalities not observed” does not necessarily equal “glycocalyx truly without abnormalities.”
Consequently, the discussion object needs to be elevated from single permeability readout indicators to “interface states.” Table 9 summarizes the interface state interpretation framework and translational implications for brain endothelial glycocalyx abnormalities in AD.
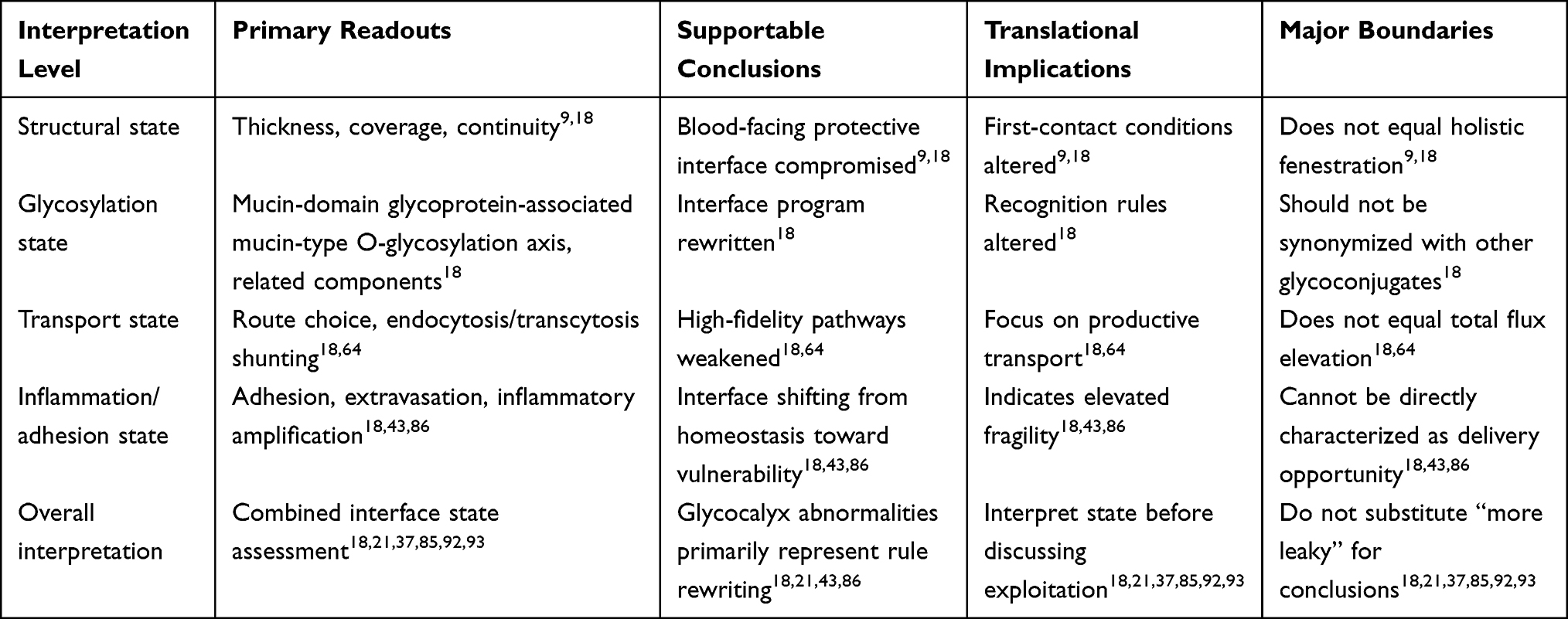 |
Table 9 Interface State Interpretation Framework and Translational Implications for Brain Endothelial Glycocalyx Abnormalities in AD |
For brain endothelial glycocalyx abnormalities, at least four classes of mutually related yet translationally distinct interface phenotypes can be distinguished. The first class is structural phenotypes, namely glycocalyx thinning, coverage decline, and continuity impairment. Studies have demonstrated that brain capillary glycocalyx under physiological conditions already possesses relatively high coverage and longer fiber structures, suggesting it undertakes stronger pre-positioned sieving functions in BBB than peripheral organs.9 Under aging backgrounds, both brain endothelial glycocalyx thickness and area significantly decline.18 Such structural thinning implies that plasma proteins, circulating inflammatory mediators, and cell surface adhesion molecules more easily approach endothelial membranes.9,18 However, it does not automatically equate to immediate tight junction collapse, nor can it be directly extrapolated as “safely exploitable delivery opportunity.”9,18 The second class is glycosylation phenotypes, namely glycosylation program rewriting, particularly mucin-type O-glycosylation downregulation. Related studies demonstrate downregulation of C1galt1, B3gnt3, and other pathways related to glycan extension in aged cerebrovascular endothelium, while C1GALT1 and various mucin-domain glycoproteins are simultaneously reduced in human AD brain microvasculature.18 The significance of this change extends beyond mere “glycan reduction” to qualitative rewriting of luminal interface molecular grammar: mucin-domain glycoproteins constitute not merely passive gel layers, but participate in cell signaling, cell-cell interactions, and membrane morphology maintenance.18 Therefore, O-glycosylation decline likely causes qualitative changes in interface recognition rules, exclusion rules, and response rules rather than merely quantitative thickness loss.18 The third class is transport phenotypes, namely shifts from relatively specific trans-endothelial transport toward low-selectivity endocytosis/pinocytosis spectra. Aged BBB has been demonstrated to shift from receptor-mediated transcytosis (RMT) dominant states toward more caveolar transcytosis-dependent nonspecific trans-endothelial entry.64 Under glycocalyx imbalance backgrounds, ALPL and caveolin-1 (CAV1) upregulation also suggest glycocalyx abnormalities may reside in the same pathological chain as this transport spectrum shift.18 From a delivery perspective, this point requires particularly cautious interpretation: more entry does not automatically equal higher therapeutic index, because when entry depends on enhanced low-specificity pinocytosis/endocytosis, drugs, plasma proteins, inflammatory mediators, and potential toxicity burdens may synchronously increase.18,64 In contrast, redirecting BBB delivery through transport engineering rather than passively relying on pathological damage may more favorably improve brain distribution and reduce risk while overall BBB structure remains relatively intact.69 The fourth class is inflammation/adhesion phenotypes, namely leukocyte approach to endothelium, plasma protein extravasation, and decreased inflammatory amplification thresholds. Glycocalyx resides at the blood-facing first line, with its integrity determining the distance and probability of circulating cells and proteins approaching endothelial membranes.18,43 In related models, whether brain endothelial-specific downregulation of C1galt1 or exogenous cleavage of mucin-domain glycocalyx, both can induce albumin and IgG extravasation, tight junction abnormalities, and neuroinflammation; under more severe injury conditions, this can further progress to cerebral hemorrhage.18 This suggests glycocalyx abnormalities represent not merely “pore enlargement,” but rather a process beginning at the blood-facing side and progressively lowering thresholds for inflammation and injury entry.18,43
Based upon these four phenotype classes, it can be further understood why “local destabilization” does not equal “global rupture,” and why not all forms of BBB abnormalities can be equated as exploitable delivery windows. Experimental studies suggest that short-term removal of mucin-domain glycoproteins can cause BBB permeability increase, but sustained injury is required before more extensive BBB dysfunction or even cerebral hemorrhage occurs.18 In population studies, early cognitive impairment and early AD can exhibit BBB leakage in hippocampus and other regions.2,29,92 However, this temporal sequence has not been consistently repeated in all subsequent cohorts, with some studies instead only observing relatively clear hippocampal leakage in AD dementia stages, and sPDGFRβ showing stronger associations with age and neuroinflammation than with cognitive subgrouping.37,93 Therefore, regional hotspot leakage, pericyte injury, glycocalyx remodeling, transport spectrum shifts, and extensive barrier collapse do not possess necessary synchronous relationships.2,37,92,93 This point carries particular importance when coexistent with CAA, microhemorrhages, or amyloid-related imaging abnormalities (ARIA) susceptibility backgrounds. CAA is characterized by vascular wall amyloid-beta deposition, small vessel remodeling, and hemorrhage tendencies, constituting important underlying pathology for lobar hemorrhage in aged brains.82,94 ARIA, particularly hemorrhagic ARIA-H, indicates that under anti-amyloid immunotherapy backgrounds, vulnerable balances exist among vascular wall stress-bearing capacity, perivascular inflammation, and amyloid clearance dynamics.82 Under such circumstances, any phenomenon appearing as “more entry” should first be interpreted as interface plasticity alteration or vascular wall homeostasis reserve decline; the latter typically represents not opportunity expansion, but safety boundary reduction.
It must be emphasized that brain endothelial glycocalyx in AD is no longer merely a speculative concept, yet current-stage human evidence should still be expressed cautiously. Existing studies have observed C1GALT1 and mucin-domain glycoprotein staining declines in AD group brain microvascular samples, and observed downregulation of endothelial mucin-type O-glycan biosynthetic pathways in independent human brain single-nuclear RNA sequencing datasets.18 This renders brain endothelial glycocalyx abnormalities sufficient to be regarded as strong mechanistic candidates in AD-BBB research.18 However, causal manipulations remain primarily completed at animal levels, with brain endothelial-specific downregulation of C1galt1 causing albumin and IgG leakage; conversely, restoring core 1 mucin O-glycans can improve aged mouse BBB function, alleviate neuroinflammation, and ameliorate cognitive deficits.18 Therefore, current brain endothelial glycocalyx abnormalities have entered the core mechanistic layer of AD-BBB research, yet “human causal closure loop” remains uncompleted; animal functional causal results should not be directly extrapolated as human clinical conclusions.
In summary, under this framework, brain endothelial glycocalyx abnormalities in AD should not be simply equated with “BBB already opened,” but more accurately stated as glycocalyx abnormalities primarily meaning BBB blood-facing interface rule rewriting. Consequently, so-called “opportunities” do not equate to any form of BBB permeability elevation, but specifically refer to interface imbalance states still possessing reversibility, dominated by endothelial glycocalyx, not yet evolving into hemorrhagic vascular fragility. Correspondingly, subsequent discussion priorities are no longer general judgments of whether BBB is more leaky, but further distinctions: which glycocalyx abnormalities can still be incorporated into repair and transport engineering frameworks, and which abnormalities should rather be interpreted as risk stratification and contraindication identification signals.
When Brain Endothelial Glycocalyx Abnormalities Constitute Treatment/Delivery Opportunities versus Merely Indicating Vascular Fragility
Whether AD brain endothelial glycocalyx abnormalities should be defined as treatment/delivery opportunities depends not upon whether the blood-brain barrier is “more leaky,” but rather upon whether the corresponding vascular substrate still belongs to plastic, reversible interface imbalance, or has already entered structurally fragile and hemorrhage-prone states. Based upon previous characterizations of brain endothelial glycocalyx and BBB dysfunction heterogeneity,18,20 the key to so-called “glycocalyx opportunity windows” in AD lies not in seeking locations with largest barrier gaps, but in recognizing which patients and stages remain dominated by endothelial interface imbalance, and whether such imbalance still possesses repairability and translational significance.
The first layer of basis supporting “opportunity validity” is that in certain populations, BBB abnormalities can appear earlier than, or at least partially independent of, classical amyloid-beta/tau readouts. DCE-MRI studies demonstrate that hippocampal BBB permeability can elevate in aged populations, further worsening in mild cognitive impairment (MCI), correlating with BBB-associated pericyte injury indicators.31 Subsequent human studies further suggest that early cognitive function decline individuals already exhibit hippocampal and parahippocampal gyrus capillary injury and BBB disruption, and this change does not attach to cerebrospinal fluid (CSF) amyloid-beta or tau alterations.2 Additional studies demonstrate that apolipoprotein E ε4 (APOE4) carriers exhibit hippocampal and medial temporal lobe BBB disruption even at pre-symptomatic cognitive stages, with baseline CSF sPDGFRβ elevation predicting subsequent cognitive decline, this predictive effect statistically independent of amyloid-beta and tau burdens.31 These results collectively suggest that in considerable individual subsets, BBB destabilization is not merely a late-stage concomitant phenomenon, but may enter disease courses earlier.2,29,31,95 However, this conclusion should not be absolutized. Another population study demonstrated that hippocampal BBB disruption is primarily observed in dementia stages rather than MCI stages, with sPDGFRβ more obviously increasing with age and neuroinflammation, without significant correspondence with overall AD biomarker positivity status or APOE status.37 Therefore, at early AD stages, BBB abnormalities do not manifest as homogeneous changes experienced by all patients, but more tend to constitute vascular phenotypes enriched in specific subgroups across the aging-APOE4-MCI-early AD continuum.
The second layer, and more critically, basis supporting “opportunity validity” lies in glycocalyx abnormalities representing not merely passive concomitant changes, but possessing causal plasticity and a certain degree of reversibility. Shi et al’s research on brain endothelial glycocalyx structural and compositional changes suggests that glycocalyx abnormalities represent not “already leaked, therefore exploitable,” but rather “blood-facing interface imbalance can still be upstream-reshaped, and this reshaping can reciprocally influence BBB, inflammation, and partial cognitive readouts”.18 However, it must be equally emphasized that current strongest “glycocalyx reversibility” evidence derives primarily from aging models rather than specifically typical AD pathology load-designed treatment models; the more circumspect current conclusion is that brain endothelial glycocalyx may constitute a key axis for AD vascular pathology, yet remains insufficient to be regarded as a mature target fully validated in AD treatment models.
This judgment receives lateral support from multiple parallel recent studies. Different studies, though not all directly pointing to glycocalyx, converge from different sections toward the same conclusion: that “endothelial interface repair” itself may be one of the main axes for disease course intervention. For example, endothelial ANGPT2 elevation in patient AD endothelium correlates with disease severity, with endothelial-specific deletion of Angpt2 in 5xFAD mice alleviating amyloid-beta burden and improving cognition, while its overexpression exacerbates plaque load and memory damage.96 In APP/PS1 models and in vitro brain endothelial systems, death receptor 6 (DR6) overexpression can rescue amyloid-beta-induced barrier dysfunction through Wnt/β-catenin and JNK pathways, and upregulate claudin-5, glucose transporter 1 (Glut-1), and ZO-1.97 During natural aging, gap junction protein 43 (CX43) decline in Cdh5+ cerebrovascular cells constitutes an important node in BBB aging networks, with NAD+ supplementation or PARP1 inhibition capable of reversing related injuries.98 Human AD cerebrovascular cell single-nuclear transcriptome analysis demonstrates that endothelial cells are enriched for AD susceptibility genes, amyloid-beta increase accompanies BBB injury, angiogenic failure, vascular inflammatory activation, aging and apoptosis, accordingly proposing that reducing vascular inflammatory activation and restoring effective angiogenesis may alleviate early AD vascular dysfunction.34 These results do not possess uniform evidentiary strength, nor can they substitute for one another, but their directional consistency suggests: in at least certain AD/aging-associated cerebrovascular phenotypes, what is truly worth exploiting is not “gaps,” but repairable endothelial interfaces.34,96–98
Therefore, conditions under which brain endothelial glycocalyx abnormalities constitute treatment/delivery opportunities should be more strictly defined as following scenarios: first, abnormalities appear early and superficially, primarily manifesting as blood-facing interface imbalance and endothelial plasticity decline, rather than already entering vascular wall structural fragility stages; second, related changes still possess reversibility, meaning glycocalyx/glycosylation repair can improve BBB integrity, alleviate inflammation, and bring system-level benefits.18 Third, delivery design does not premise upon “exploiting pathological leakages,” but can rewrite entry rules upon relatively intact BBB, thereby improving effective brain parenchymal exposure and maintaining acceptable safety boundaries.20,69 This also implies that for AD, so-called delivery benefits cannot be defined by “whether permeability is elevated,” but should be defined by “whether drugs can reach effective brain parenchymal exposure in controllable, reproducible, low-hemorrhage-risk manners, and thereby enhance therapeutic index.”
This point carries importance because the most common misjudgment in AD is precisely misreading “interface imbalance” as “channel amplification.” Core changes in aged BBB may not be simply “more leaky,” but rather trans-endothelial transport rule rewriting. Previous studies suggest that trans-endothelial transport can shift from ligand-specific receptor-mediated transport toward nonspecific caveolar transcytosis, while healthy brain physiological uptake of endogenous plasma proteins actually declines during aging.64 For brain endothelial glycocalyx, this implies that after O-glycosylation abnormalities, what is first rewritten may be recognition and sorting logic for circulating ligands, antibodies, or carriers, rather than an abstract “leakiness” indicator.18,64 Similarly, small molecule tracer extravasation cannot be directly extrapolated as controllable brain entry for macromolecular drugs, therapeutic antibodies, or nanocarriers. Tg2576 model studies applying stable isotope-labeled [13C12]sucrose demonstrate that even with amyloid-beta deposition and CAA, BBB passive sucrose crossing remains extremely low overall; local amyloid-beta-associated tight junction morphological abnormalities do not automatically convert to elevated sucrose concentrations in vascular amyloid-beta deposition regions.85 Therefore, glycocalyx abnormalities, tight junction abnormalities, plasma protein extravasation, and therapeutic antibody exposure do not belong to the same evidentiary hierarchy, and cannot be generically compressed as “BBB more open, therefore delivery more favorable.”18,64,85 Similarly, focal, short-term, rigorously monitored artificial opening cannot be extrapolated as routine windows safely callable long-term and repeatedly. Focused ultrasound (FUS) combined with aducanumab human proof-of-concept studies indeed demonstrate the feasibility of focal BBB opening combined with anti-amyloid therapy, but this study possesses extremely small sample sizes, and implements strict risk control through excluding APOE ε4 carriers and limiting aducanumab dosages, therefore proving “focal enhancement feasible” rather than “AD possesses universally, safely, and repeatedly exploitable pathological window-opening opportunity.”84 Conversely, recent engineered trans-BBB transport studies suggest that under many scenarios, maintaining relatively intact BBB and rewriting entry rules through receptor-mediated transport may obtain higher therapeutic indices than exploiting pathological leakages. TfR-targeting anti-amyloid-beta antibodies can improve brain distribution, enhance parenchymal target engagement, and reduce ARIA-like pathology and vascular inflammation in mice.69 ATV:TREM2 research similarly does not rely upon pathological extravasation, but improves brain biodistribution and enhances TREM2 signaling through enhanced trans-BBB transcytosis.83 Such results collectively suggest that glycocalyx significance extends beyond merely “whether passive gaps appear after damage,” to whether the interface rules it represents can be repaired or rewritten. Relative to “opportunity validity,” core scenarios where glycocalyx abnormalities merely indicate fragility rather than exploitable windows are vascular substrates already dominated by CAA, hemorrhage-prone, or inflammation-prone states. For this, the most important clinical reference is not general BBB leakage, but amyloid-related imaging abnormalities (ARIA) under anti-amyloid immunotherapy backgrounds. Nature Reviews Neurology in 2025 has defined ARIA as one of the most influential adverse reactions for anti-amyloid immunotherapy, noting that imaging manifestations, risk factors, neuropathology, and animal model evidence collectively point to the central position of CAA.82 This implies that once AD cerebrovascular pathology has shifted from “endothelial interface imbalance” to “vascular walls themselves becoming primary lesions,” then “more entry” no longer automatically means “more favorable therapy,” because barrier abnormalities at this stage primarily represent declining vascular wall stress-bearing capacity rather than rising delivery efficiency.82 Differences in amyloid-beta antibody binding to human CAA fibrils among different drugs also provide directional support: when therapeutic targets couple more strongly with vascular wall amyloid substrates, clinical benefits and vascular side effects often may amplify simultaneously.99 It must be emphasized that ARIA incidence rates among different drugs are not head-to-head comparisons, and cannot be interpreted as strict risk rankings, but their overall direction suggests that CAA presence substantially rewrites the meaning of interface abnormalities, bringing them closer to vascular fragility characterization rather than delivery advantages.
Against this background, baseline microbleeds (cerebral microbleeds, CMBs), cortical superficial siderosis (cSS), convexity subarachnoid hemorrhage (cSAH), APOE ε4 homozygous backgrounds, and previous/potential ARIA susceptibility should not be understood as “minor, acceptable concomitant abnormalities,” but rather as visible edges of occult microvascular disease and fragile substrates. FDA prescription information regarding lecanemab and donanemab both indicate that baseline presence of at least 2 microbleeds or at least 1 cSS location has been identified as ARIA risk factors, while >4 microbleeds, previous larger cerebral hemorrhage, obvious cSS, or vasogenic edema and other imaging manifestations are typically regarded as exclusion criteria.100–105 More importantly, case pathology demonstrates that clinically observed MRI microbleeds may represent only the lowest visible expression of microvascular disease burden, not reflecting true scope; in a lecanemab-related fatal β-amyloid-related arteritis case, 4 cortical microbleeds and possible CAA evidence existed before treatment, with rapid progression to extensive ARIA-E/ARIA-H and eventual death post-treatment.106 From a population perspective, CMB prevalence co-enriches with age, amyloid pathology, cognitive status, and APOE ε4 copy number.107 Therefore, among AD true vascular substrates, microbleeds should not be interpreted as “higher endothelial permeability,” but should be prioritized as “vascular wall integrity has been eroded.”
ARIA and related vasculitis-like reactions further demonstrate that certain BBB abnormalities, the more obvious they are, the less they should be treated as delivery windows. Although most ARIA events are mild or asymptomatic, approximately 5% can be severe enough to cause hospitalization, permanent disability, or even death.82 Autopsy and multi-omics studies suggest that some lecanemab-related ARIA contain not merely passive leakage, but may be accompanied by obvious vasculitic injuries, cytotoxic CD8+ T cell clonal expansion, glycolytic reprogramming, and vascular tropism immune characteristics.106,108 These results are insufficient to alone rewrite clinical stratification standards, but they clearly demonstrate: when fragile vascular substrates encounter superposition of antigen clearance, amyloid-beta mobilization, and immune activation, outcomes involve not merely “barrier more open,” but possibly “vasculitic injuries and hemorrhage risks amplified.”82,106,108 This clinical chain and Shi et al’s mechanistic results echo each other directionally: the extreme endpoint of brain endothelial glycocalyx/O-glycosylation abnormalities is not “easier drug delivery,” but BBB dysfunction, inflammatory amplification, and cerebral hemorrhage.18 Therefore, the terminal boundary of glycocalyx abnormalities is not a larger opportunity window, but danger signals after threshold collapse.
In summary, brain endothelial glycocalyx abnormalities can be regarded as treatment/delivery opportunities only under conditions of “still early, still superficial, still reversible.” Conversely, when abnormalities are superimposed with CAA, baseline microbleeds, cSS/cSAH, APOE ε4 high-risk backgrounds, ARIA susceptibility, or vasculitis-like reaction tendencies, their primary meaning is no longer “drugs more easily enter,” but rather “vasculature more easily develops problems.” Based upon this stratification, this review further discusses translational thresholds and strategy sequencing: when repair should be prioritized, and when cautious exploitation may be permitted.
Translational Thresholds and Strategy Sequencing: When to Prioritize Repair, When Cautious Exploitation May Be Permitted
As previously indicated, the translational significance of AD brain endothelial glycocalyx abnormalities does not depend upon whether the BBB exhibits higher “leakiness,” but rather upon whether the corresponding vascular substrate still belongs to plastic, reversible interface imbalance, or has already entered stages characterized by structural fragility, inflammatory amplification, and hemorrhage susceptibility. In other words, what truly requires answering is not “whether drugs more easily enter after glycocalyx damage,” but rather: does this abnormality still belong to upstream-dominated, reversible, and reshapeable blood-facing pathology, or has it already entered dangerous stages characterized by vascular wall structural fragility, inflammatory amplification, and hemorrhage susceptibility. Only under the former scenario can glycocalyx abnormalities constitute therapeutic targets, and even under strictly constrained conditions provide clues for delivery design; once coupled with CAA, microbleeds, or ARIA susceptibility, their primary meaning shifts from “more entry” to “higher risk.” Thus, the translational significance of AD brain endothelial glycocalyx abnormalities should be established upon comprehensive interpretation of interface states, vascular substrates, and safety boundaries; accordingly, distinctions can be made regarding whether they are more suitable to be regarded as repair targets, delivery clues, or high-risk states inappropriate for further exploitation.
Minimum Validity Conditions for Repair-Type Research
To define brain endothelial glycocalyx abnormalities as repair targets worth prioritizing in AD, the first threshold is that evidence must fall upon brain endothelium itself, and cannot miswrite peripheral circulating glycocalyx shedding as brain interface pathology. Syndecan-1 (Sdc-1), heparan sulfate (HS), and hyaluronic acid (HA) and other molecules do not possess brain specificity; their elevations can only first indicate systemic endothelial stress, not directly proving brain endothelial glycocalyx as the primary lesion site for BBB destabilization. The methodological representativeness of Shi et al lies precisely in evidence simultaneously falling upon brain microvascular imaging, acute brain microvascular isolation staining, brain endothelial transcriptomics, and endothelial-specific AAV manipulation levels, thereby more clearly distinguishing “brain endothelial glycocalyx abnormality” and “systemic endothelial injury” as non-equivalent propositions.18,109–111 Therefore, any repair-type research relying solely upon peripheral blood shedding biomarker elevation to rise to the level of “brain glycocalyx repair” still possesses insufficient evidentiary chains.
The second threshold requires that research cannot vaguely generalize “barrier damage” to summarize BBB phenotypes, but must specifically clarify which layer glycocalyx abnormalities have rewritten in the BBB. As previously noted, BBB destabilization does not possess only one form, manifesting potentially as paracellular leakage, transcellular transport spectrum reprogramming, enhanced leukocyte adhesion, decreased inflammatory thresholds, or fluid mechanical perception and shear response imbalances.18,20,21,43,64,86 For brain endothelial glycocalyx, its pathological consequences may not be immediately forming a “gap,” but more likely first rewriting blood-facing interface structural order, molecular recognition rules, and membrane response logic, subsequently influencing BBB function, inflammatory recruitment, and later-stage hemorrhage phenotypes.18 Therefore, glycocalyx-directed repair should not be characterized as vague “improving barrier integrity,” but should be more precisely defined as restoring glycocalyx shielding and anti-adhesion functions, correcting abnormal transcellular transport shifts, alleviating inflammatory adhesion amplification, and reconstructing interfaces with mechanosensation closer to physiological states.
The third threshold requires that disease stage and vascular substrate must be simultaneously stratified, rather than merely using “AD patients” as a homogeneous label. Existing human evidence suggests that hippocampal and medial temporal lobe BBB-associated changes can appear in aging, APOE4 carriers, MCI, and even early AD spectra, but other studies demonstrate that relatively clear hippocampal leakage is primarily observed in AD dementia stages, with sPDGFRβ more obviously associated with age and neuroinflammation.2,31,37 Therefore, so-called “repairable interfaces” do not synchronously exist in all AD patients, but more likely enrich in specific vascular phenotypes across the aging-APOE4-MCI-early AD continuum. Correspondingly, repair-type research should at minimum distinguish two substrate classes: one primarily characterized by endothelial interface imbalance, pericyte stress, and local BBB functional abnormalities, not yet entering obvious hemorrhagic fragility states of early spectra; the other being high-risk vascular spectra already merged with CAA, microbleeds, cSS/cSAH, or anti-amyloid immunotherapy-related fragility.2,20,37 Without first distinguishing vascular substrates, so-called “glycocalyx repair” can easily conceptually point toward two completely different pathological objects simultaneously.
The fourth threshold requires that repair-type research cannot remain merely at glycocalyx molecule or BBB indicator improvements, but must establish “interface improvement—brain outcome improvement” dual-endpoint closure. In other words, if an intervention can only restore glycan-related molecular expression, reduce local leakage, or improve single BBB readout indicators, but cannot synchronously alleviate neuroinflammation, improve brain network function, or bring behavioral/cognitive benefits, then its translational significance remains incomplete. The importance of Shi et al lies not merely in demonstrating that core 1 mucin-type O-glycan restoration can improve BBB function, but further showing that it can reduce neuroinflammation and alleviate cognitive deficits, thereby connecting “interface repair” and “brain outcome” as a relatively complete functional chain.18 For AD, such closure is particularly necessary because BBB repair itself is not an endpoint, but merely one link in systematic brain pathology intervention chains.20
The fifth threshold lies in avoiding “interface centralism.” While brain endothelial glycocalyx abnormalities may represent key amplifiers in AD vascular pathology, their repair cannot be characterized as a “master key” sufficient to independently resolve AD complex disease courses. Increasingly more studies suggest that cerebrovascular endothelial interface repair may indeed be one of the important axes for disease course intervention: for example, ANGPT2 upregulation correlates with AD brain endothelial abnormalities and disease severity, with endothelial-specific deletion of Angpt2 alleviating amyloid-beta burden and improving cognition.96 DR6 overexpression can rescue amyloid-beta-induced barrier dysfunction in APP/PS1 models and in vitro brain endothelial systems, and improve multiple barrier-related molecular expressions.97 CX43 decline in cerebrovascular cells constitutes an important node in BBB aging networks, with NAD+ supplementation or PARP1 inhibition capable of reversing related injuries.98 Human AD cerebrovascular single-nuclear transcriptomes also suggest that endothelial inflammatory activation, aging, apoptosis, and angiogenic imbalance may collectively participate in vascular dysfunction.34 The common direction of these evidences is not “interface determines everything,” but rather suggesting that brain endothelial interface repair should be incorporated into AD comprehensive treatment frameworks, considered synergistically with amyloid-beta/tau-targeted therapies, anti-inflammatory interventions, vascular risk factor management, and trans-BBB transport platforms, rather than being isolatedly elevated as a single central strategy.
Additional Safety Thresholds for Exploitation/Delivery-Type Research
Compared with repair-type research, if one further asserts that AD brain endothelial glycocalyx abnormalities constitute “exploitation-type” or “delivery-type” opportunities, then required evidentiary thresholds should be markedly higher. The core does not lie in proving certain molecules “enter more,” but in proving that such entry can be converted into controllable, reproducible, low-hemorrhage-risk effective brain parenchymal exposure, thereby enhancing therapeutic indices rather than synchronously amplifying inflammatory burdens, vascular toxicity, or local hemorrhage risks.20,64,69,83–85 Therefore, delivery-type propositions must logically pass at least three additional screenings.
First, obvious hemorrhage-prone substrates must be excluded. AD is not merely neuronal pathology, but often superimposes CAA, lobar microbleeds, cortical superficial siderosis (cSS), and varying degrees of ARIA susceptibility.82,99–107 Under these fragile vascular backgrounds, “more entry” often primarily means declining vascular wall stress-bearing capacity and contracting safety margins, rather than stably exploitable pharmacokinetic advantages. Particularly under anti-amyloid immunotherapy backgrounds, ARIA has been explicitly regarded as one of the most influential adverse reactions, with imaging, neuropathology, and animal model evidence all pointing to the central role of CAA.82 Thus, once glycocalyx abnormalities are superimposed with CAA, baseline microbleeds, cSS/cSAH, APOE ε4 high-risk backgrounds, or previous/potential ARIA susceptibility, their most important clinical meaning should shift from “window” to “fragility marker.” Under such conditions, continued emphasis on exploitation can easily misjudge safety boundary decline as delivery opportunity expansion.
Second, exploitation-type research must utilize real cargo, and cannot substitute single tracer or low-dose tracer molecule results for actual exposure under therapeutic doses and therapeutic configurations. Brain delivery success highly depends upon molecular size, valency, affinity, dosage, administration frequency, and distribution patterns within brain parenchyma.112 Therefore, local extravasation of small molecule tracers, slow extravasation of local imaging contrast agents, or even extravasation of certain plasma proteins cannot be directly extrapolated as controllable, optimized brain parenchymal exposure for therapeutic antibodies, bispecific molecules, or nanocarriers.64,85 This is also why the result that [13C12]sucrose passive flux remains extremely low overall in Tg2576 models85 constitutes an important restriction to the narrative that “pathological window-opening naturally favors delivery”: even with amyloid-beta deposition, CAA, or local tight junction morphological abnormalities, different cargo classes do not automatically obtain therapeutically meaningful entry advantages. Conversely, TfR-targeting anti-amyloid-beta cargo studies suggest that improving brain parenchymal distribution through engineered trans-BBB transport under conditions of relatively intact overall BBB structure is more likely to simultaneously improve target engagement and reduce ARIA-like pathology.69
Third, any delivery scheme exploiting pathological interface abnormalities should be compared head-to-head with engineered trans-BBB transport strategies, rather than defaulting that “pathological gaps” are naturally superior to transport engineering. For a period within the BBB field, a common misjudgment involved directly translating “interface imbalance” as “channel amplification,” yet more common changes under aging and AD backgrounds may not be simply “more leaky,” but rather trans-endothelial transport rule rewriting, manifesting as weakened RMT advantages, enhanced nonspecific caveolar transcytosis, lost selectivity, and uncertain therapeutic indices.18,64 Under such circumstances, even if certain pathological backgrounds do have more molecules entering, this does not indicate such entry possesses superior therapeutic value. In contrast, engineered TfR shuttles, ATV:TREM2 and other strategies demonstrate that under conditions not relying upon pathological extravasation, entry rules can be rewritten to improve brain distribution and control vascular side reactions.69,83 Thus, for AD, exploitation-type research that cannot prove non-inferiority in effective exposure, safety, and reproducibility compared with transport engineering should not prematurely rise to priority conversion pathways.
Additionally, it should be emphasized that single or small-sample proof-of-concept studies cannot be directly extrapolated as long-term, repeatedly, safely callable platform strategies. Focused ultrasound (FUS) combined with aducanumab human studies indeed demonstrate the feasibility of focal BBB opening combined with anti-amyloid therapy,84 but this result is established under strict screening and rigorous monitoring conditions with extremely small sample sizes, and cannot be used to derive that “AD possesses a universally callable, long-term, repeatedly exploitable pathological window-opening platform.”84 Thus, for exploitation/delivery-type research, “whether it can be done short-term” and “whether it can be converted to routine pathways” remain questions at two different levels.
Biomarkers and Patient Stratification Frameworks
Based upon the above thresholds, if AD brain endothelial glycocalyx abnormalities are to enter clinical translational contexts, they cannot rely upon single readout indicators, but must construct a multi-level patient stratification framework. This is because glycocalyx abnormalities essentially represent rewrites of BBB blood-facing interface rules, and such rewrites can manifest as either focal, low-grade, chronic interface imbalances, or further progress toward inflammatory amplification, plasma protein extravasation, and even hemorrhagic fragility.18,20,43,64,86 Therefore, any single peripheral blood indicator is insufficient to independently represent brain glycocalyx states, and even more insufficient to alone adjudicate whether patients should enter “repair” or “exploitation” pathways.
More reasonable approaches should establish combinatorial interpretations from at least four levels. The first level is BBB/NVU imaging, including DCE-MRI or other BBB quantitative methods, to capture regional, low-grade, chronic leakage burdens and their brain region distributions.2,29,31,37,92 The second level is cerebrospinal fluid or fluid biomarker levels, such as QAlb, sPDGFRβ, etc., to indicate BBB/NVU-related abnormalities and pericyte injury backgrounds, but their interpretations must return to specific disease course and brain region contexts rather than being directly equated with “elevated permeability.”37,64,92,93 The third level is vascular fragility levels, including baseline microbleeds, cSS, cSAH, CAA burden, and ARIA risk backgrounds—this level is particularly critical when determining “whether exploitation can be discussed.”82,100–107 The fourth level is clinical staging and treatment exposure contexts, meaning at minimum distinguishing aging, preclinical AD, MCI, early AD dementia, and anti-amyloid therapy exposure states, because identical glycocalyx/BBB readout indicators carry different risk-benefit implications across different stages.2,20,29,31,37,82,95
Under this framework, peripheral shedding biomarkers such as Sdc-1, HA, and HS are more appropriately positioned as auxiliary signals for systemic endothelial stress or interface stress, rather than independent diagnostic bases for brain glycocalyx injury.109–111 Only when imaging, CSF/NVU biomarkers, vascular fragility readouts, and clinical staging contexts are jointly incorporated can “brain endothelial glycocalyx abnormality” potentially convert from a mechanistic concept to a truly discriminative clinical stratification tool.
Strategy Sequencing: Why AD Should Adhere to Repair-First
Synthesizing existing evidence, the most important translational value of AD brain endothelial glycocalyx abnormalities does not lie in providing a pathological delivery window that can be directly called upon, but rather in serving as a discriminator for BBB blood-facing interface states, helping distinguish which patients remain at stages dominated by plastic, reversible, endothelial interface imbalance, and which patients have already entered stages characterized by vascular wall fragility exposure, inflammatory amplification, and elevated hemorrhage risk. The key task for the former is repairing glycocalyx/glycosylation, reducing inflammatory adhesion thresholds, reconstructing transport and mechanical response rules closer to physiology, thereby improving effective brain parenchymal exposure while maintaining BBB structural integrity; the key task for the latter is no longer improving entry, but identifying risks, contracting exposure, and avoiding further triggering ARIA, vasculitis-like injuries, or hemorrhagic outcomes. More reasonable strategy sequencing in AD should be: repair-first, transport-engineering-second, opening/exploitation-last. This is not because the latter two are absolutely infeasible under any conditions, but because brain endothelial glycocalyx abnormalities essentially represent dynamic interface pathology rather than naturally favorable structural leaks. Only when interface abnormalities remain early, superficial, reversible, and without obvious CAA, microbleeds, cSS/cSAH, or ARIA high-risk backgrounds can delivery systems potentially discuss “exploitation” issues within strict safety boundaries; once abnormalities are coupled with fragile vascular substrates, their translational significance should retreat from “opportunity” back to “contraindication identification.”
It is precisely in this sense that the most promising direction in AD is not actively amplifying pathological gaps, but further rewriting entry rules through engineered trans-BBB transport upon the foundation of BBB blood-facing interface repair. Existing studies suggest that TfR shuttles, ATV:TREM2 and other pathways represent not reliance upon pathological gaps, but rather improving parenchymal target engagement, brain distribution, and control of ARIA-like pathology upon foundations of relatively intact overall BBB structure, thereby enhancing therapeutic indices.69,83 This pathway conceptually connects with glycocalyx-directed repair-first logic without conflict: the former is responsible for restoring or maintaining interface homeostasis, the latter responsible for rewriting entry rules upon this basis, rather than treating the most fragile vascular substrates as default channels. Integrating the “opportunity-fragility” interpretation framework, research thresholds, and strategy sequencing reveals that only when related abnormalities still belong to plastic, reversible interface imbalances and are not coupled with obviously high-risk vascular substrates can cautious translational exploitation be discussed; once entering structural fragility and hemorrhage-prone states, their meaning shifts from “opportunity” to “risk identification” (Table 10).
 |
Table 10 Opportunity-Fragility Stratification, Translational Thresholds, and Strategy Sequencing for Brain Endothelial Glycocalyx Abnormalities in AD |
Therefore, AD brain endothelial glycocalyx abnormalities should not be generalized as “the worse, the more favorable” delivery shortcuts. They are first BBB repair and patient stratification targets, and second, under conditions of sufficient evidence and controllable safety boundaries, provide design bases for engineered trans-BBB delivery; as for direct exploitation of pathological gaps, they should remain at the last position, and should not be characterized as routine conversion pathways for AD. In other words, brain endothelial glycocalyx in AD functions more like an interface adjudicator determining when repair is worthwhile, when transport can be designed, and when BBB-targeting interventions must be halted, rather than a universally callable therapeutic shortcut.
Limitations
This review is subject to several limitations inherent to the current evidence base and review scope.
First, the strongest mechanistic and interventional evidence for brain endothelial glycocalyx dysfunction in AD derives primarily from murine aging models and endothelial-specific genetic manipulations (eg., C1galt1 deletion/restoration).18,47,48 Direct demonstration that identical O-glycosylation defects drive BBB collapse in human AD remains incomplete. Consequently, all causal chains proposed here should be interpreted as cross-species hypotheses requiring prospective human validation.
Second, human evidence currently relies heavily on post-mortem brain microvascular staining and single-nuclear transcriptomic reanalysis.18,34 These approaches cannot capture longitudinal glycocalyx dynamics within the same subject, nor can they resolve luminal glycocalyx ultrastructure with the fidelity achievable in vivo. The “invisibility” of glycocalyx under conventional fixation introduces additional observational bias.
Third, preclinical repair strategies—particularly adeno-associated virus (AAV)-mediated C1GALT1/B3GNT3 delivery—face unresolved translational barriers, including brain endothelial tropism, immunogenicity, and scalable manufacturing for human CNS delivery.54,56 Exogenous biomimetic glycocalyx engineering remains at the proof-of-concept stage and lacks chronic safety data in neurodegenerative disease contexts.
Fourth, the four-dimensional interface-state framework proposed herein awaits prospective clinical stratification. No existing human cohort has simultaneously acquired glycocalyx-specific readouts, multimodal BBB imaging (eg., DCE-MRI), fluid biomarkers, and cognitive trajectories in a longitudinal design. Therefore, the framework currently serves as a conceptual stratification tool rather than a validated clinical algorithm. To bridge these gaps, future clinical strategies should prioritize same-subject, multimodal, longitudinal designs. Advancements in dynamic contrast-enhanced MRI protocols, novel PET tracers targeting glycosylation pathways or endothelial activation markers, and multiplex fluid biomarker panels could enable longitudinal tracking of glycocalyx alterations, BBB dysfunction, and cognitive decline within individual patients, thereby moving the field from cross-sectional inference to prospective mechanistic validation.
Finally, this review does not systematically address peripheral glycocalyx shedding biomarkers (eg., syndecan-1, heparan sulfate) as direct surrogates for brain endothelial pathology, given their lack of CNS specificity.109–111 Future studies should prioritize same-subject, multimodal, longitudinal designs to close these gaps.
Conclusion
In summary, brain endothelial glycocalyx in AD should no longer be regarded as a peripheral accessory structure of the BBB, but positioned as a key translational interface residing at the blood-facing frontline, connecting vascular pathology, barrier homeostasis, and brain delivery design. Existing evidence suggests that AD-associated glycocalyx abnormalities align more closely with interface remodeling centered on luminal protective mucin-domain glycoproteins and mucin-type O-glycosylation program impairment, rather than uniform holistic detachment; among these, the brain endothelial O-glycosylation axis has formed the currently most convincing mechanistic chain, indicating that glycocalyx abnormalities are not merely associated with BBB leakage, inflammatory amplification, and neurofunctional abnormalities, but may reside upstream of BBB destabilization. Based upon this, the primary translational positioning of glycocalyx in AD should be as a pathogenic interface for priority repair, rather than a delivery “gap” directly exploitable, because its pathological significance lies not in simply increasing brain entry flux, but in front-positionedly rewriting BBB starting contact conditions, receptor recognition environments, trans-endothelial pathway selection, and brain distribution allocation. Concurrently, current evidence does not support the existence of a unified, brain-wide, static “glycocalyx delivery window” in AD; the translational significance of glycocalyx abnormalities is highly dependent upon brain regions, vascular segments, disease stages, and accompanying vascular substrates, particularly in contexts merged with CAA, microbleeds, or ARIA susceptibility, where they first indicate vascular fragility rather than delivery advantages. Therefore, future research should shift from single judgments of “whether BBB leaks” toward multidimensional interface stratification of glycocalyx structure, glycosylation, transport, and inflammation/adhesion phenotypes, and further establish same-subject, multimodal, and longitudinal evidentiary chains between glycocalyx alterations, BBB functional abnormalities, and clinical outcomes in human AD. Overall, the most rational strategy sequencing at current evidence levels is repair-first, transport-engineering-second, and exploitation-last. In AD, the glycocalyx functions not as a universally callable delivery shortcut, but as an interface arbiter: it determines when repair is warranted, when transport can be engineered, and when BBB-targeting interventions must be halted to avoid vascular catastrophe.
Abbreviations
5xFAD, five familial Alzheimer’s disease mutations (mouse model); Aβ, amyloid-beta; AAV, adeno-associated virus; AD, Alzheimer’s disease; ALCAM, activated leukocyte cell adhesion molecule; ALPL, alkaline phosphatase, liver/bone/kidney; ANGPT-2, angiopoietin-2; ANKS1A, ankyrin repeat and SAM domain-containing protein 1A; APOE4, apolipoprotein E ε4; APP, amyloid precursor protein; APP/PS1, amyloid precursor protein/presenilin 1 (mouse model); ARIA, amyloid-related imaging abnormalities; ATV, transport vehicle; B3GNT3, UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 3; BBB, blood-brain barrier; C1GALT1, core 1 beta1,3-galactosyltransferase 1; CAA, cerebral amyloid angiopathy; CAV1, caveolin-1; CBF, cerebral blood flow; CBV, cerebral blood volume; CD8+, cluster of differentiation 8 positive; CD44, cluster of differentiation 44; CD68, cluster of differentiation 68; Cdh5, cadherin-5 (VE-cadherin); CLDN5, claudin-5; CMBs, cerebral microbleeds; CNS, central nervous system; CSF, cerebrospinal fluid; cSAH, convexity subarachnoid haemorrhage; cSS, cortical superficial siderosis; CX43, connexin 43 (gap junction protein 43); DCE-MRI, dynamic contrast-enhanced magnetic resonance imaging; DR6, death receptor 6; ECM, extracellular matrix; FUS, focused ultrasound; FUT11, fucosyltransferase 11; GAGs, glycosaminoglycans; Galnt2, polypeptide N-acetylgalactosaminyltransferase 2; Galnt10, polypeptide N-acetylgalactosaminyltransferase 10; GLUT1, glucose transporter 1; HA, hyaluronic acid; HAS2, hyaluronan synthase 2; HS, heparan sulfate; HSPGs, heparan sulfate proteoglycans; Iba1, ionized calcium-binding adapter molecule 1; ICAM-1, intercellular adhesion molecule-1; IgG, immunoglobulin G; K^trans^, volume transfer constant; LPS, lipopolysaccharide; LRP1, low-density lipoprotein receptor-related protein 1; MCI, mild cognitive impairment; NAD+, nicotinamide adenine dinucleotide (oxidized form); NeuN, neuronal nuclei; NVU, neurovascular unit; PARP1, poly(ADP-ribose) polymerase 1; PDGFRβ, platelet-derived growth factor receptor-β; PSEN1, presenilin 1; PVS, perivascular space; QAlb, cerebrospinal fluid/serum albumin ratio; RAGE, receptor for advanced glycation end-products; RMT, receptor-mediated transcytosis; SDC1, syndecan-1; sPDGFRβ, soluble platelet-derived growth factor receptor-β; StcE, *E. coli* secreted and surface-associated mucinase; Tg2576, transgenic mouse model expressing human APP with Swedish mutation; TfR, transferrin receptor; TfR1, transferrin receptor 1; TREM2, triggering receptor expressed on myeloid cells 2; TSG-6, tumor necrosis factor-stimulated gene-6; UEA1, *Ulex europaeus* agglutinin 1; VCAM-1, vascular cell adhesion molecule-1; VEGF-A, vascular endothelial growth factor A; Wnt, Wingless-related integration site (signaling pathway); ZO-1, zonula occludens-1.
Acknowledgments
The authors declare that Figures 1–7 were created by the authors using Adobe Illustrator through manual drawing and digital composition. No artificial intelligence–assisted illustration tools (e.g., Midjourney, Adobe Firefly, DALL-E, or Stable Diffusion) were used in the generation of these figures. All schematic illustrations were subsequently reviewed and scientifically validated by the authors to ensure accuracy and consistency with the published literature.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (No.81503257); Inner Mongolia Major science and technology project (No.2021ZD0017); Liaoning Provincial Science and Technology Programme Joint Programme (Applied Basic Research Project) (2023JH2/101700206); Liaoning University of Traditional Chinese Medicine-Natural Science University Key Project (2021LZY047); Liaoning Provincial Department of Education university basic scientific research project reserve project (LJ212410162055); National Key Research and Development Program (No.2018YFC1706903). Liaoning Province Science and Technology Plan Joint Plan (Natural Science Foundation-Doctoral Research Start-up Project) (2024-BSLH-006).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–43. doi:10.1038/nrneurol.2017.188
2. Nation DA, Sweeney MD, Montagne A, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–276. doi:10.1038/s41591-018-0297-y
3. Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151–3169. doi:10.1084/jem.20171406
4. Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–738. doi:10.1038/nrn3114
5. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5(5):347–360. doi:10.1038/nrn1387
6. Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–1078. doi:10.1016/j.cell.2015.10.067
7. Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21–78. doi:10.1152/physrev.00050.2017
8. Yoon JH, Lee ES, Jeong Y. In vivo imaging of the cerebral endothelial glycocalyx in mice. J Vasc Res. 2017;54(2):59–67. doi:10.1159/000457799
9. Ando Y, Okada H, Takemura G, et al. Brain-specific ultrastructure of capillary endothelial glycocalyx and its possible contribution for blood brain barrier. Sci Rep. 2018;8(1):17523. doi:10.1038/s41598-018-35976-2
10. Kutuzov N, Flyvbjerg H, Lauritzen M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood-brain barrier. Proc Natl Acad Sci U S A. 2018;115(40):E9429–E9438. doi:10.1073/pnas.1802155115
11. Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, Oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–359. doi:10.1007/s00424-007-0212-8
12. Tarbell JM, Pahakis MY. Mechanotransduction and the glycocalyx. J Intern Med. 2006;259(4):339–350. doi:10.1111/j.1365-2796.2006.01620.x
13. Weinbaum S, Tarbell JM, Damiano ER. The structure and function of the endothelial glycocalyx layer. Annu Rev Biomed Eng. 2007;9:121–167. doi:10.1146/annurev.bioeng.9.060906.151959
14. Santa-Maria AR, Walter FR, Figueiredo R, et al. Flow induces barrier and glycocalyx-related genes and negative surface charge in a lab-on-a-chip human blood-brain barrier model. J Cereb Blood Flow Metab. 2021;41(9):2201–2215. doi:10.1177/0271678X21992638
15. Ribeiro MMB, Domingues MM, Freire JM, Santos NC, Castanho MARB. Translocating the blood-brain barrier using electrostatics. Front Cell Neurosci. 2012;6:44. doi:10.3389/fncel.2012.00044
16. Hervé F, Ghinea N, Scherrmann JM. CNS delivery via adsorptive transcytosis. AAPS J. 2008;10(3):455–472. doi:10.1208/s12248-008-9055-2
17. O’Hare N, Millican K, Ebong EE. Unraveling neurovascular mysteries: the role of endothelial glycocalyx dysfunction in Alzheimer’s disease pathogenesis. Front Physiol. 2024;15:1394725. doi:10.3389/fphys.2024.1394725
18. Shi SM, Suh RJ, Shon DJ, et al. Glycocalyx dysregulation impairs blood-brain barrier in ageing and disease. Nature. 2025;639(8056):985–994. doi:10.1038/s41586-025-08589-9
19. Li X, Cai Q, Wilson BA, et al. Mechanobiological modulation of blood-brain barrier permeability by laser stimulation of endothelial-targeted nanoparticles. Nanoscale. 2023;15(7):3387–3397. doi:10.1039/d2nr05062e
20. Searson PC, Banks WA. Strategies for blood-brain barrier rejuvenation and repair. Nat Rev Drug Discov. 2026. doi:10.1038/s41573-025-01364-5
21. Chagnot A, Montagne A. The blood-brain barrier. Curr Biol. 2025;35(20):R1010–R1015. doi:10.1016/j.cub.2025.06.061
22. Friedman A, Prager O, Serlin Y, Kaufer D. Dynamic modulation of the blood-brain barrier in the healthy brain. Nat Rev Neurosci. 2025;26(12):749–764. doi:10.1038/s41583-025-00976-5
23. Stoddart P, Satchell SC, Ramnath R. Cerebral microvascular endothelial glycocalyx damage, its implications on the blood-brain barrier and a possible contributor to cognitive impairment. Brain Res. 2022;1780:147804. doi:10.1016/j.brainres.2022.147804
24. Walter FR, Santa-Maria AR, Mészáros M, Veszelka S, Dér A, Deli MA. Surface charge, glycocalyx, and blood-brain barrier function. Tissue Barriers. 2021;9(3):1904773. doi:10.1080/21688370.2021.1904773
25. Dancy C, Heintzelman KE, Katt ME. The glycocalyx: the importance of sugar coating the blood-brain barrier. Int J Mol Sci. 2024;25(15):8404. doi:10.3390/ijms25158404
26. Iş Ö, Wang X, Reddy JS, et al. Gliovascular transcriptional perturbations in Alzheimer’s disease reveal molecular mechanisms of blood brain barrier dysfunction. Nat Commun. 2024;15(1):4758. doi:10.1038/s41467-024-48926-6
27. Galea I. The blood-brain barrier in systemic infection and inflammation. Cell Mol Immunol. 2021;18(11):2489–2501. doi:10.1038/s41423-021-00757-x
28. Wu D, Chen Q, Chen X, Han F, Chen Z, Wang Y. The blood-brain barrier: structure, regulation, and drug delivery. Signal Transduct Target Ther. 2023;8(1):217. doi:10.1038/s41392-023-01481-w
29. Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi:10.1016/j.neuron.2014.12.032
30. van de Haar HJ, Jansen JFA, van Osch MJP, et al. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging. 2016;45:190–196. doi:10.1016/j.neurobiolaging.2016.06.006
31. Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–76. doi:10.1038/s41586-020-2247-3
32. Van Hulle C, Ince S, Okonkwo OC, et al. Elevated CSF angiopoietin-2 correlates with blood-brain barrier leakiness and markers of neuronal injury in early Alzheimer’s disease. Transl Psychiatry. 2024;14(1):3. doi:10.1038/s41398-023-02706-w
33. Yang AC, Vest RT, Kern F, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603(7903):885–892. doi:10.1038/s41586-021-04369-3
34. Tsartsalis S, Sleven H, Fancy N, et al. A single nuclear transcriptomic characterisation of mechanisms responsible for impaired angiogenesis and blood-brain barrier function in Alzheimer’s disease. Nat Commun. 2024;15(1):2243. doi:10.1038/s41467-024-46630-z
35. Reid MM, Menon S, Liu H, et al. Human brain vascular multi-omics elucidates disease-risk associations. Neuron. 2025;113(19):3143–3161.e5. doi:10.1016/j.neuron.2025.07.001
36. Sun N, Akay LA, Murdock MH, et al. Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer’s disease. Nat Neurosci. 2023;26(6):970–982. doi:10.1038/s41593-023-01334-3
37. Preis L, Villringer K, Brosseron F, et al. Assessing blood-brain barrier dysfunction and its association with Alzheimer’s pathology, cognitive impairment and neuroinflammation. Alzheimer’s Res Ther. 2024;16(1):172. doi:10.1186/s13195-024-01529-1
38. Ziegler KC, Askarova A, Gergian C, et al. The brain neurovascular epigenome and its association with dementia. Neuron. 2026;114(2):268–286.e9. doi:10.1016/j.neuron.2025.10.001
39. Wiseman S. Sugar-coating the BBB. Nat Neurosci. 2025;28(4):709. doi:10.1038/s41593-025-01939-w
40. Smyth LCD, Murray HC, Hill M, et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol Commun. 2022;10(1):38. doi:10.1186/s40478-022-01347-2
41. Reed MJ, Damodarasamy M, Pathan JL, et al. Increased hyaluronan and TSG-6 in association with neuropathologic changes of Alzheimer’s disease. J Alzheimers Dis. 2019;67(1):91–102. doi:10.3233/JAD-180797
42. McMillan IO, Gearing M, Wang L. Vascular heparan sulfate and amyloid-β in Alzheimer’s disease patients. Int J Mol Sci. 2024;25(7):3964. doi:10.3390/ijms25073964
43. Weinbaum S, Zhang X, Han Y, Vink H, Cowin SC. Mechanotransduction and flow across the endothelial glycocalyx. Proc Natl Acad Sci U S A. 2003;100(13):7988–7995. doi:10.1073/pnas.1332808100
44. Zhu J, Li X, Yin J, Hu Y, Gu Y, Pan S. Glycocalyx degradation leads to blood-brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J Cereb Blood Flow Metab. 2018;38(11):1979–1992. doi:10.1177/0271678X17726062
45. Al-Ahmad AJ, Patel R, Palecek SP, Shusta EV. Hyaluronan impairs the barrier integrity of brain microvascular endothelial cells through a CD44-dependent pathway. J Cereb Blood Flow Metab. 2019;39(9):1759–1775. doi:10.1177/0271678X18767748
46. DeOre BJ, Partyka PP, Fan F, Galie PA. CD44 mediates shear stress mechanotransduction in an in vitro blood-brain barrier model through small GTPases RhoA and Rac1. FASEB J. 2022;36(5):e22278. doi:10.1096/fj.202100822RR
47. Xia L, Ju T, Westmuckett A, et al. Defective angiogenesis and fatal embryonic hemorrhage in mice lacking core 1-derived O-glycans. J Cell Biol. 2004;164(3):451–459. doi:10.1083/jcb.200311112
48. Fu J, Gerhardt H, McDaniel JM, et al. Endothelial cell O-glycan deficiency causes blood/lymphatic misconnections and consequent fatty liver disease in mice. J Clin Invest. 2008;118(11):3725–3737. doi:10.1172/JCI36077
49. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91(9):e867–e877. doi:10.1212/WNL.0000000000006082
50. Chen J, Dai AX, Tang HL, et al. Increase of ALCAM and VCAM-1 in the plasma predicts the Alzheimer’s disease. Front Immunol. 2023;13:1097409. doi:10.3389/fimmu.2022.1097409
51. Salian VS, Tang X, Thompson KJ, et al. Molecular mechanisms underlying amyloid beta peptide mediated upregulation of vascular cell adhesion molecule-1 in Alzheimer disease. J Pharmacol Exp Ther. 2024;391(3):430–440. doi:10.1124/jpet.124.002280
52. Cruz Hernández JC, Bracko O, Kersbergen CJ, et al. Neutrophil adhesion in brain capillaries reduces cortical blood flow and impairs memory function in Alzheimer’s disease mouse models. Nat Neurosci. 2019;22(3):413–420. doi:10.1038/s41593-018-0329-4
53. Ali M, Falkenhain K, Njiru BN, et al. VEGF signalling causes stalls in brain capillaries and reduces cerebral blood flow in Alzheimer’s mice. Brain. 2022;145(4):1449–1463. doi:10.1093/brain/awab387
54. Chen X, Wolfe DA, Bindu DS, et al. Functional gene delivery to and across brain vasculature of systemic AAVs with endothelial-specific tropism in rodents and broad tropism in primates. Nat Commun. 2023;14(1):3345. doi:10.1038/s41467-023-38582-7
55. Velazquez-Rivera E, Dey O, Kim NS, et al. Specific targeting of brain endothelial cells using enhancer AAV vectors. Neuron. 2025;113(10):1562–1578.e6. doi:10.1016/j.neuron.2025.03.031
56. Hordeaux J, Yuan Y, Clark PM, et al. The GPI-linked protein LY6A drives AAV-PHP.B transport across the blood-brain barrier. Mol Ther. 2019;27(5):912–921. doi:10.1016/j.ymthe.2019.02.013
57. Siren EMJ, Luo HD, Tam F, et al. Prevention of vascular-allograft rejection by protecting the endothelial glycocalyx with immunosuppressive polymers. Nat Biomed Eng. 2021;5(10):1202–1216. doi:10.1038/s41551-021-00777-y
58. Wardzala CL, Clauss ZS, Kramer JR. Principles of glycocalyx engineering with hydrophobic-anchored synthetic mucins. Front Cell Dev Biol. 2022;10:952931. doi:10.3389/fcell.2022.952931
59. Fu L, Kim HN, Sterling JD, Baker SM, Lord MS. The role of the cell surface glycocalyx in drug delivery to and through the endothelium. Adv Drug Deliv Rev. 2022;184:114195. doi:10.1016/j.addr.2022.114195
60. Ramnath RD, Butler MJ, Newman G, et al. Blocking matrix metalloproteinase-mediated syndecan-4 shedding restores the endothelial glycocalyx and glomerular filtration barrier function in early diabetic kidney disease. Kidney Int. 2020;97(5):951–965. doi:10.1016/j.kint.2019.09.035
61. Zou Z, Li L, Li Q, et al. The role of S100B/RAGE-enhanced ADAM17 activation in endothelial glycocalyx shedding after traumatic brain injury. J Neuroinflammation. 2022;19(1):46. doi:10.1186/s12974-022-02412-2
62. Deane R, Du Yan S, Submamaryan RK, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–913. doi:10.1038/nm890
63. Balistreri CR, Di Giorgi L, Monastero R. Focus of endothelial glycocalyx dysfunction in ischemic stroke and Alzheimer’s disease: possible intervention strategies. Ageing Res Rev. 2024;99:102362. doi:10.1016/j.arr.2024.102362
64. Yang AC, Stevens MY, Chen MB, et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature. 2020;583(7816):425–430. doi:10.1038/s41586-020-2453-z
65. Todorov-Völgyi K, González-Gallego J, Müller SA, et al. Proteomics of mouse brain endothelium uncovers dysregulation of vesicular transport pathways during aging. Nat Aging. 2024;4(4):595–612. doi:10.1038/s43587-024-00598-z
66. Lee J, Lee H, Lee H, et al. ANKS1A regulates LDL receptor-related protein 1 (LRP1)-mediated cerebrovascular clearance in brain endothelial cells. Nat Commun. 2023;14(1):8463. doi:10.1038/s41467-023-44319-3
67. Chen J, Xiang P, Duro-Castano A, et al. Rapid amyloid-β clearance and cognitive recovery through multivalent modulation of blood-brain barrier transport. Signal Transduct Target Ther. 2025;10(1):331. doi:10.1038/s41392-025-02426-1
68. Moyer TC, Hoffman BA, Chen W, et al. Highly conserved brain vascular receptor ALPL mediates transport of engineered AAV vectors across the blood-brain barrier. Mol Ther. 2025;33(8):3902–3916. doi:10.1016/j.ymthe.2025.04.046
69. Pizzo ME, Plowey ED, Khoury N, et al. Transferrin receptor-targeted anti-amyloid antibody enhances brain delivery and mitigates ARIA. Science. 2025;389(6760):eads3204. doi:10.1126/science.ads3204
70. Xing M, Song W. Improving Alzheimer’s disease immunotherapy. Science. 2025;389(6760):571–572. doi:10.1126/science.adz8959
71. Khoury N, Pizzo ME, Discenza CB, et al. Fc-engineered large molecules targeting blood-brain barrier transferrin receptor and CD98hc have distinct central nervous system and peripheral biodistribution. Nat Commun. 2025;16(1):1822. doi:10.1038/s41467-025-57108-x
72. Vega MR, Hansen HH, Jensen CS, et al. Transferrin receptor-binding blood-brain barrier shuttle enhances brain delivery and plaque-clearing efficacy of a therapeutic anti-Aβ antibody. Fluids Barriers CNS. 2025;22(1):121. doi:10.1186/s12987-025-00737-7
73. Wells RC, Akkapeddi P, Chan D, et al. Dual targeting of transferrin receptor and CD98hc enhances brain exposure of large molecules. Cell Rep. 2025;44(8):116038. doi:10.1016/j.celrep.2025.116038
74. Gao J, Xia Z, Gunasekar S, Jiang C, Karp JM, Joshi N. Precision drug delivery to the central nervous system using engineered nanoparticles. Nat Rev Mater. 2024;9(8):567–588. doi:10.1038/s41578-024-00695-w
75. Johnsen KB, Burkhart A, Thomsen LB, Andresen TL, Moos T. Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 2019;181:101665. doi:10.1016/j.pneurobio.2019.101665
76. Yamamoto K, Scilabra SD, Bonelli S, et al. Novel insights into the multifaceted and tissue-specific roles of the endocytic receptor LRP1. J Biol Chem. 2024;300(8):107521. doi:10.1016/j.jbc.2024.107521
77. Tang K, Tang Z, Niu M, et al. Allosteric targeted drug delivery for enhanced blood-brain barrier penetration via mimicking transmembrane domain interactions. Nat Commun. 2025;16(1):3410. doi:10.1038/s41467-025-58746-x
78. Bridges CA, Fu L, Yeow J, et al. The interplay between endothelial glycocalyx maturity and both the toxicity and intracellular uptake of charged nanoparticles. Acta Biomater. 2025;196:293–306. doi:10.1016/j.actbio.2025.03.012
79. Zhang W, Zhu D, Tong Z, et al. Influence of surface ligand density and particle size on the penetration of the blood-brain barrier by porous silicon nanoparticles. Pharmaceutics. 2023;15(9):2271. doi:10.3390/pharmaceutics15092271
80. Porkoláb G, Mészáros M, Szecskó A, et al. Synergistic induction of blood-brain barrier properties. Proc Natl Acad Sci U S A. 2024;121(21):e2316006121. doi:10.1073/pnas.2316006121
81. Cox A, Andreozzi P, Dal Magro R, et al. Evolution of nanoparticle protein Corona across the blood-brain barrier. ACS Nano. 2018;12(7):7292–7300. doi:10.1021/acsnano.8b03500
82. Greenberg SM, Bax F, van Veluw SJ. Amyloid-related imaging abnormalities: manifestations, metrics and mechanisms. Nat Rev Neurol. 2025;21(4):193–203. doi:10.1038/s41582-024-01053-8
83. van Lengerich B, Zhan L, Xia D, et al. A TREM2-activating antibody with a blood-brain barrier transport vehicle enhances microglial metabolism in Alzheimer’s disease models. Nat Neurosci. 2023;26(3):416–429. doi:10.1038/s41593-022-01240-0
84. Rezai AR, D’Haese PF, Finomore V, et al. Ultrasound blood-brain barrier opening and aducanumab in Alzheimer’s disease. N Engl J Med. 2024;390(1):55–62. doi:10.1056/NEJMoa2308719
85. Nozohouri E, Noorani B, Patel D, Ahn Y, Zoubi S, Bickel U. Assessing blood-brain barrier (BBB) integrity in an Alzheimer’s disease mouse model: is the BBB globally or locally disrupted? Fluids Barriers CNS. 2025;22(1):79. doi:10.1186/s12987-025-00685-2
86. Konig S, Jayarajan V, Wray S, Kamm R, Moeendarbary E. Mechanobiology of the blood-brain barrier during development, disease and ageing. Nat Commun. 2025;16(1):7233. doi:10.1038/s41467-025-61888-7
87. Garcia-Polite F, Martorell J, Del Rey-Puech P, et al. Pulsatility and high shear stress deteriorate barrier phenotype in brain microvascular endothelium. J Cereb Blood Flow Metab. 2017;37(7):2614–2625. doi:10.1177/0271678X16672482
88. Yates AK, Murray H, Kjar A, et al. Substrate stiffness and shear stress collectively regulate the inflammatory phenotype in cultured human brain microvascular endothelial cells. Fluids Barriers CNS. 2025;22(1):73. doi:10.1186/s12987-025-00683-4
89. Korte N, Barkaway A, Wells J, et al. Inhibiting Ca2+ channels in Alzheimer’s disease model mice relaxes pericytes, improves cerebral blood flow and reduces immune cell stalling and hypoxia. Nat Neurosci. 2024;27(11):2086–2100. doi:10.1038/s41593-024-01753-w
90. Wälchli T, Ghobrial M, Schwab M, et al. Single-cell atlas of the human brain vasculature across development, adulthood and disease. Nature. 2024;632(8025):603–613. doi:10.1038/s41586-024-07493-y
91. Miners JS, Schulz I, Love S. Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab. 2018;38(1):103–115. doi:10.1177/0271678X17690761
92. van de Haar HJ, Burgmans S, Jansen JFA, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology. 2016;281(2):527–535. doi:10.1148/radiol.2016152244
93. Vrillon A, Ashton NJ, Bouaziz-Amar E, et al. Dissection of blood-brain barrier dysfunction through CSF PDGFRβ and amyloid, tau, neuroinflammation, and synaptic CSF biomarkers in neurodegenerative disorders. EBioMedicine. 2025;115:105694. doi:10.1016/j.ebiom.2025.105694
94. Kozberg MG, Munting LP, Hanlin LH, et al. Vasomotion loss precedes impaired cerebrovascular reactivity and microbleeds in cerebral amyloid angiopathy. Brain Commun. 2025;7(3):fcaf186. doi:10.1093/braincomms/fcaf186
95. Barisano G, Montagne A, Kisler K, Schneider JA, Wardlaw JM, Zlokovic BV. Blood-brain barrier link to human cognitive impairment and Alzheimer’s disease. Nat Cardiovasc Res. 2022;1(2):108–115. doi:10.1038/s44161-021-00014-4
96. Lee E, Kim S, Zhu CL, et al. Angiopoietin-2 aggravates Alzheimer’s disease by promoting blood-brain barrier dysfunction and neuroinflammation. Cell Rep. 2026;45(1):116621. doi:10.1016/j.celrep.2025.116621
97. Huang X, Qi J, Su Y, et al. Endothelial DR6 in blood-brain barrier malfunction in Alzheimer’s disease. Cell Death Dis. 2024;15(4):258. doi:10.1038/s41419-024-06639-0
98. Zhan R, Meng X, Tian D, et al. NAD+ rescues aging-induced blood-brain barrier damage via the CX43-PARP1 axis. Neuron. 2023;111(22):3634–3649.e7. doi:10.1016/j.neuron.2023.08.010
99. Söderberg L, Johannesson M, Gkanatsiou E, et al. Amyloid-beta antibody binding to cerebral amyloid angiopathy fibrils and risk for amyloid-related imaging abnormalities. Sci Rep. 2024;14(1):10868. doi:10.1038/s41598-024-61691-2
100. Cummings J, Apostolova L, Rabinovici GD, et al. Lecanemab: appropriate use recommendations. J Prev Alzheimer’s Dis. 2023;10(3):362–377. doi:10.14283/jpad.2023.30
101. Rabinovici GD, Selkoe DJ, Schindler SE, et al. Donanemab: appropriate use recommendations. J Prev Alzheimer’s Dis. 2025;12(5):100150. doi:10.1016/j.tjpad.2025.100150
102. Zimmer JA, Ardayfio P, Wang H, et al. Amyloid-related imaging abnormalities with donanemab in early symptomatic Alzheimer disease: secondary analysis of the TRAILBLAZER-ALZ and ALZ 2 randomized clinical trials. JAMA Neurol. 2025;82(5):461–469. doi:10.1001/jamaneurol.2025.0065
103. Cogswell PM, Andrews TJ, Barakos JA, et al. Alzheimer disease anti-amyloid immunotherapies: imaging recommendations and practice considerations for monitoring of amyloid-related imaging abnormalities. AJNR Am J Neuroradiol. 2025;46(1):24–32. doi:10.3174/ajnr.A8469
104. van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med. 2023;388(1):9–21. doi:10.1056/NEJMoa2212948
105. Sims JR, Zimmer JA, Evans CD, et al. Donanemab in early symptomatic Alzheimer disease: the TRAILBLAZER-ALZ 2 randomized clinical trial. JAMA. 2023;330(6):512–527. doi:10.1001/jama.2023.13239
106. Solopova E, Romero-Fernandez W, Harmsen H, et al. Fatal iatrogenic cerebral β-amyloid-related arteritis in a woman treated with lecanemab for Alzheimer’s disease. Nat Commun. 2023;14(1):8220. doi:10.1038/s41467-023-43933-5
107. Oomens JE, van Gils V, Vos SJB, et al. Cerebral microbleeds and amyloid pathology estimates from the amyloid biomarker study. JAMA Network Open. 2025;8(1):e2455571. doi:10.1001/jamanetworkopen.2024.55571
108. Johnson LA, Saito K, Pallerla AV, et al. Clonal expansion of cytotoxic CD8+ T cells in lecanemab-associated ARIA. Nat Commun. 2026;17(1):2180. doi:10.1038/s41467-026-68921-3
109. French SR, Meyer BP, Arias JC, Levendovzsky SR, Weinkauf CC. Biomarkers of blood-brain barrier and neurovascular unit integrity in human cognitive impairment and dementia. Alzheimers Dement. 2025;21(3):e70104. doi:10.1002/alz.70104
110. Hahn RG, Patel V, Dull RO. Human glycocalyx shedding: systematic review and critical appraisal. Acta Anaesthesiol Scand. 2021;65(5):590–606. doi:10.1111/aas.13797
111. Inoda A, Suzuki K, Tomita H, Okada H. Glycocalyx shedding as a clinical biomarker in critical illness. Exp Mol Pathol. 2025;144:104997. doi:10.1016/j.yexmp.2025.104997
112. Bonvicini G, Singh S, Sandersjöö L, Sehlin D, Syvänen S, Andersson KG. The effects of dose, valency, and affinity on TfR-mediated brain delivery in vivo. Fluids Barriers CNS. 2025;22(1):36. doi:10.1186/s12987-025-00643-y
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Recent Advancements in Lipid Nanoparticles-Based Phytoactives Delivery Systems for Neurodegenerative Diseases

Dirir AM, Ali A, Hachem M
International Journal of Nanomedicine 2025, 20:10279-10300
Published Date: 25 August 2025