Back to Journals » International Journal of Nanomedicine » Volume 11
Bolaamphiphile-based nanocomplex delivery of phosphorothioate gapmer antisense oligonucleotides as a treatment for Clostridium difficile
Authors Hegarty J, Krzeminski J, Sharma A, Guzman-Villanueva D, Weissig V, Stewart D
Received 31 March 2016
Accepted for publication 7 June 2016
Published 1 August 2016 Volume 2016:11 Pages 3607—3619
DOI https://doi.org/10.2147/IJN.S109600
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
John P Hegarty,1 Jacek Krzeminski,2 Arun K Sharma,2 Diana Guzman-Villanueva,3 Volkmar Weissig,3 David B Stewart Sr1
1Deparment of Surgery, Pennsylvania State University College of Medicine Hershey, PA, USA; 2Department of Pharmacology, Penn State Hershey Cancer Institute, Hershey, PA, USA; 3Department of Pharmaceutical Sciences, College of Pharmacy, Nanomedicine Center of Excellence in Translational Research, Midwestern University, Glendale, AZ, USA
Abstract: Despite being a conceptually appealing alternative to conventional antibiotics, a major challenge toward the successful implementation of antisense treatments for bacterial infections is the development of efficient oligonucleotide delivery systems. Cationic vesicles (bolasomes) composed of dequalinium chloride (“DQAsomes”) have been used to deliver plasmid DNA across the cardiolipin-rich inner membrane of mitochondria. As cardiolipin is also a component of many bacterial membranes, we investigated the application of cationic bolasomes to bacteria as an oligonucleotide delivery system. Antisense sequences designed in silico to target the expression of essential genes of the bacterial pathogen, Clostridium difficile, were synthesized as 2'-O-methyl phosphorothioate gapmer antisense oligonucleotides (ASO). These antisense gapmers were quantitatively assessed for their ability to block mRNA translation using luciferase reporter and C. difficile protein expression plasmid constructs in a coupled transcription–translation system. Cationic bolaamphiphile compounds (dequalinium derivatives) of varying alkyl chain length were synthesized and bolasomes were prepared via probe sonication of an aqueous suspension. Bolasomes were characterized by particle size distribution, zeta potential, and binding capacities for anionic oligonucleotide. Bolasomes and antisense gapmers were combined to form antisense nanocomplexes. Anaerobic C. difficile log phase cultures were treated with serial doses of gapmer nanocomplexes or equivalent amounts of empty bolasomes for 24 hours. Antisense gapmers for four gene targets achieved nanomolar minimum inhibitory concentrations for C. difficile, with the lowest values observed for oligonucleotides targeting polymerase genes rpoB and dnaE. No inhibition of bacterial growth was observed from treatments at matched dosages of scrambled gapmer nanocomplexes or plain, oligonucleotide-free bolasomes compared to untreated control cultures. We describe the novel application of cationic bolasomes to deliver ASOs into bacteria. We also report the first successful in vitro antisense treatment to inhibit the growth of C. difficile.
Keywords: cationic bolaamphiphiles, dequalinium derivatives, nanocomplex, antisense, gapmers, Clostridium difficile, bacteria
Introduction
Clostridium difficile is a gram-positive, spore-forming, anaerobic bacterium whose pathogenic strains result in a toxin-mediated colitis due to the production of large clostridial and ADP-ribosyltransferase toxins.1 The most important causal factor for the development of C. difficile infection (CDI) is exposure to conventional antibiotics,2 due to their indiscriminate mechanism of action that results in widespread eradication of bacteria, leading to an intestinal dysbiosis, which provides a selective advantage toward colonization of the gut with C. difficile.3 Certain strains, including ribotypes 027 and 078, are associated with particularly severe forms of CDI,4,5 greater amounts of bacterial toxin production,6 as well as a relative resistance to the effects of antibiotic treatment, potentially resulting in an increase of these subpopulations of C. difficile.7 The frequent inefficacy and relapse of CDI in response to conventional antibiotics is due in large measure to the same mechanism by which non-difficile-directed antibiotics create CDI – nonspecific bacterial killing resulting in an alteration to gut-associated communities, which provides a selective advantage to pathogenic ribotypes of C. difficile.
Given frequent CDI relapse rates and unpredictable responses associated with conventional antibiotics, there is considerable interest in the development of alternative CDI therapies. In addition to the prospect of higher rates of treatment success, alternative therapies may potentially allow for treatment targets beyond bacterial killing, such as inhibition of toxin production and spore formation, which are associated with the CDI disease state and the spread of CDI, respectively. One alternative treatment modality is the use of “antisense antibiotics,” which can prevent the expression of bacterial genes essential for growth or virulence through posttranscriptional mechanisms.8–12 This approach could potentially provide species-specific targeting of bacteria, while also providing multiple molecular targets beyond bacterial killing.
While antisense antibacterial therapies offer these potential advantages, a major hurdle to their development is that free oligonucleotides are poorly taken up by bacteria due to electrostatic charge as well as size discrimination imposed by the bacterial cell envelope. Antisense approaches have wide application in eukaryotic cells due to the availability of numerous oligonucleotide delivery strategies; however, delivery into bacteria has received far less attention. Antisense bacterial studies have principally relied upon covalent conjugation of the molecules to arginine-rich cell-penetrating peptides to achieve entry into bacteria. Previous publications13–17 suggest that this approach may be more effective in gram-negative species, with fewer reports of successful treatment of gram-positive organisms and with no reports of successful treatment of C. difficile.18
We recently presented our initial work investigating the use of cell-penetrating peptides to deliver antisense morpholinos into C. difficile.19 In considering alternative delivery systems that might more efficiently transport cargo into this bacterium, we noted that cationic lipids and polymers can be utilized to condense antisense oligonucleotides (ASOs) through loss of their chiral secondary structure, which may improve cellular entry.20,21 Along these lines, nanoparticulate systems of various types are being developed to deliver antibacterial agents to microorganisms.22–25 In particular, cationic bolaamphiphile nanovesicles (bolasomes) have recently been used to deliver plasmid DNA into eukaryotic cells more effectively than the gold standard of cationic polymers, polyethylenimine.26
Cationic bolaamphiphiles are amphiphilic molecules with hydrophilic charge centers separated by an internal hydrophobic chain. The supramolecular self-organization of cationic bolaamphiphiles in an aqueous environment can produce monolayer bolasomes with a high packing density. Of particular interest was use of dequalinium bolasomes to deliver plasmid DNA into the cardiolipin-rich mitochondria of eukaryotic cells.27 Dequalinium (1,1′-decamethylene bis[4-aminoquinaldinium chloride]; DQA) is a cationic, quaternary ammonium, single-chain bolaamphiphile with two delocalized charge centers (Figure 1A). This structure is similar to that of the monolayer bolalike membrane lipids of Archaebacteriae, with the difference being the latter has two hydrophobic chains instead of one. Weissig et al28 discovered that upon sonication in aqueous solution, DQA assembles into nanometer-scaled bolasomes (DQAsomes) with diameters ranging from 70 to 700 nm. These DQAsomes efficiently bind and condense DNA through electrostatic interaction with the cationic charge centers and primary 4-amino groups of its quinolinium ring system, thereby protecting the bound DNA against nuclease activity (Figure 2).28 It was determined that DQAsome–DNA complexes become destabilized upon contact with intact mitochondria, with a resulting DNA accumulation within the mitochondrial matrix.27 The relative lipophilicity of these structures, in part due to delocalization of their positive charge centers, is suggested to reduce the free energy change involved in transitioning from an aqueous to a hydrophobic environment, which may promote the accumulation of DNA in the mitochondrial matrix in response to the electrochemical gradient potential of the inner mitochondrial membrane.29 Structure–activity studies of derivatives of DQA have demonstrated that replacing its methyl group with an aliphatic ring system to generate acridinium polar groups (cyclohexyl-DQA) conferred superior bolasome-forming properties, with high stability, a much narrower size distribution, and lower cell toxicity.30 Cyclohexyl-DQA forms stable vesicles at 3.6 mM, indicating a lower critical vesicle concentration than DQA, and it is has been suggested that the bulky aliphatic residues attached to the heterocyclic ring structure may favor self-association of the planar ring system by sterically preventing the free rotation of the hydrophilic head of the amphiphile around the axis of the CH2 moiety.31,32
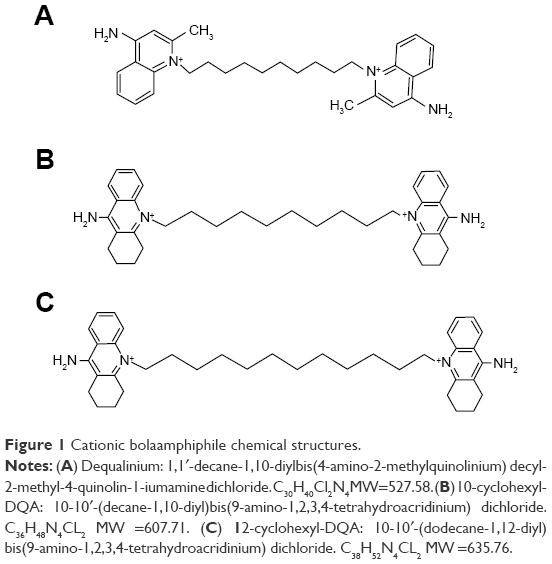 | Figure 1 Cationic bolaamphiphile chemical structures. |
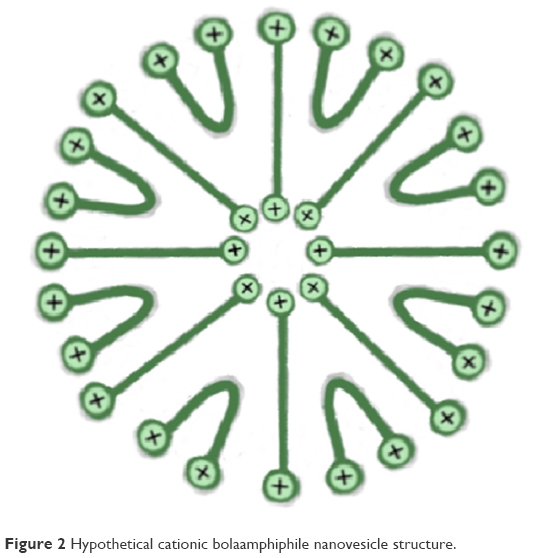 | Figure 2 Hypothetical cationic bolaamphiphile nanovesicle structure. |
Of particular interest was the finding that DNA is selectively released from DQAsome complexes in the presence of cardiolipin-rich [1,3-bis(sn-3′-phosphatidyl)-sn-glycerol] liposomes; DNA remains tightly associated in the presence of anionic lipids other than cardiolipin.29 Cardiolipin has a key role in the organization of both mitochondrial and most bacterial membranes, but it is not present in eukaryotic cell membranes.33 Notably, cardiolipin is known to be a significant negatively charged polar glycerophospholipid component of the C. difficile cell plasma membrane.34 In this work, we investigated the use of mitochondriotropic, cationic cyclohexyl-DQA bolasomes, with alkyl chain lengths of 10 and 12, as an ASO delivery strategy for C. difficile. The anionic ASO chosen to form complexes with cationic bolasomes for this study were chimeric 2′-O-methyl flanked phosphorothioate ASO gapmers.35 These “second-generation” ASOs consist of a central phosphorothioate core (the nonbridging oxygen in the native phosphodiester replaced with sulfur) susceptible to binding of RNase H; this region is flanked by 2′-alkyl-modified nucleotides at the ribose sugar moieties to increase binding stability. These alkyl modifications provide protection from nuclease attack, with an increased affinity to target mRNA and with an improved lipophilic character favorable for lipid bilayer diffusion.36 Gapmer annealing to complementary mRNA can disrupt translation due to steric blocking of ribosome read-through and by recognition and degradation of the RNA–gapmer duplexes by cellular RNase H.10,37
Materials and methods
C. difficile isolation
Culture media and microbiological supplies were purchased from Becton Dickinson (Franklin Lakes, NJ, USA) and Anaerobe Systems (Morgan Hill, CA, USA). A hypervirulent ribotype 027 C. difficile clinical isolate (HMC001) was obtained by screening of samples from a Milton S. Hershey Medical Center Institution Review Board-approved tissue bank storing patient-derived stool samples from consented hospitalized patients. Stool–saline suspensions were treated with an equal volume of ethyl alcohol for 30 minutes and then cultured on CDC anaerobe 5% sheep blood/phenylethyl alcohol agar. Following anaerobic incubation (85% N2, 10% CO2, 5% H2; Anoxomat, Advanced Instruments Inc. Norwood, MA, USA) at 37°C for 48 hours, subcultured presumptive isolates were identified using RapID ANA II metabolic panels. HMC001 was maintained on trypticase soy agar with 5% sheep blood, and stocks were banked at −80°C in trypticase soy broth containing 15% glycerol.
DNA extraction and isolate typing
Genomic DNA from clinical isolate HMC001 was extracted from a phosphate buffer-washed cell pellet by 0.15 mm garnet bead mechanical lysis (M-Bio Laboratories, Carlsbad, CA, USA), followed by DNA clarification on a silica column.38 The concentration of the purified DNA was determined by ultraviolet (UV) spectrophotometry (NanoDrop 2000; Thermo Fisher Scientific, Wilmington, DE, USA) and the sample quality was assessed by Qubit dsDNA BR-fluorometer assay (Life Technologies, Grand Island, NY, USA). The C. difficile ribotype was determined by polymerase chain reaction (PCR) amplification of the 16S-23S rRNA intergenic-spacer region (16S and 23S primers; Table 1) by resolution of the amplicons through 2.5% MetaPhor high-resolution agarose gels (Lonza, Allendale, NJ, USA) in 0.5× chilled Tris-borate-EDTA buffer at 5 V/cm for 4 hours, followed by briefly staining with 0.25 μg/mL ethidium bromide.39 The appropriate ribotype was assigned by matching the gel banding pattern (up to 1,000 bp) to known PCR ribotype profiles. Toxinotype for the major C. difficile toxins was confirmed by PCR as previously described.40
 | Table 1 PCR primers |
Whole-genome sequencing
To ensure precision in the design of the gapmer ASOs, we sequenced the genome of the ribotype 027 HMC001 isolate through creation of a shotgun library using Nextera XT chemistry41 as per manufacturer’s directions (Illumina Inc., San Diego, CA, USA). In brief, transposon/transposase-fragmented genomic DNA was tagged with amplification adaptors in a 55°C tagmentation reaction. Following 12 cycles of PCR amplification using adaptor-targeted primers, uniform sequence lengths were obtained by magnetic bead normalization (AMPure XP beads; Beckman Coulter Genomics, Danvers, MA, USA) and the size range of the library determined on a high-sensitivity microfluidic DNA chip processed with a 2,100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). In preparation for cluster generation of the paired-end 150 bp sequencing run, the concentration of amplicons was determined by fluorometry before equimolar pooling, dilution in hybridization buffer, and heat denaturation. Genome sequencing was performed on an Illumina MiSeq system. Trimmed paired reads were assembled with a minimum quality-filtering Phred score of Q30.
Design of phosphorothioate gapmer ASOs
5′-Untranslated region (5′UTR) sequences from mRNA transcripts of five genes known to be essential for C. difficile growth42 were obtained from our HMC001 assembled genomic contig data using SeqMan NGen software (DNAStar, Madison, WI, USA) referencing an annotated C. difficile genome assembly (NCBI accession PRJNA38039). Candidate 25-mer ASO sequences ideally targeting accessible loop structures43 within the 5′UTR near the start codons were chosen through predictive in silico modeling (RNAstructure v5.8; Mfold v3.6)44,45 of mRNA transcript local secondary structures (Figure 3). Selected antisense sequences were evaluated for intraorganismal specificity using NCBI BLAST46 with screening of potential off-target binding near other known initiation codons. Chimeric 25-mer phosphorothioate gapmer ASOs flanked by six terminal 2-O-methyl bases (Table 2) targeting five C. difficile genes and a scrambled control were synthesized, desalted, and polyacrylamide gel-purified (Integrated DNA Technologies, Coralville, IA, USA). Oligonucleotide stocks were prepared at 250 μM in nuclease-free water and maintained at −20°C.
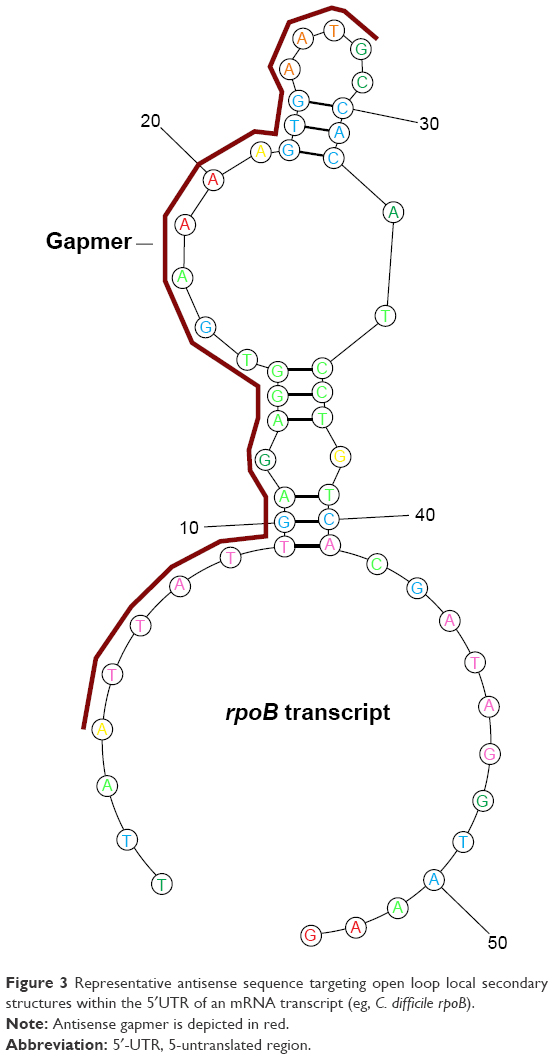 | Figure 3 Representative antisense sequence targeting open loop local secondary structures within the 5′UTR of an mRNA transcript (eg, C. difficile rpoB). |
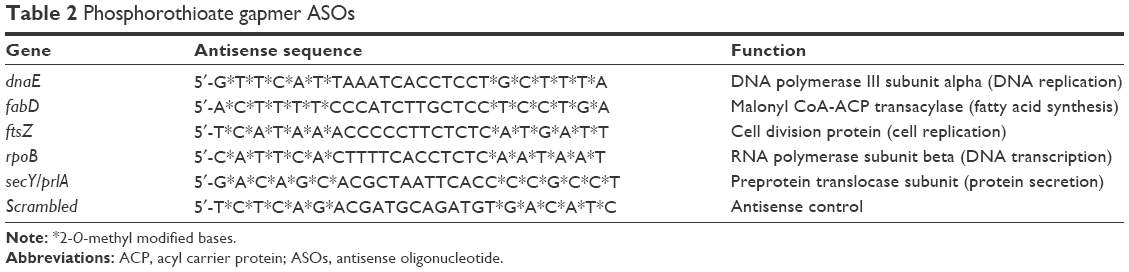 | Table 2 Phosphorothioate gapmer ASOs |
Luciferase reporter plasmids
A set of luciferase reporter plasmids were constructed for quantifying the degree of translation inhibition of each phosphorothioate gapmer in a cell-free transcription–translation system. Specifically, a region of the 5′UTR of the firefly luciferase (FLuc) gene sequence (spanning the corresponding gapmer binding locations) from a reporter plasmid under control of a constitutive recA/ΔlexA promoter (pSF-OXB20-FLuc, Oxford Genetics UK) was modified to match the 5′-UTRs of the five candidate essential C. difficile genes.
Construction of the initial reporter plasmid utilized an overhang-extension PCR (OE-PCR) strategy to generate a FLuc gene with its proximal 5′UTR modified to match the concordant 37 bp sequence of the C. difficile dnaE gene. High-performance liquid chromatography (HPLC)-purified OE-PCR primers (Integrated DNA Technologies; dnaE_F and dnaE_R; Table 1) were used to amplify the modified 1691 bp FLuc sequence using a high-fidelity DNA polymerase (Q5 Hot Start; NEB, Ipswich, MA, USA). The OE-PCR product with the substituted 5′UTR sequence extended from the plasmid HindIII restriction site located upstream of the FLuc ATG start codon through the FLuc 3′-terminal XbaI site (Figure 4A). HindIII/XbaI restriction enzyme digestion of the column-purified (MinElute; Qiagen, Valencia, CA, USA) OE-PCR amplicon generated a dnaE 5′UTR-FLuc insert with disparate cloning termini that was then purified by excision from a 1.2% agarose gel. Separate HindIII/XbaI digestion of the plasmid expelled the native FLuc sequence and the linearized vector fragment was dephosphorylated with alkaline phosphatase to prevent annealing and gel-purified. The insert was then cloned in-context to the luciferase start codon of the linearized vector using T4 DNA ligase. Ligation reaction products were transformed by heat-shock (42°C, 40 seconds) into competent Escherichia coli cells (XL1-Blue, Agilent Genomics, Santa Clara, CA, USA) and transformants selected by 37°C overnight incubation on yeast extract/tryptone (YT) agar plates containing 50 μg/mL kanamycin (Km). Km-resistant colonies were cultured in YT-Km broth for plasmid DNA isolations (PureLink; Invitrogen, Wilmington, DE, USA). The sequence integrity of the completed reporter plasmid, pCdiff5′UTR-FLuc-dnaE (Figure 4A), was confirmed by ABI 3130XL capillary sequencing (5′UTR-F; Table 1; BigDye Terminator chemistry v3.1; Applied Biosystems, Carlsbad, CA, USA).
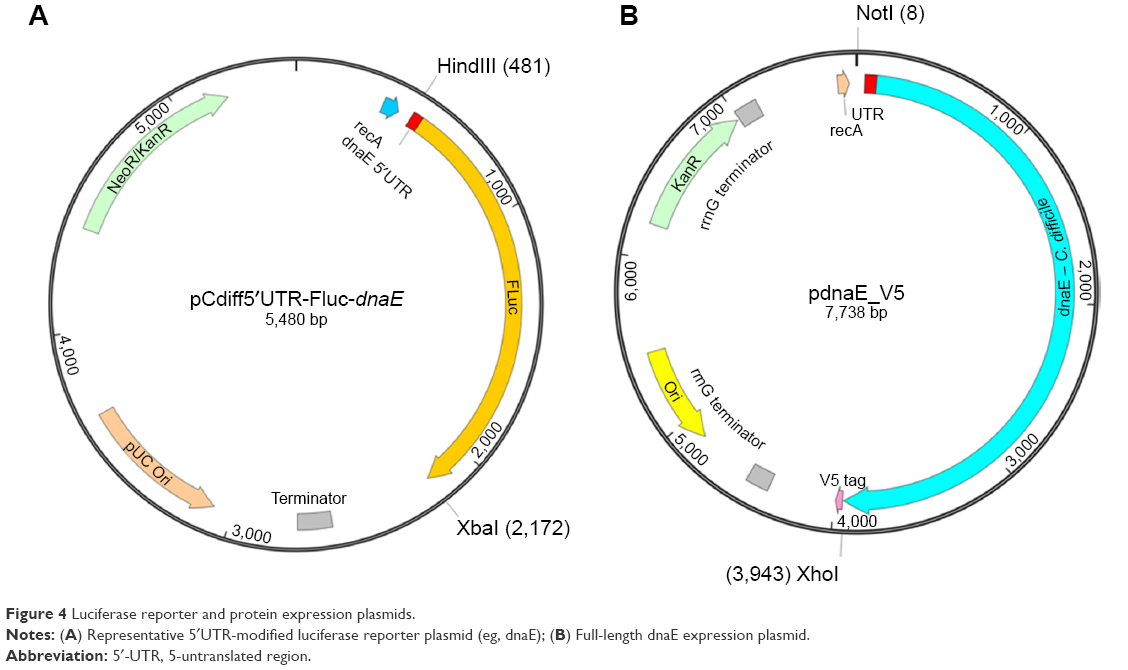 | Figure 4 Luciferase reporter and protein expression plasmids. |
Reporter plasmids for the remaining gapmer ASOs (fabD, ftsZ, rpoB, and secY) were constructed through site-directed mutagenesis of pCdiff5′UTR-FLuc-dnaE by designing adjacent PCR primers with the desired 5′UTR substitution split between the primer 5′-ends (site-directed mutagenesis primers; Table 1). Whole plasmid mutagenesis high-fidelity PCR generated linear double-stranded plasmid with the desired 5′UTR-substitution. The initial methylated plasmid template was selectively removed by DpnI Type IIM endonuclease digestion followed by phosphorylation of the 5′-termini with T4 polynucleotide kinase. The plasmid was recircularized through T4 DNA ligase intramolecular ligation, transformed into competent XL1-Blue cells, and then incubated on YT-Km plates. Plasmid DNA purified from broth cultures of Km-resistant colonies were capillary sequenced to confirm the integrity of the substituted 5′UTR regions for each completed reporter construct.
S30 cell-free mRNA translation
The effect of antisense gapmers on FLuc mRNA translation was determined in 50 μL coupled transcription–translation reactions using a prokaryotic S30 E. coli cytoplasmic extract (S30 Circular DNA System; Promega Corp, Madison WI, USA).47,48 Reactions were programmed with 250 ng of the appropriate plasmid in 96-well chimney microplates (CellStar; Greiner Bio-One, Monroe, NC, USA) with gapmer ASOs tested at serial concentrations from 0.01 to 5 μM. Following incubation for 45 minutes at 35°C, the reaction mixtures were cooled on ice and mixed with an equal volume of luciferin buffer containing ATP, coenzyme A, and Mg2+. Luciferase light emission, proportional to the amount of translated FLuc protein product, was immediately measured as relative luminosity units (RLU) using a luminometer with an integration time of 5 seconds with Gen5 software (Synergy 2, BioTek, Winooski, VT, USA). The mean and standard error of three measurements per sample were calculated and the inhibition of FLuc mRNA translation determined by dividing the normalized RLUs of the antisense reactions by the RLUs of untreated controls minus background fluorescence.
dnaE protein expression and immunoblotting
Observations from luciferase reporter data were further validated by cloning the 4 kb coding sequence of the C. difficile dnaE gene, including 70 nucleotides of its proximal 5′UTR sequence, into a protein expression plasmid. The PCR-amplified sequence (dnaE-Full-NotI, dnaE-Full-XhoI; Table 1) was NotI/XhoI digested, column purified, and cloned into a restriction site adjacent to a 3′-V5 epitope (GKPIPNPLLGLDST) tag and then transformed into XL1-Blue cells. Plasmid DNA extracted from Km-resistant colonies was confirmed for the appropriate sequence substitution by capillary sequencing, yielding the final expression construct (pdnaE_V5; Figure 4B). The dnaE and scrambled gapmer ASOs at serial concentrations ranging from 0.01 to 1 μM were evaluated in cell-free reactions, as described earlier, with the reactions programmed with pdnaE_V5.
Polyethylene glycol was removed from the completed dnaE expression reactions by precipitation of the proteins with 20 volumes of chilled acetone and incubation on ice for 15 minutes. Proteins were pelleted (12,000 rcf, 5 minutes, 4°C) and then air-dried for 45 minutes. Dried pellets were resuspended in 100 μL of lithium dodecyl sulfate–glycerol reducing buffer (pH 8.4) followed by centrifugation at 10,000 rcf to remove particulates. Aliquots (10 μL/lane) were heated at 70°C for 10 minutes and loaded along with molecular weight marker spaced in triplicate (Precision Plus Protein™ Dual Color Standards, Bio-Rad, Hercules, CA, USA) on 12% Bis-Tris Novex gels to ensure even transfer to PVDF membrane. The 138 kDa dnaE protein bands resolved by 10% Bis-Tris polyacrylamide gel electrophoresis in a MOPS/SDS buffer system (Life Technologies Grand Island, NY, USA) at 175 V for 2 hours. Proteins were wet-transferred in buffer containing 12% methanol onto 0.45 μm polyvinylidene difluoride membranes (Immobilon; Millipore, Billerica, MA, USA) by electroblotting at 100 V for 1 hour at 4°C. Immunoblotting was initiated by blocking the membranes for 1 hour in tris-buffered saline with Tween 20 (TBST) buffer (20 mM Tris-HCL pH 7.6, 150 mM NaCl, 0.1% Tween 20) containing 5% nonfat milk. Blocked membranes were labeled overnight in 0.5% nonfat milk containing 1:2,000 mouse anti-V5 epitope IgG2a monoclonal antibody (R960-25; Thermo Fisher Scientific, Wilmington, DE, USA). The anti-V5-labeled membranes were washed three times in TBST and then secondarily labeled for 1 hour with 50 ng/mL goat anti-mouse IgG horseradish peroxide (HRP)-conjugate antibody (sc-2005; Santa Cruz Biotech) in 0.5% nonfat milk. The HRP antibody-labeled membranes were washed five times in TBST to remove nonbound conjugate antibody and then incubated for 5 minutes in an HRP chemiluminescent substrate (Super Signal Pico; Pierce Biotechnology, Rockford, IL, USA). Protein bands were detected by 5–15-minute exposures of the membranes to autoradiography film (HyBlot CL; Denville Scientific, South Plainfield, NJ, USA). dnaE protein bands (138 kDa) were quantified by densitometry using ImageJ software (NIH, Bethesda, MD, USA).
Synthesis and purification of cyclohexyl-DQA bolaamphiphile compounds
N,N′-(Decane-1,10-diyl)-bis(9-amino-1,2,3,4-tetrahydroacridinium)dichloride [10-cyclohexyl-DQA, 3]. A mixture of 1,2,3,4-tetrahydroacridin-9-amine (0.53 g, 2.67 mmol) and 1,10-dichlorodecane (0.26 g, 1.23 mmol) was gradually heated to 160°C over 30 minutes. Molten mixture heated continuously at 185°C –195°C solidifies after the first hour. After heating at that temperature for another 4 hours, the solid mass was cooled and dissolved in boiling methanol (10 mL). Acetone (25 mL) was added, and then the mixture set aside to form a precipitate (0.28 g, 28%). The material was further purified by chromatography on alumina using a gradient of CH3OH in CH2Cl2 as solvents and finally recrystallized from CH3OH to give 3 (Figure 1B) as a white solid. 1H NMR (500 MHz, CDCl3) δ 1.30–1.38 (m, 8H), 1.45–1.52 (m, 4H), 1.71–1.77 (m, 4H), 1.82–1.85 (m, 4H), 1.87–1.91 (m, 4H), 2.60 (t, 4H, J=6.5 Hz), 4.48 (t, 4H, J=8.0 Hz), 7.70 (dd, 2H, J=7.5 and 7.5 Hz), 7.98 (dd with fine splitting, 2H, J=8.0 and 6.5 Hz), 8.13 (d, 2H, J=9.0 Hz), 8.60 (d with fine splitting, 2H, J=8.5 Hz), 8.96 (br s, 4H, NH2). HRMS m/z 536.7 (M-2Cl).
N,N′-(Dodecane-1,12-diyl)-bis(9-amino-1,2,3,4-tetrahydroacridinium)dichloride [12-cyclohexyl-DQA, 4]. A mixture of 1,2,3,4-tetrahydroacridine-9-amine (0.4 g, 2.01 mmol) and 1,12-dichlorododecane (0.24 g, 1.00 mmol) was slowly heated to 160°C, over 40 minutes under N2, when it melted into a brown stirrable liquid. Heating was continued and the mixture solidified when the temperature reached 175°C (1 hour). The reaction mixture was maintained at 190°C–200°C for another 3 hours and was then cooled, wetted with CH3OH (5 mL), and abraded. The solid was filtered off and dissolved in a mixture of CH2Cl2 and CH3OH for chromatographical purification on alumina (neutral, Brockmann I) using CH2Cl2:CH3OH (4:1) as an eluent. The product thus obtained was further purified by recrystallization from CH3OH/Et2O to yield 4 (Figure 1C) as a white solid. 1H NMR (500 MHz, CDCl3) δ 1.24–1.36 (m, 12H), 1.44–1.50 (m, 4H), 1.69–1.76 (m, 4H), 1.80–1.85 (m, 4H), 1.87–1.91 (m, 4H), 2.60 (t, 4H, J=6.0 Hz), 4.48 (t, 4H, J=8.0 Hz), 7.71 (dd, 2H, J=8.0 and 7.5 Hz), 7.98 (dd with fine splitting, 2H, J=8.0 and 7.0 Hz), 8.13 (d, 2H, J=9.0 Hz), 8.57 (d with fine splitting, 2H, J=8.5 Hz), 8.76 (br s, 4H, NH2). HRMS m/z 564.7 (M-2Cl).
Purity determination of compounds 3 and 4: the purity of compounds 3 and 4 was determined by analytical HPLC using Zorbax SB-C8 3.0 mm ×25 cm column. Isocratic elutions using solvent mixtures of A = water +0.5% sodium hexanesulfonate +0.5% orthophosphoric acid, and B = CH3OH +0.5% sodium hexanesulfonate +0.5% orthophosphoric acid at the ratio of A:B/25:75 and the rate of 1 mL/minute were performed and the compounds were found to be ≥98% pure. Retention times (Rt) at the aforementioned conditions were 3.92 minutes for compound 3 and 6.26 minutes for compound 4.
Preparation of cyclohexyl-DQA bolasomes
Cyclohexyl-DQA bolasomes were prepared as previously described.49 Briefly, the compound was dissolved in 10 mL methanol in a round-bottom flask and then the solvent was evaporated with a rotary evaporator. The resulting thin-film residue was hydrated at 10 mM by vortexing in ultrapure water. Bolasomes were formed by pulse sonication for 45 minutes on ice using an ultrasonic processor with a high-intensity probe maintained at a 12 μm amplitude (XL-2000; Misonix, Farmingdale, NY, USA). Bolasome preparations were centrifuged (2,000 rcf, 10 minutes), followed by 0.45 μm surfactant-free cellulose acetate filtration to remove particulates and stored at 4°C. Concentrations of bolasome preparations (1:250 dilution in methanol) were measured by fluorescence spectroscopy (245 nm excitation, 370 nm emission; 10 nm bandwidth) against prepared standards (linear between 1 and 10 μM).
Characterization of cyclohexyl-DQA bolasomes
The mean particle hydrodynamic diameters (z-average), size distribution (polydispersity index), and zeta potentials were measured at 25°C with a Zetasizer Nano series instrument (Malvern Instruments Inc., Westborough, MA, USA) by dynamic light scattering and laser Doppler velocimetry. The results were averaged from four measurements of each sample preparation.
Preparation of bolasome–gapmer nanocomplexes
Nanocomplexes of bolasomes and antisense phosphorothioate gapmers were prepared by mixing the appropriate phosphorothioate gapmer and cationic bolasomes in 25 mM MES (2-(N-morpholino)ethanesulfonic acid; pH 5.5)-buffered solutions at room temperature with incubation for 20 minutes before use. For cell treatment experiments, bolasomes were mixed at a 40:1 molar ratio with appropriate gapmer to yield the nanocomplex working preparations.
Bolasome binding capacities
The amount of bolasomes for each compound required to fully bind 25-mer phosphorothioate gapmer oligonucleotide was determined using an unsymmetrical cyanine dye-exclusion assay.50 The fluorescent single-stranded nucleic acid dye, Oligreen (Thermo Fisher Scientific, Wilmington, DE, USA), does not effectively bind to single-stranded phosphorothioate bases following cationic complexation, leading to a large decrease in fluorescence emission intensity compared with free oligonucleotide. Binding was measured by exclusion of the dye from oligonucleotide binding through a stepwise mixing of bolasomes into triplicate MES-buffered 2 μg/mL gapmer ASO solutions in black 96-well optical bottom microplates (Nunc; Thermo Fisher Scientific, Wilmington, DE, USA). Following a 20 minute incubation to stabilize the fluorescent signals, readings (485 nm excitation, 520 nm emission; 20 nm bandwidth) were taken and complexation curves for each experiment created.
Bolasome–gapmer complexation assays
The complexation and retention of phosphorothioate antisense gapmers by cyclohexyl-DQA bolasomes was determined by gel electrophoresis of complexes prepared with 1 μg of gapmer and bolasomes over a range of N/P molar ratios (where N represents compound unprotonated bolaamphiphile amine groups and P represents anionic oligonucleotide phosphate groups). Complexation was assessed following migration of the nanocomplexes through 6% Metaphor agarose Tris-borate-EDTA gels with 0.5× SYBR Green II at 5 V/cm. Full retention of 25-mer gapmer was documented as disappearance of free oligo fluorescence upon epi-illumination using a SYBR Green filter (Bio Doc-IT System, Upland, CA, USA).
Nanocomplex minimum inhibitory concentrations (MIC)
Empty bolasomes and gapmer ASO nanocomplexes were tested for dose effects on the growth of C. difficile. For nanocomplexes, the broth microdilution method51 was used to determine the lowest concentration (MIC) that completely inhibited the growth of C. difficile. Specifically, antisense complexes were tested for C. difficile growth inhibition in triplicate 200 μL BHIS cultures (prereduced brain–heart infusion broth supplemented with L-cysteine, hemin, and vitamin K1) inoculated 1:10 with a log phase culture (1×106 [colony-forming units] CFU/mL) of C. difficile in optical bottom 96-well microplates (μClear 655087; Greiner Bio-One, Monroe, NC, USA). Nanocomplex preparations were evaluated using a twofold dilution series (100–800 nM final gapmer doses). Following anaerobic culture at 37°C for 24 hours, growth of cultures was assessed at OD590 nm on a UV-Visible spectrophotometer microplate reader. The triplicate MIC experiments were repeated using a fresh ribotype 027 log-phase culture to confirm observations. MIC values for nanocomplex-treated cultures were interpreted as the doses where growth was completely inhibited (<0.1 log growth) as compared to initial inoculum OD590 nm absorbance values.
Experiments examining the effects of 12-cyclohexyl-DQA gapmer nanocomplex and equivalent empty bolasome treatments on the growth of log-phase C. difficile were repeated and quantified by determining viable counts of CFU for both the initial culture inoculums and the triplicate cultures following 24 hour incubation. CFUs were determined by serial tenfold dilutions of the initial inoculum and of the 24 hour cultures in prereduced BHIS broth. Aliquots of serial dilutions were spread plated on prereduced TSA-II blood agar plates and the colonies counted on plates containing between 20 and 300 CFU following 48 hours anaerobic incubation at 37°C.
Results
Luciferase reporter S30 translation assays
The dose effect of individual 25-mer gapmer ASOs (0.01–5 μM) targeting sites within the 5′UTR transcription initiation region of five essential C. difficile genes was quantified within cell-free S30-coupled transcription–translation reactions. Treatments for all five gapmer ASOs resulted in a >50% decrease in the production of FLuc protein at 1 μM, with three gapmers (rpoB, ftsZ, and dnaE) achieving 60%–80% reductions in FLuc protein at 0.1 μM and a ≥90% reduction at 1 μM (Figure 5). A scrambled 25-mer antisense gapmer sequence had negligible effect (<10% at 5 μM) on the amount of translated FLuc mRNA protein product.
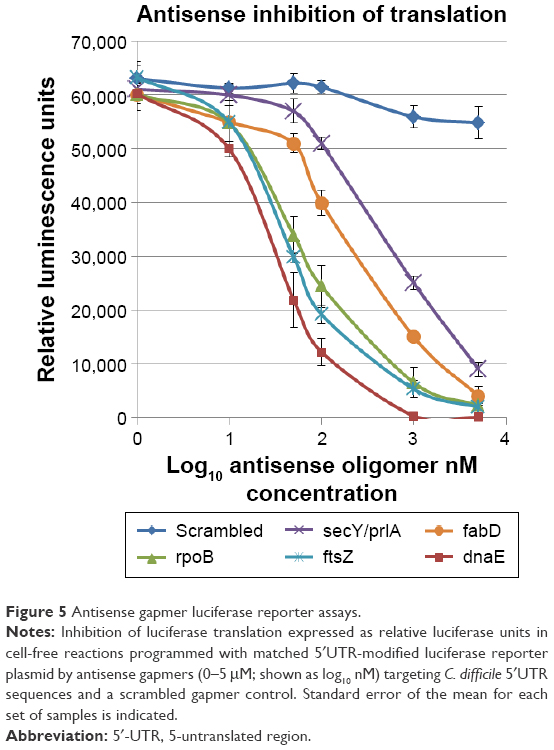 | Figure 5 Antisense gapmer luciferase reporter assays. |
dnaE expression immunoblotting
Coupled transcription–translation reactions programmed with a dnaE expression plasmid were used to examine the dose effect (0.01–1 μM) of the dnaE gapmer on the translational yield of dnaE protein. Protein was precipitated from dnaE expression plasmid translation reactions treated in triplicate with serial doses of dnaE-gapmer ASO and a scrambled gapmer ASO control. HRP immunoblotting of V5-tagged proteins showed a clear dose effect for the antisense gapmer on the yield of dnaE protein, with essentially complete silencing of mRNA translation at 0.5 μM (Figure 6). Limited inhibition was observed on dnaE protein production for treatments with scrambled gapmer, without an apparent dose effect.
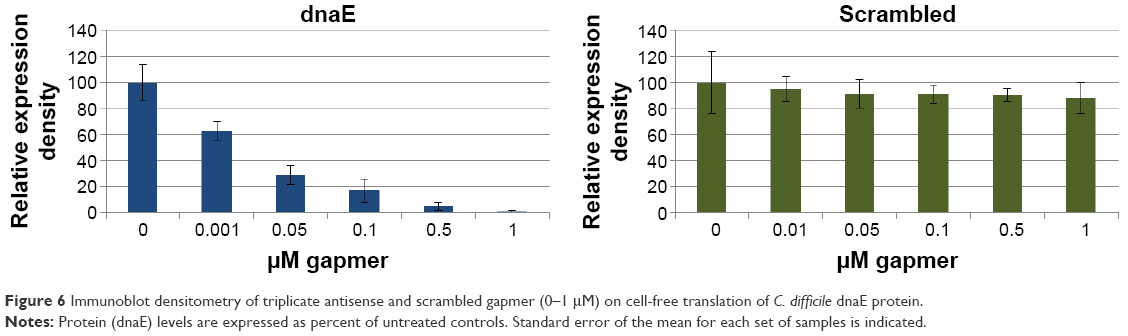 | Figure 6 Immunoblot densitometry of triplicate antisense and scrambled gapmer (0–1 μM) on cell-free translation of C. difficile dnaE protein. |
Synthesis of cyclohexyl-DQA bolaamphiphile compounds
The compounds 10-cyclohexyl-DQA 3 and 12-cyclohexyl-DQA 4 were synthesized according to a previously reported procedure,52 with modifications as outlined in Figure 7. Briefly, tricyclic aromatic amine 1 was heated together with alkyl dichloride 2 at an elevated temperature without any solvent to affect the quaternization of heterocyclic nitrogens, therefore linking the two acridine moieties with an alkyl chain. Both the compounds were purified by column chromatography on alumina and characterized on the basis of 1H NMR and mass spectral data. The purity of the compounds was determined by analytical HPLC and was found to be ≥98%.
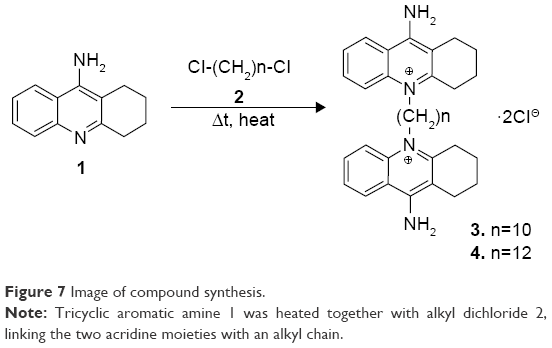 | Figure 7 Image of compound synthesis. |
Characterization of cyclohexyl-DQA bolasomes
The cyclohexyl-DQA bolasomes were characterized with respect to their size distribution and zeta potential (Table 3). The results are in perfect agreement with previously published data.53 Oligreen dye-exclusion assays were performed to determine the weight amount of bolasomes for each compound required for binding of 2 μg of ASO. Following the subtraction of background fluorescence, complete binding of 25-mer gapmer was achieved upon addition of 4 μg of either bolasome preparation per microgram of phosphorothioate oligonucleotide gapmer (Figure 8A). The nanocomplex N/P ratio should preferably be sufficiently high to complex the anionic oligonucleotide cargo and provide a net positive charge to promote interaction with the negatively charged cells, while minimizing the complexed vesicle size and any toxicity issues.26,54 Complexation and retention of 25-mer gapmer ASO during agarose gel electrophoresis with bolasome preparations of both compounds resulted in the complete loss of free oligonucleotide fluorescence approaching an N/P ratio of 5 (Figure 8B).
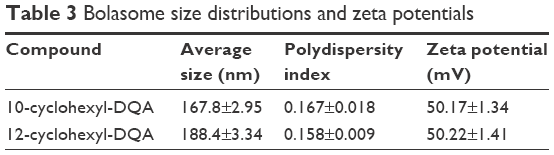 | Table 3 Bolasome size distributions and zeta potentials |
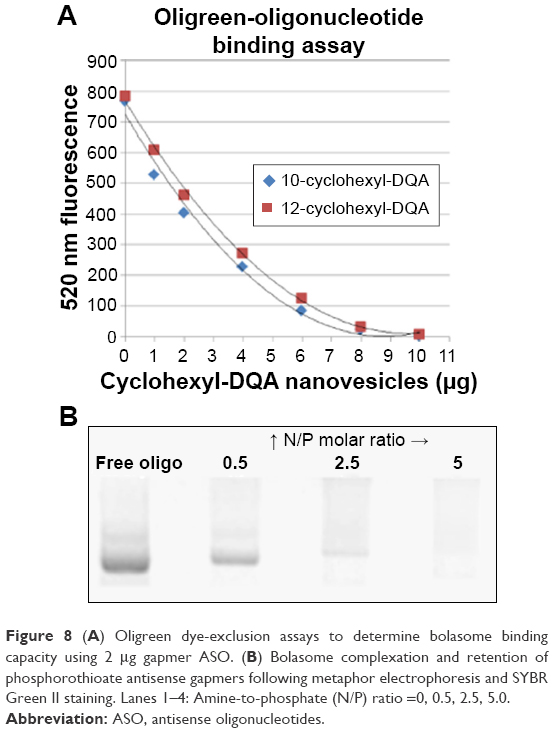 | Figure 8 (A) Oligreen dye-exclusion assays to determine bolasome binding capacity using 2 μg gapmer ASO. (B) Bolasome complexation and retention of phosphorothioate antisense gapmers following metaphor electrophoresis and SYBR Green II staining. Lanes 1–4: Amine-to-phosphate (N/P) ratio =0, 0.5, 2.5, 5.0. |
Nanocomplex MICs
No inhibition of C. difficile ribotype 027 growth in 24 hour triplicate anaerobic BHIS cultures was observed based on 590 nm optical densities values following treatment with empty bolasomes as compared to untreated control cultures, including the highest dose (9.6 μM bolasomes complexing 800 μM gapmer) evaluated in the antisense nanocomplex experiments. Further testing of empty bolasomes at higher doses demonstrated significant inhibition of growth starting at a lower concentration (12 μM) for 10-cyclohexyl-DQA bolasomes compared to 12-cyclohexyl-DQA bolasomes (18 μM). No inhibition of C. difficile growth was observed following treatment with nanocomplexes with a scrambled 25-mer phosphorothioate gapmer sequence, including the highest tested dose (800 nM gapmer ASO). In contrast, 10-cyclohexyl-DQA and 12-cyclohexyl-DQA nanocomplexes of gapmer ASOs targeting the expression of four C. difficile genes (dnaE, fabD, ftsZ, and rpoB) reached MIC values in 24 hour triplicate BHIS broth cultures at or below a dose of 400 μM (antisense gapmer). The lowest MIC (200 nM antisense gapmer) was observed for gapmers targeting the C. difficile polymerase genes rpoB and dnaE.
The effects of 12-cyclohexyl-DQA gapmer nanocomplexes and equivalent empty bolasome treatments on the growth of log-phase C. difficile cultures were replicated and further measured using quantitative CFU counts in triplicate. CFU results are expressed as average CFU/mL growth normalized to initial inoculum CFU counts (Figure 9).
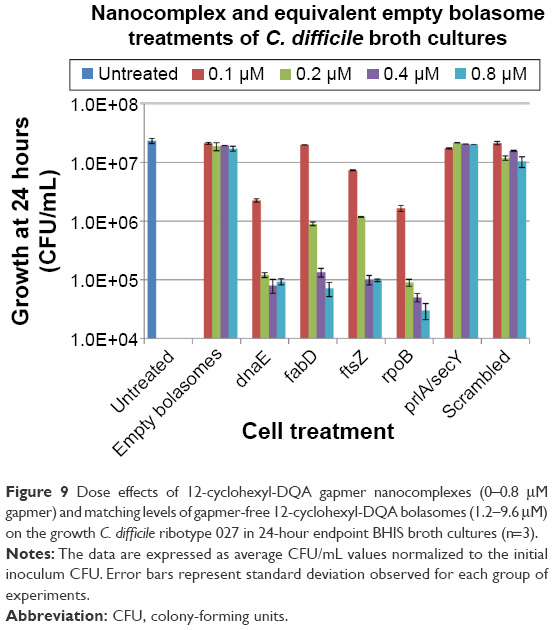 | Figure 9 Dose effects of 12-cyclohexyl-DQA gapmer nanocomplexes (0–0.8 μM gapmer) and matching levels of gapmer-free 12-cyclohexyl-DQA bolasomes (1.2–9.6 μM) on the growth C. difficile ribotype 027 in 24-hour endpoint BHIS broth cultures (n=3). |
Discussion
The application of antisense inhibition to bacterial cells is an attractive alternative to conventional antibiotics but presents a particular challenge in requiring efficient delivery of antisense molecules across bacterial cell envelopes. Compared to the many presently available nonviral systems for use with eukaryotic cells, there are few demonstrated carriers for antisense delivery into bacteria. The uptake of ASOs by bacteria can often be improved by conjugation to a membrane-penetrating peptide. The efficacy of this approach is dependent on the species of bacteria, with more success reported for gram-negative species. In our previous work, evaluation of a panel of cell-penetrating peptides with C. difficile demonstrated limited cellular delivery following conjugation to antisense morpholinos.19
In this work, we investigated the use of mitochondriotropic, cyclohexyl-DQA bolasomes as a phosphorothioate gapmer ASO delivery system for C. difficile. Gapmer ASO targeting the expression of five essential C. difficile genes was quantitatively assessed for dose-related knockdown of protein expression using luciferase reporter and protein expression plasmids in coupled transcription–translation reactions. Cationic acridinium bolasomes were characterized for their particle size distributions, zeta potentials, and binding capacity to anionic gapmer ASO. Bolasome–gapmer nanocomplexes or equivalent amounts of empty bolasomes were tested for dose effects in 24 hour anaerobic log phase C. difficile broth cultures. No significant inhibition of bacterial growth was observed from treatments at matched dosages with scrambled gapmer (100–800 nM) nanocomplexes or empty bolasomes (1.2–9.6 μM). In contrast, nanocomplex MICs in 24-hour broth cultures were reached at nanomolar doses for gapmer ASOs targeting four C. difficile genes.
The lowest nanocomplex MICs (200 nM antisense gapmer) were observed for gapmers targeting the C. difficile polymerase genes rpoB and dnaE. In contrast to dnaE (DNA polymerase III subunit alpha) where silencing inhibits DNA replication, the silencing of rpoB (RNA polymerase subunit beta) expression would inhibit DNA transcription, and thereby should function in a cyclic synergy with the ASO, making rpoB a particularly attractive gene target for further study.55 A potential limitation in the use of cationic bolasomes with bacteria is their intrinsic antimicrobial activity at high concentrations. We are currently synthesizing additional cyclohexyl-DQA derivatives with longer alkyl chains and/or modified polar head groups in an attempt to identify delivery compounds with decreased antimicrobial activity.
In summary, we report the first in vitro antisense treatment to inhibit the growth of the gram-positive pathogen, C. difficile. We also describe the novel application of cationic acridinium bolasomes as an ASO delivery system for a bacterium, with potential application to other species. The efficient delivery of antisense molecules into bacteria can allow the advantageous targeting of virulence functions essential for infection, disease, and recurrence. Antisense antibacterials might also be utilized as an adjuvant therapy in combination with conventional antibiotic treatment, potentially increasing treatment effectiveness and decreasing the likelihood for development of resistance.
Acknowledgments
The authors thank Dr Jon Moulton, GeneTools, LLC, for helpful discussions on the cellular delivery of ASOs. This study was supported by a grant from the Clinical and Translational Science Institute of the Pennsylvania State University.
Disclosure
The authors declare no conflicts of interests in this work.
References
Davies AH, Roberts AK, Shone CC, Acharya KR. Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem J. 2011;436(3):517–526. | ||
McFarland LV, Elmer GW, Surawicz CM. Breaking the cycle: treatment strategies for 163 cases of recurrent Clostridium difficile disease. Am J Gastroenterol. 2002;97(7):1769–1775. | ||
Cammarota G, Ianiro G, Gasbarrini A. Fecal microbiota transplantation for the treatment of Clostridium difficile infection: a systematic review. J Clin Gastroenterol. 2014;48(8):693–702. | ||
Steglich M, Nitsche A, Von Müller L, et al. Tracing the spread of Clostridium difficile ribotype 027 in Germany based on bacterial genome sequences. PLoS One. 2015;10(10):e0139811. | ||
Gilliland C, Weaver J, Stevenson M, et al. Association of Clostridium difficile ribotype 078 with detectable toxin in human stool specimens. J Med Microbiol. 2015;64(11):1341–1345. | ||
Carlson PE, Walk ST, Bourgis AE, et al. The relationship between phenotype, ribotype, and clinical disease in human Clostridium difficile isolates. Anaerobe. 2013;24:109–116. | ||
Archbald-Pannone LR, Boone JH, Carman RJ, Lyerly DM, Guerrant RL. Clostridium difficile ribotype 027 is most prevalent among inpatients admitted from long-term care facilities. J Hosp Infect. 2014;88(4):218–221. | ||
Bai H, Xue X, Hou Z, Zhou Y, Meng J, Luo X. Antisense antibiotics: a brief review of novel target discovery and delivery. Curr Drug Discov Technol. 2010;7(2):76–85. | ||
Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. 2007;3(9):541–548. | ||
Rasmussen LC, Sperling-Petersen HU, Mortensen KK. Hitting bacteria at the heart of the central dogma: sequence-specific inhibition. Microb Cell Fact. 2007;6(1):24. | ||
Dinan AM, Loftus BJ. (Non-)translational medicine: targeting bacterial RNA. Front Genet. 2013;4(Nov):230. | ||
Ji Y, Lei T. Antisense RNA regulation and application in the development of novel antibiotics to combat multidrug resistant bacteria. Sci Prog. 2013;96(1):43–60. | ||
Järver P, Coursindel T, Andaloussi SE, Godfrey C, Wood MJ, Gait MJ. Peptide-mediated cell and in vivo delivery of antisense oligonucleotides and siRNA. Mol Ther Nucleic Acids. 2012;1(6):e27. | ||
Hansen AM, Bonke G, Larsen CJ, Yavari N, Nielsen PE, Franzyk H. Antibacterial peptide nucleic acid – antimicrobial peptide (PNA-AMP) conjugates: antisense targeting of fatty acid biosynthesis. Bioconjug Chem. 2016;27(4):863–867. | ||
Geller BL, Marshall-Batty K, Schnell FJ, McKnight MM, Iversen PL, Greenberg DE. Gene-silencing antisense oligomers inhibit Acinetobacter growth in vitro and in vivo. J Infect Dis. 2013;208(10):1553–1560. | ||
Wang H, Meng J, Jia M, et al. oprM as a new target for reversion of multidrug resistance in Pseudomonas aeruginosa by antisense phosphorothioate oligodeoxynucleotides. FEMS Immunol Med Microbiol. 2010;60(3):275–282. | ||
Meng J, Da F, Ma X, et al. Antisense growth inhibition of methicillin-resistant Staphylococcus aureus by locked nucleic acid conjugated with cell-penetrating peptide as a novel FtsZ inhibitor. Antimicrob Agents Chemother. 2015;59(2):914–922. | ||
Patenge N, Pappesch R, Krawack F, et al. Inhibition of growth and gene expression by PNA-peptide conjugates in streptococcus pyogenes. Mol Ther Nucleic Acids. 2013;2(April):e132. | ||
Hegarty JP, Sangster W, Harris LR, Durdach M, Stewart DB. Targeted silencing of essential C. difficile genes using antisense morpholino-peptide conjugates. Presented at: 2015 American Society for Microbiology. 115th General Meeting; 2015; New Orleans, LA; 011:51. | ||
Zhao XB, Pan F, Zhang ZQ, et al. Nanostructure of polyplexes formed between cationic diblock copolymer and antisense oligodeoxynucleotide and its influence on cell transfection efficiency. Biomacromolecules. 2007;8(11):3493–3502. | ||
Chen Z, Hu Y, Meng J, et al. Efficient transfection of phosphorothioate oligodeoxyribonucleotides by lipofectamine 2000 into different bacteria. Curr Drug Deliv. 2015;12:1–8. | ||
Zhang L, Pornpattananangku D, Hu C-MJ, Huang C-M. Development of nanoparticles for antimicrobial drug delivery. Curr Med Chem. 2010;17(6):585–594. | ||
Khameneh B, Diab R, Ghazvini K, Fazly Bazzaz BS. Breakthroughs in bacterial resistance mechanisms and the potential ways to combat them. Microb Pathog. 2016;95:32–42. | ||
Forier K, Raemdonck K, De Smedt SC, Demeester J, Coenye T, Braeckmans K. Lipid and polymer nanoparticles for drug delivery to bacterial biofilms. J Control Release. 2014;190:607–623. | ||
Hong CA, Nam YS. Functional nanostructures for effective delivery of small interfering RNA therapeutics. Theranostics. 2014;4(12):1211–1232. | ||
Khan M, Ang CY, Wiradharma N, et al. Diaminododecane-based cationic bolaamphiphile as a non-viral gene delivery carrier. Biomaterials. 2012;33(18):4673–4680. | ||
Lyrawati D, Trounson A, Cram D. Expression of GFP in the mitochondrial compartment using DQAsome-mediated delivery of an artificial mini-mitochondrial genome. Pharm Res. 2011;28(11):2848–2862. | ||
Weissig V, Lasch J, Erdos G, Meyer H, Rowe T, Hughes J. DQAsomes: a novel potential drug and gene delivery system made from dequalinium. Pharm Res. 1998;15(2):334–337. | ||
Weissig V, Lizano C, Torchilin VP. Selective DNA release from DQAsome/DNA complexes at mitochondria-like membranes. Drug Deliv. 2000;7(1):1–5. | ||
Weissig V, Lizano C, Ganellin C, Torchilin V. DNA binding cationic bolasomes with delocalized charge center – a structure-activity relationship study. STP Pharma Sci. 2001;11(1):91–96. | ||
Weissig V. From serendipity to mitochondria-targeted nanocarriers. Pharm Res. 2011;28(11):2657–2668. | ||
Grinberg S, Stern A, Guidotti M, et al. Steric environment around acetylcholine head groups of bolaamphiphilic nanovesicles influences the release rate of encapsulated compounds. Int J Nanomedicine. 2014;9:561. | ||
Schlame M. Cardiolipin synthesis for the assembly of bacterial and mitochondrial membranes. J Lipid Res. 2008;49(8):1607–1620. | ||
Guan Z, Katzianer D, Zhu J, Goldfine H. Clostridium difficile contains plasmalogen species of phospholipids and glycolipids. Biochim Biophys Acta. 2014;1841(10):1353–1359. | ||
Yoo BH, Bochkareva E, Bochkarev A, Mou T-C, Gray DM. 2′-O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004;32(6):2008–2016. | ||
Niemietz C, Chandhok G, Schmidt H. Therapeutic oligonucleotides targeting liver disease: TTR amyloidosis. Molecules. 2015;20(10):17944–17975. | ||
Chan JH, Lim S, Wong WF. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33(5–6):533–540. | ||
Stewart DB, Berg AS, Hegarty JP. Single nucleotide polymorphisms of the tcdC gene and presence of the binary toxin gene predict recurrent episodes of Clostridium difficile infection. Ann Surg. 2014;260(2):299–304. | ||
Stubbs SL, Brazier JS, O’Neill GL, Duerden BI. PCR targeted to the 16S-23S rRNA gene intergenic spacer region of Clostridium difficile and construction of a library consisting of 116 different PCR ribotypes. J Clin Microbiol. 1999;37(2):461–463. | ||
Rupnik M, Avesani V, Janc M, Von Eichel-Streiber C, Delmée M. A novel toxinotyping scheme and correlation of toxinotypes with serogroups of Clostridium difficile isolates. J Clin Microbiol. 1998;36(8):2240–2247. | ||
Adey A, Morrison HG, Asan, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010;11(12):R119. | ||
Dembek M, Barquist L, Boinett CJ, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. MBio. 2015;6(2):e02383. | ||
Lima WF, Vickers TA, Nichols J, Li C, Crooke ST. Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One. 2014;9(7):e101752. | ||
Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics. 2010;11(1):129. | ||
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–3415. | ||
Gish W, Altschul SF, Lipman DJ, Miller W, Myers EW. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. | ||
Shao Y, Wu Y, Chan CY, McDonough K, Ding Y. Rational design and rapid screening of antisense oligonucleotides for prokaryotic gene modulation. Nucleic Acids Res. 2006;34(19):5660–5669. | ||
Zaher HS, Green R. In vitro synthesis of proteins in bacterial extracts. Methods Enzymol. 2014;539:3–15. | ||
Weissig V. DQAsomes as the prototype of mitochondria-targeted pharmaceutical nanocarriers: preparation, characterization, and use. Methods Mol Biol. 2015;1265:1–11. | ||
Márquez-Miranda V, Peñaloza JP, Araya-Durán I, et al. Effect of terminal groups of dendrimers in the complexation with antisense oligonucleotides and cell uptake. Nanoscale Res Lett. 2016;11(1):66. | ||
European Society of Clinical Microbiology and Infectious Diseases. Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clin Microbiol Infect. 2003;9(8):1–7. | ||
Jerchel D, Ost W, Klaus T. Quaternary derivatives of 1,2,3,4-tetra-hydro-9-amino-acridine. US patent 3519631; 1970. | ||
Weissig V, Torchilin VP. Mitochondriotropic cationic vesicles: a strategy towards mitochondrial gene therapy. Curr Pharm Biotechnol. 2000;1(4):325–346. | ||
Zhao Q-Q, Chen J-L, Lv T-F, et al. N/P ratio significantly influences the transfection efficiency and cytotoxicity of a polyethylenimine/chitosan/DNA complex. Biol Pharm Bull. 2009;32(4):706–710. | ||
Bai H, Zhou Y, Hou Z, Xue X, Meng J, Luo X. Targeting bacterial RNA polymerase: promises for future antisense antibiotics development. Infect Disord Drug Targets. 2011;11(2):175–187. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.