Back to Journals » International Journal of Nanomedicine » Volume 12
Bitistatin-functionalized fluorescent nanodiamond particles specifically bind to purified human platelet integrin receptor αIIbβ3 and activated platelets
Authors Marcinkiewicz C, Gerstenhaber JA, Sternberg M, Lelkes PI ![]() , Feuerstein G
, Feuerstein G
Received 7 February 2017
Accepted for publication 4 April 2017
Published 15 May 2017 Volume 2017:12 Pages 3711—3720
DOI https://doi.org/10.2147/IJN.S134128
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Cezary Marcinkiewicz,1,2 Jonathan A Gerstenhaber,1 Mark Sternberg,2 Peter I Lelkes,1 Giora Feuerstein1,2
1Department of Bioengineering, College of Engineering, Temple University, Philadelphia, 2Debina Diagnostic, Inc., Newton Square, PA, USA
Abstract: Thromboembolic events (TEE) underwrite key causes of death in developed countries. While advanced imaging technologies such as computed tomography scans serve to diagnose blood clots during acute cardiovascular events, no such technology is available in routine primary care for TEE risk assessment. Here, we describe an imaging platform technology based on bioengineered fluorescent nanodiamond particles (F-NDPs) functionalized with bitistatin (Bit), a disintegrin that specifically binds to the αIIbβ3 integrin, platelet fibrinogen receptor (PFR) on activated platelets. Covalent linkage of purified Bit to F-NDP was concentration-dependent and saturable, as validated by enzyme-linked immunosorbent assay using specific anti-Bit antibodies. F-NDP–Bit interacted with purified PFR, either in immobilized or soluble form. Lotrafiban, a nonpeptide, αIIbβ3 receptor antagonist, specifically blocked F-NDP–Bit–PFR complex formation. Moreover, F-NDP–Bit specifically binds to activated platelets incorporated into a clot generated by thrombin-activated rat platelet-rich plasma (PRP). Our results suggest that engineered F-NDP–Bit particles could serve as noninvasive, “real-time” optical diagnostics for clots present in blood vessels.
Keywords: carbon nanoparticles, blood clots, imaging, platelet fibrinogen receptor, fluorescence, disintegrin, thromboembolic complications, thrombosis
Introduction
Thromboembolic events (TEE) in cerebral and coronary vessels are key causes of death from strokes and heart attacks in developed countries.1,2 Advanced technologies such as computed tomography (CT) scans, magnetic resonance imaging (MRI), and angiography routinely serve to guide treatment options and prognostic outcomes in acute strokes and heart attacks.3 However, diagnostic technologies for acute TEE have drawbacks that prelude their routine utilization for primary disease risk assessment. For example, MRI requires specialized facilities, specialty-trained medical teams, and is restrictive in terms of its costs. CT scans and angiography require hospital-based facilities and highly trained technical and medical personnel to acquire and interpret the complex imaging information. Furthermore, most current imaging technologies in use for assessing TEE are indirect – that is, deducing the consequences rather than localizing blood clots per se. Altogether, the ability to assess the risk of TEE as part of the general health management for the primary prevention of vascular thrombosis risks remains a significant, unmet medical need. Taken together, detection of blood clots in critical vascular/cardiac locations (eg, cardiac chambers) as well as sites of internal bleeding poses challenges due to the lack of rapid, ambulatory, and noninvasive and “real-time” diagnostic capabilities.
In recent years, significant advances have been made in bioengineering functional nanoparticles, specifically in the design of new fabrics, matrixes, and particles compatible with biological constituents including blood and living tissues.4 Of particular interest are carbon-based particles, such as nanodiamond particles (NDPs), which have been functionalized for diverse applications,5 such as drug discovery,6–8 drug delivery,9,10 and imaging of pathological sites in vivo.9,11–13 More recently, special attention is focused on NDPs that can be excited optically to emit fluorescent light (F-NDPs) at various wavelengths, especially in the near infrared (NIR) range.11,12 The innate ability of these new-generation NIR-emitting F-NDPs to report from within biological tissues with limited interference from endogenous fluorophores has already been demonstrated in studies, where certain elements of blood clots, such as fibrin, were detected in vivo.14 Furthermore, functionalization of nanoparticles that specifically interact with platelets has been shown in vitro.15
Bitistatin (Bit) belongs to the family of snake venom disintegrin proteins, which contain an arginyl-glycyl-aspartic (RGD) motif in their active sites, rendering these disintegrins cross-reactive with integrins, including the platelet fibrinogen receptor (PFR), αIIbβ3 integrin.16 A radiolabeled form of this disintegrin has been used for the detection of deep vein thrombosis in a canine model.17,18 Data from an initial clinical trial, assessing the biodistribution of [99m]Tc-labeled Bit, suggest that this disintegrin did not induce clots nor did it change the levels of circulating platelets.19 These recent findings support the hypothesis that F-NDP-functionalized with Bit (F-NDP–Bit) could serve for imaging of intravascular blood clots via extracorporeal NIR fluorescence scans.
In this report, we present in vitro feasibility data for bioengineering and characterization of F-NDPs that were functionalized with carboxyl groups (–COOH) and covalently coupled to Bit. We anticipate that this in vitro “proof-of-concept” study will lead to the development of in vivo imaging capabilities based on the ability of F-NDP–Bit to specifically identify activated platelets embedded in blood clots in select intravascular locations.
Materials and methods
Antibodies and other reagents
Lyophilized snake venom of Bitis arietans was purchased from Latoxan Serpentarium (Valence, France). Polyclonal rabbit serum against the purified, native whole molecule of Bit was developed commercially (custom-made by Millipore, Inc., Billerica, MA, USA). Polyclonal IgG was purified using a purification kit based on a Protein-G column (Pierce Biotech., Rockford, IL, USA) and validated by enzyme-linked immunosorbent assay (ELISA) and Western blotting. Polyclonal antibody against fibrinogen receptor (αIIb subunit) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-rabbit IgG (whole molecule) alkaline phosphatase (AP)-conjugated and p-nitrophenyl phosphate (pNPP), a colorimetric substrate for AP, were purchased from Sigma, Inc. (St Louis, MO, USA). Fibrinogen receptor, purified from human platelets, was purchased from Millipore, Inc. Lotrafiban was a gift from GlaxoSmithKline Pharm. (King of Prussia, PA, USA). Pierce protein coupling reagents: EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride), Sulfo-NHS (N-hydroxysulfosuccinimide), and MES buffer (-[N-morpholino]ethane sulfonic acid) were purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Nanodiamond particles
Fluorescent nanodiamond particles (F-NDPs), chemically surface-functionalized with carboxyl groups (–COOH), were purchased from Adamas Nanotechnologies (Raleigh, NC, USA). The green fluorescence (induced by excitation at λ=485 nm) of F-NDPs is based on N–V–N color centers incorporated into the diamond lattice. This strategy eliminates photobleaching/photoblinking, compared with, respectively, organic dyes or quantum dots.20 The median size of the F-NDPs used for these experiments was ~700 nm (Figure S1). Microscopic observation of F-NDPs coupled to the proteins (Bit or bovine serum albumin [BSA]) was carried out at 400× magnification (fluorescein channel) on an Olympus IX81 inverted fluorescence microscope and showed no tendency for aggregation.
Purification and validation of native Bit
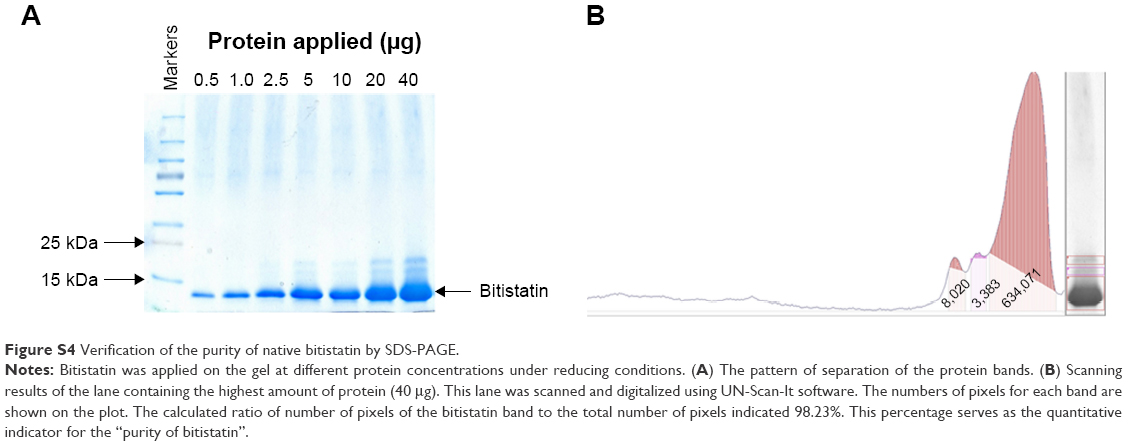
Bit was purified from B. arietans venom according to a previously described procedure21 with some minor modifications. Briefly, lyophilized snake venom was dissolved in 0.1% TFA (30 mg/mL). Insoluble material was pelleted by centrifugation at 14,000 rpm for 5 minutes at room temperature (RT). The supernatant was fractionated by reverse-phase high-performance liquid chromatography (HPLC) on a C18 column (250×10 mm; Vydac, Hesperia, CA, USA). The column was eluted with a linear acetonitrile gradient 0%–80% over 45 minutes at a flow rate of 2 mL/min (Figure S2). Separation was monitored at 230 nm; fractions containing Bit (~22 minutes retention time) were collected manually and lyophilized in a Speed-Vac system. The lyophilized fractions were dissolved in water, and protein concentrations determined using the BCA assay (Pierce, Rockford, IL, USA). The crude Bit preparation (5 mg protein) was re-chromatographed using the same HPLC system but eluting the column with a shallower acetonitrile gradient (20%–80% over 120 minutes). The main peak containing purified Bit (Figure S3) was lyophilized and dissolved in deionized water to prepare the stock solution (8–10 mg/mL) used for coupling to F-NDPs. The purity of Bit, as tested by sodium dodecyl sulfate – polyacrylamide gel electrophoresis, was found to be >98%, based on the digitalized intensity (HP ScanJet G3110 and software UN-scan-It gel version 6.1 by Silk Scientific Corp., Orem, UT, USA) of the major bands at maximal concentration (Figure S4).
Coupling of proteins to F-NDPs
Bit, or BSA as control, was coupled to the carboxyl group on the surface of carboxyl-functionalized F-NDPs. The coupling procedure was based on using EDC as a hetero-bifunctional cross-linker.22 Briefly, 1 mg of F-NDPs was added to 1 mL of MES buffer (pH =6.0) containing 2 mM EDC and 5 mM Sulfo-NHS. The activation of F-NDPs was carried out for 15 minutes at RT. EDC was then quenched by the addition of β-mercaptoethanol (20 mM final concentration). Activated F-NDPs were pelleted by centrifugation at 10,000× g for 5 minutes at RT. Bit or BSA (1 mg of protein) in 1 mL of coupling buffer (phosphate-buffered saline [PBS]) was added to the F-NDP pellet and incubated for 2 hours at RT. The reaction was quenched by adding hydroxylamine (10 mM final concentration). F-NDP–Bit or F-NDP–BSA was separated from the reaction mixture by centrifugation as above and suspended in PBS at the required concentration.
Semi-ELISA assay
Protein-conjugated F-NDPs (1 mg/mL) were incubated with 3% BSA in PBST (PBS and 0.05% Tween-20) for 1 hour at 37°C to block unoccupied IgG-binding sites. After removing the blocking agent by centrifugation (washing three times), F-NDPs were suspended in PBST and applied to the bottom of a U-shaped 96-well plate (200 μL per well). The plate was centrifuged (1,000 rpm at RT) for 5 minutes. Pellets containing F-NDPs were re-suspended in 100 μL of PBST containing the rabbit anti-Bit polyclonal antibody (5 μg/mL). The plate was incubated for 1 hour at 37°C. F-NDPs were washed three times by centrifugation (1,000 rpm at RT) for 5 minutes, and goat anti-rabbit IgG AP conjugate was added at 1:2,000 dilution in PBST. The suspension was incubated for 1 hour at 37°C. F-NDPs were washed three times with PBST and two times with PBS by centrifugation and suspended in 200 μL per well of pNPP. The plate was incubated for 30 minutes and centrifuged (1,000 rpm at RT), as above. Supernatants were transferred to a flat-bottomed 96-well plate (Falcon; Corning Inc., Corning, NY, USA) (100 μL per well) and absorbance was read using an ELISA plate reader (BioTek, Vinooski, VT, USA; EL ×800) at 405 nm.
The functional semi-ELISA assay was performed using purified human PFR (Millipore, Inc., Billerica, MA, USA). F-NDP–Bit or F-NDP–BSA (1 mg/mL) was blocked with 3% BSA in PBST for 1 hour at 37°C. The blocking agent was removed by centrifugation, and F-NDPs were applied to the bottom of a U-shaped 96-well plate in HBSS containing Ca2+ and Mg2+, as described above. PFR was dissolved at different concentrations in HBSS with Ca2+ and Mg2+ and preincubated with lotrafiban (10 mg/mL) or the HBSS vehicle (control) for 15 minutes at 37°C, under gentle agitation. After the preincubation step, aliquots of the above reaction mixtures were added to the wells of the 96-well plate containing F-NDP–Bit and incubated for 60 minutes at 37°C. Thereafter, F-NDP–Bit–PFR complexes were washed three times with PBST as above, and primary AP-conjugated polyclonal antibody against PFR was added in PBST at a dilution of 1:1,000. The semi-ELISA experiment was then completed and evaluated under the same conditions as described above for anti-Bit.
Adhesion of F-NDPs to immobilized fibrinogen receptor
PFR was immobilized on 96-well plates at different concentrations by overnight incubation at 4°C in PBS. The plate and F-NDP–Bit or F-NDP–BSA were blocked with 3% BSA in PBS by incubation for 1 hour at RT. HBSS with Ca2+ and Mg2+, supplemented with lotrafiban23,24 (4.67 μmol/mL) or vehicle control, was added to the wells and preincubated for 15 minutes at 37°C under mild agitation. F-NDP–Bit was added to the wells (6 mg/mL) and incubation was continued for 60 minutes at 37°C. Unbound F-NDPs were removed by intensive washing (six times) with PBS by vacuum aspiration. Finally, 100 μL of PBS was added to the wells, and fluorescence was read using a fluorescence plate reader (BioTek; FL ×800) at 485 nm (excitation ) and 530 (emission).
To quantify F-NDP adhesion by fluorescence microscopy, PFR was immobilized on glass eight-well chamber slides at different concentrations, as described above for the 96-well plates. Slides and F-NDPs were blocked with 3% BSA, as above. F-NDP–Bit and F-NDP–BSA were applied to the slides (250 μg/mL) in HBSS containing Ca2+ and Mg2+. Chamber slides were incubated for 1 hour at 37°C. Wells were washed intensively (six times) with PBS by vacuum aspiration, and cover slides were topped in mounting buffer (Vector Laboratories; Burlingame, CA, USA). The slides were visualized in a fluorescence microscope (Olympus IX81; Center Valley, PA, USA) at 400× magnification (fluorescein channel). The number of F-NDP/mm2 was calculated from digital images using ImageJ software. Competitive studies with lotrafiban were performed according to the procedure described above for 96-well plate adhesion.
Platelet-rich plasma clot assay
Platelet-Rich Plasma (PRP) was generated from rat blood collected from deeply anesthetized (ketamine) Fischer rats by cardiac puncture. All animal procedures were performed according to the guidelines of the US Animal Welfare Act and approved by the Institutional Animal Care & Use Committee at Temple University. Blood was aspirated into sodium citrate (3.8%) containing syringes. PRP was obtained by centrifugation (1,250 rpm at RT for 20 minutes). Clots were generated by adding thrombin (1 U/mL) to PRP. After 30 minutes, the clots were washed three times with HBSS containing Ca2+ and Mg2+ using gravitational sedimentation and decantation of supernatant. Clots were manually dissected into small pieces (ca. 2–3 mm), placed in tubes containing 2 mL of F-NDP–Bit or F-NDP–BSA (500 μg/mL), and incubated for 15 or 60 minutes at 37°C with mild agitation. Competitive studies with lotrafiban were performed by preincubation of clots with lotrafiban (4.67 μg/mL) for 15 minutes at 37°C with mid-agitation before adding F-NDP–Bit. Clots were washed (six times) as described above, transferred onto glass slides, and covered with glass coverslips. Clot-associated fluorescence was visualized and quantified in a fluorescence microscope (Olympus IX81 with 100× magnification) and the IVIS 50 Imaging System (PerkinElmer, Inc., Akron, OH, USA).
Statistical analysis
Unless stated otherwise, each experiment was performed independently three times in triplicate. Data are represented as mean ± SD. Statistical analyses were done by the Student’s t-test using SigmaPlot® 12 SPSS (Systat Software, Inc., San Jose, CA, USA). P<0.05 was considered significant.
Results
Preparation of F-NDP–Bit
Proteins (Bit or albumin) were covalently coupled to the COOH activated surface of fluorescent nanoparticles, as described in Materials and Methods section. Coupling efficiency was evaluated by immune detection of Bit on the surface of F-NDP (NDP–Bit) using a semi-ELISA assay (Figure 1). The maximal (saturated) concentration of Bit was estimated as 1 mg per 1 mg NDP. As control, F-NDPs were coupled to the same amount of BSA. As depicted in Figure 1, the anti-Bit polyclonal antibody did not detect F-NDP–BSA, even at the highest concentrations used.
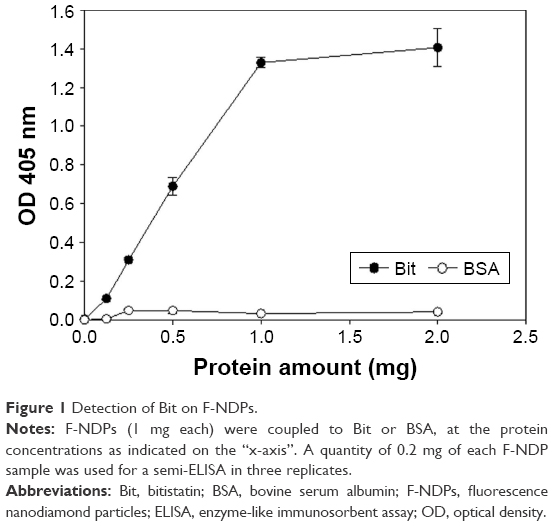 | Figure 1 Detection of Bit on F-NDPs. |
Characterization of F-NDP–Bit interaction with purified PFR
Quantification of the binding of the soluble fibrinogen receptor was based on a functional “sandwich” semi-ELISA (see Materials and Methods section). In this case, immune detection of F-NDP–Bit/αIIbβ3 integrin complex was performed using a polyclonal antibody against the αIIb integrin subunit. As seen in Figure 2, binding of soluble PFR to F-NDP–Bit was dose-dependent, whereas the same soluble PFR did not bind to the control F-NDP–BSA. Lotrafiban (23.35 μmol/mL) abrogated the interaction of F-NDP–Bit with PFR.
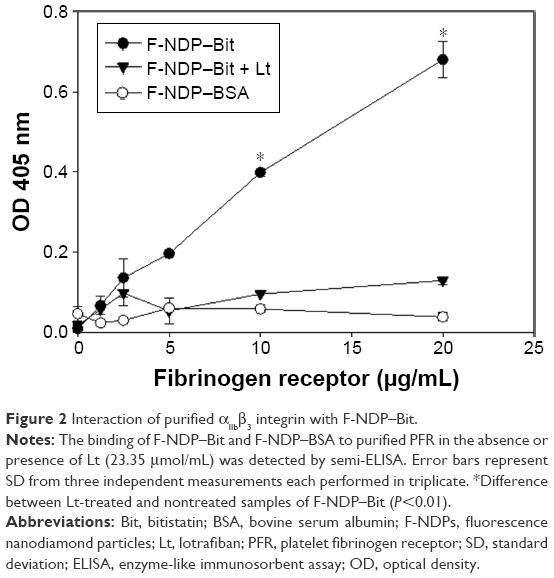 | Figure 2 Interaction of purified αIIbβ3 integrin with F-NDP–Bit. |
Subsequently, we evaluated the adhesion of F-NDP–Bit to immobilized PFR. As seen in Figure 3, adhesion of F-NDP–Bit increased linearly with increasing concentrations of immobilized PFR, whereas adhesion of the BSA-coupled F-NDPs (F-NDP–BSA) was negligible. When the assay system was preincubated for 15 minutes with lotrafiban before F-NDP–Bit was added, the interaction of F-NDP–Bit with immobilized PFR was completely abolished.
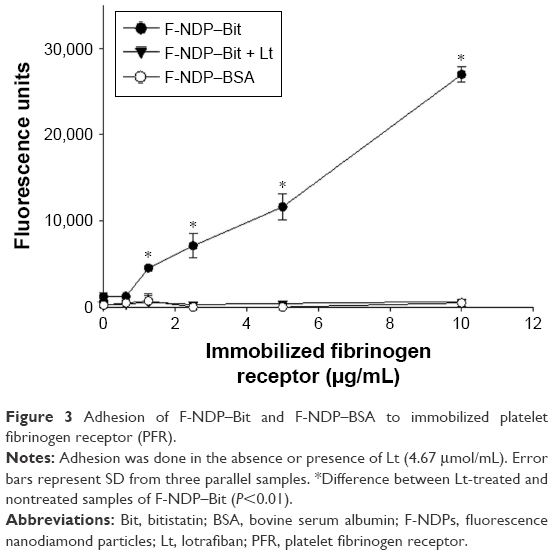 | Figure 3 Adhesion of F-NDP–Bit and F-NDP–BSA to immobilized platelet fibrinogen receptor (PFR). |
The specific interaction of F-NDP–Bit with PFR was further confirmed by fluorescence microscopy (Figure 4). Quantification of the adhesion, performed in eight-well chamber slides, revealed dose-dependent binding of F-NDP–Bit to immobilized PFR. At the highest concentration of PFR, quantitative image analysis indicated that approximately 22,000 F-NDP–Bit molecules bound to the immobilized receptor per mm2, which was ~10 times higher than the number of nonspecifically bound F-NDP–BSA. Similar to the adhesion assay performed in 96-well plates, lotrafiban blocked binding of F-NDP–Bit to PFR immobilized on glass chamber slides by >90%.
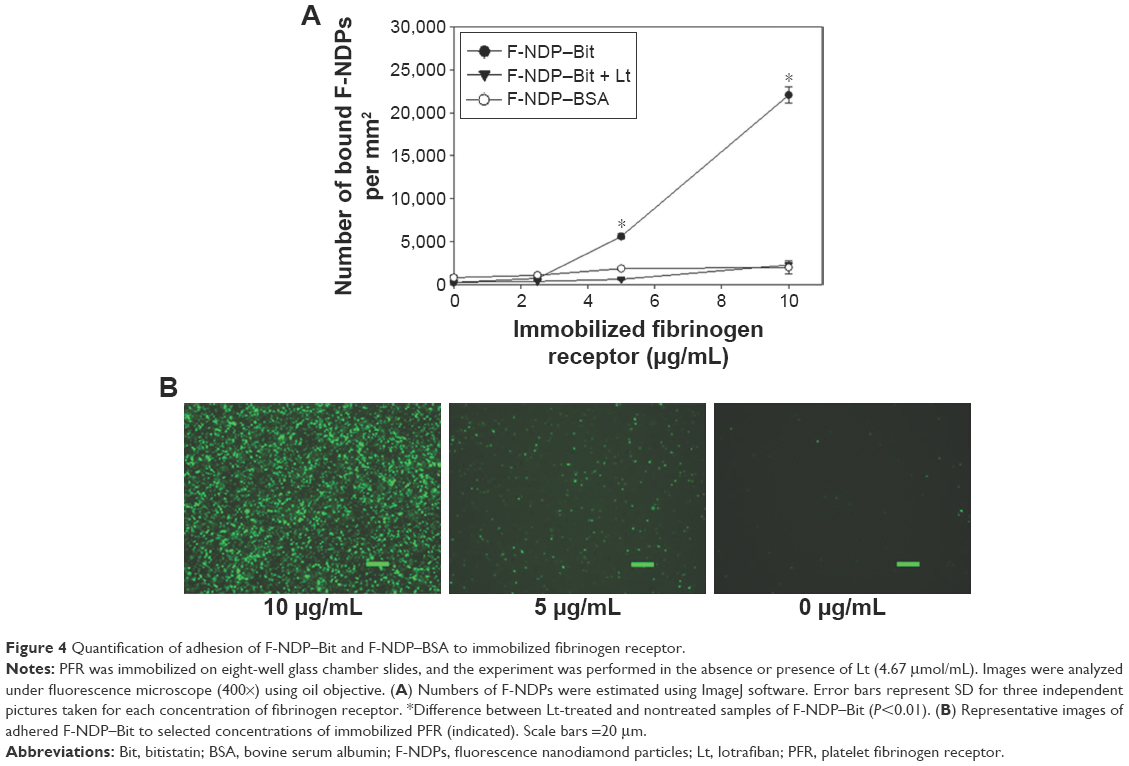 | Figure 4 Quantification of adhesion of F-NDP–Bit and F-NDP–BSA to immobilized fibrinogen receptor. |
Characterization of F-NDP–Bit interaction with thrombin-induced rat PRP clot
Finally, we studied the specificity of the interaction of F-NDP–Bit with the fibrinogen receptor present on activated platelets in a preformed PRP clot (Figure 5). In two separate experiments, clots were incubated with F-NDP–Bit for 15 or 60 minutes, respectively. Irrespective of the duration of incubation, binding of F-NDP–Bit was always 4–5 fold higher than that of the nonspecific control particles (F-NDP–BSA). Furthermore, preincubation of the clots with lotrafiban reduced F-NDP–Bit binding to the level of the control F-NDP–BSA.
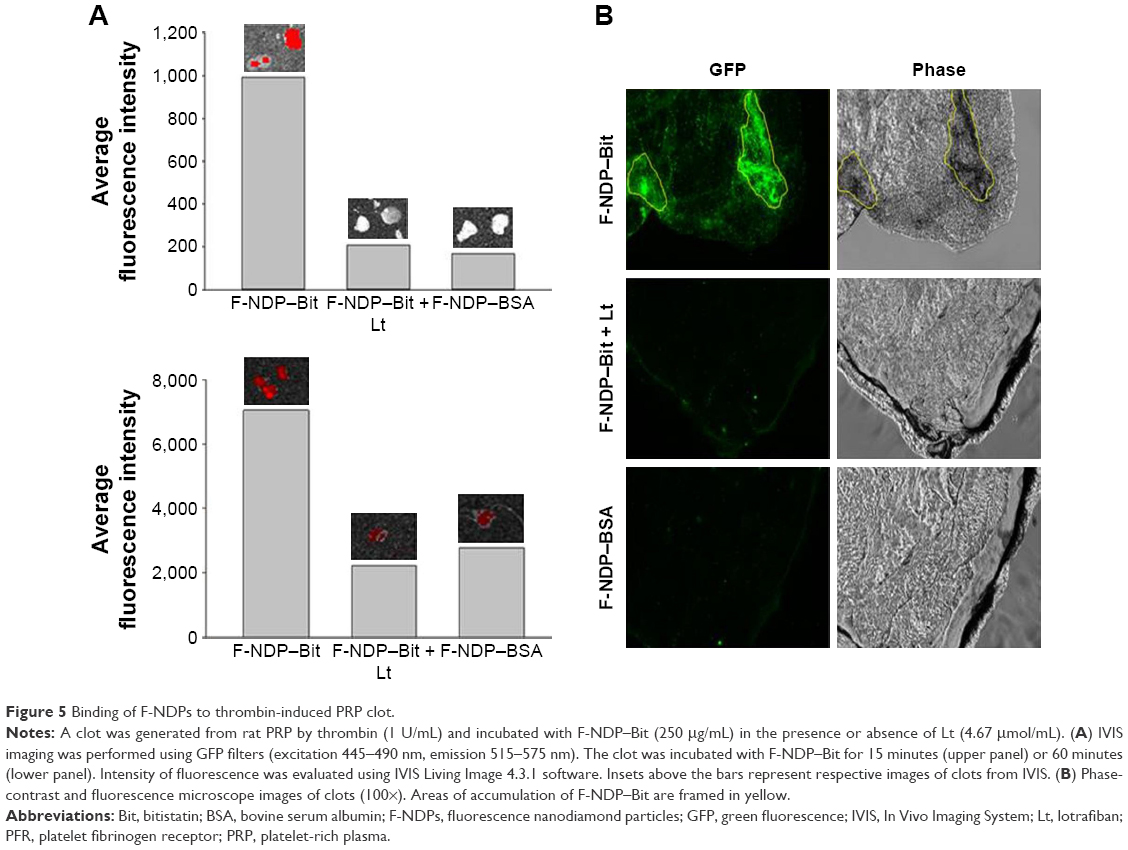 | Figure 5 Binding of F-NDPs to thrombin-induced PRP clot. |
Discussion
The experiments described in this article were designed as feasibility studies in support of our long-term objective, that is, to develop a fluorescence-based imaging platform technology that can ultimately serve as a diagnostic tool for real-time in vivo monitoring of blood clots in humans. The technology chosen to fulfill this objective builds on bioengineering of NDPs that emit fluorescence in response to excitation in a safe energy range (visible light) concomitant with marked resistance to photobleaching.11,25 If realized, optical imaging could revolutionize risk assessment of TEE by prospective mapping of vascular lesions and their propensity for blood clot formation within the patients’ circulatory system. This, in turn, could aid in the proactive management of risks for heart attacks, strokes, and venous TEE.
The studies reported in here required de novo engineering of F-NDPs with covalently coupled ligands that can specifically identify key elements of blood clots, such as activated platelets. The goal of this approach is to “load” target pathology sites with a critical mass of F-NDPs to generate a signal bright enough to be detected by extra-corporeal optical recoding. As our targeting agent, we used Bit, a disintegrin, which specifically binds to the ligand-binding pocket of the PFR through an RGD motif.16 The coupling methods used to covalently bind Bit to the F-NDPs apparently preserved Bit’s biological activity and specificity (Figures 2–5), indicating that coupling through the carboxyl group present on the F-NDPs does not interfere with the active conformation of this disintegrin and that the integrin-binding loop which contains the RGD sequence is fully accessible to fibrinogen receptor. Therefore, Bit is an appropriate ligand to specifically bind to PFR associated with activated platelets encased in a clot.
The specificity of the interaction of F-NDP–Bit with the PFR was verified by competition studies with lotrafiban. Lotrafiban is a nonpeptide, small molecular compound, which potently binds αIIbβ3 integrin. Similar compounds from the “Fiban class” are used in acute critical care medicine to prevent clot formation, such as in TEE.26 In all assays used in our studies, lotrafiban abrogated the interaction of F-NDP–Bit with the PFR in vitro. Importantly, lotrafiban also blocked F-NDP–Bit binding to a preformed PRP clot. These results strongly indicate that Bit and lotrafiban compete for the same RGD-binding site on the αIIbβ3 integrin molecule.
Our studies are distinct from past efforts, where activated platelets were labeled with Tc99 Bit for the successful detection of pulmonary emboli and deep venous thrombi in canines.17–19 We surmise that by comparison to radiolabeled molecules, the use of fluorescence would significantly simplify the production and use of a platelet-specific tag. While detection of radiolabeled imaging compounds is very efficient, radioactivity carries safety concerns for patients and for clinical personnel.3 We surmise that fluorescence-based imaging might present a better opportunity for the diagnosis of blood clots, especially of those that are found in sufficient proximity to the body surface such as in carotid artery, superficial femoral artery, or superficial venous vessels in the extremities. The high quantum yield of the nonphotobleaching fluorescence of F-NDPs could be considered a promising approach for imaging a variety of pathologies, where blood clots are suspected.
A potential caveat concerning the applicability of the technology presented in this paper is the ability to monitor fluorescence emanating from within deeply located biological targets in vivo. Living tissues have high levels of green autofluorescence related to the absorption of light by several biological compounds, including elastin, collagen, hemoglobin, and melanin.27 For the ex vivo proof-of-concept test of our new platform with a real thrombus, we used a PRP clot, which is devoid of hemoglobin. Our data (Figure 5) showed specific localization of F-NDP–Bit to the clot surface in areas that presumably contain activated platelets. This association was completely abolished by lotrafiban, confirming the specificity of the interaction of F-NDP–Bit with activated platelets ex vivo.
In summary, the experiments described in this study demonstrate that F-NDPs can be engineered to carry a targeting moiety, which could be instrumental for detecting key elements of a complex blood clot. In this study, we used, as proof of concept, green-fluorescent F-NDPs with N–V–N active centers, which is more suitable for in vitro analyses. However, for in vivo imaging aimed as a medical diagnostic of said lesions, NIR-emitting F-NDPs would likely be more advantageous.11,13 The innate ability of NIR light to penetrate biological tissues with limited interference from endogenous fluorophores has already been demonstrated in studies where certain elements of blood clots, such as fibrin, were detected in vivo.12 The NIR technology is expected to afford extracorporeal, telemetry-based, “on line” assessment of blood clots using ambulatory facilities for the accurate, large-scale assessment of cardiovascular risk of TEE. This hypothesis remains to be investigated.
Acknowledgments
Research reported in this publication was supported in part by Debina Diagnostic, Inc. Additional support was provided by a grant from the Moulder Center for Drug Discovery (CM and PIL) and a Research Bridge Funding Award (PIL) from the Office of the Vice President for Research Administration (OVPR). We are grateful to Dr Robert N Willette VP and Head of Biology, GlaxoSmithKline Pharmaceuticals, King of Prussia, PA, USA, for the generous gift of lotrafiban and to Dr Olga Shenderova (Adámas Nanotechnologies, Raleigh, NC, USA) for expert advice on F-NDPs and for the permission to use the characterization curve for the particles used in this study (Figure S1).
Disclosure
The authors report no conflicts of interest in this work.
References
Sidney S, Quesenberry CP Jr, Jaffe MG, et al. Recent trends in cardiovascular mortality in the United States and Public Health Goals. JAMA Cardiol. 2016;1(5):594–599. | ||
Guirguis-Blake JM, Evans CV, Senger CA, O’Connor EA, Whitlock EP. Aspirin for the primary prevention of cardiovascular events: a systematic evidence review for the U.S. Preventive Services Task Force. Ann Intern Med. 2016;164(12):804–813. | ||
Skovronsky DM, Feuerstein GZ. Imaging biomarkers for diagnosis, prognosis, and treatment of Alzheimer’s disease. In Wegrzyn RD, Rudolph AS, editors. Frontiers in Neuroscience. CRC Press, Boca Raton, FL, USA; 2012:103–116. | ||
Mochalin VN, Shenderova O, Ho D, Gogotsi Y. The properties and applications of nanodiamonds. Nat Nanotechnol. 2012;7(1):11–23. | ||
Wang HD, Yang Q, Niu CH. Functionalization of nanodiamond particles with N,O-carboxymethyl chitosan. Diam Relat Mater. 2010;19(5–6):441–444. | ||
Vaijayanthimala V, Lee DK, Kim SV, et al. Nanodiamond-mediated drug delivery and imaging: challenges and opportunities. Expert Opin Drug Deliv. 2015;12(5):735–749. | ||
Ho D, Wang CH, Chow EK. Nanodiamonds: the intersection of nanotechnology, drug development, and personalized medicine. Sci Adv. 2015;1(7):e1500439. | ||
Ho D, Zarrinpar A, Chow EK. Diamonds, digital health, and drug development: optimizing combinatorial nanomedicine. ASC Nano. 2016;10(10):9087–9092. | ||
Xi G, Robinson E, Mania-Farnell B, et al. Convection-enhanced delivery of nanodiamond drug delivery platforms for intracranial tumor treatment. Nanomedicine. 2014;10(2):381–391. | ||
Yan M, Du J, Gu Z, et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat Nanotechnol. 2010;5(1):48–53. | ||
Igarashi R, Yoshinari Y, Yokota H, et al. Real-time background-free selective imaging of fluorescent nanodiamonds in vivo. Nano Lett. 2012;12(11):5726–5732. | ||
Hara T, Bhayana B, Thompson B, et al. Molecular imaging of fibrin deposition in deep vein thrombosis using fibrin-targeted near-infrared fluorescence. JACC Cardiovasc Imaging. 2012;5(6):607–615. | ||
Zhang T, Cui H, Fang CY, et al. In vivo photoacoustic imaging of breast cancer tumor with HER2-targeted nanodiamonds. Proc SPIE Int Soc Opt Eng. 2013;8815; 881504. | ||
Manus LM, Mastarone DJ, Waters EA, et al. Gd(III)-nanodiamond conjugates for MRI contrast enhancement. Nano Lett. 2010;10(2):484–489. | ||
Hu CM, Fang RH, Wang KC, et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526(7571):118–121. | ||
Marcinkiewicz C. Applications of snake venom components to modulate integrin activities in cell-matrix interactions. Int J Biochem Cell Biol. 2013;45(9):1974–1986. | ||
Knight LC, Romano JE, Maurer AH. In vitro platelet binding compared with in vivo thrombus imaging using alpha(IIb)beta3-targeted radioligands. Thromb Haemost. 1998;80(5):845–851. | ||
Knight LC, Baidoo KE, Romano JE, Gabriel JL, Maurer AH. Imaging pulmonary emboli and deep venous thrombi with 99mTc-bitistatin, a platelet-binding polypeptide from viper venom. J Nucl Med. 2000;41(6):1056–1064. | ||
Knight LC, Romano JE, Bright LT, Agelan A, Kantor S, Maurer AH. Platelet binding and biodistribution of [99mTc]rBitistatin in animal species and humans. Nucl Med Biol. 2007;34(7):855–863. | ||
Chang YR, Lee HY, Chen K, et al. Mass production and dynamic imaging of fluorescent nanodiamonds. Nature Nanotechnol. 2008;3(5):284–288. | ||
Shebuski RJ, Ramjit DR, Bencen GH, Polokoff MA. Characterization and platelet inhibitory activity of bitistatin, a potent arginine-glycine-aspartic acid-containing peptide from the venom of the viper Bitis arietans. J Biol Chem. 1989;264(36):21550–21556. | ||
Grabarek Z, Gergely J. Zero-length crosslinking procedure with the use of active esters. Anal Biochem. 1990;185(1):131–135. | ||
Samanen JM, Ali FE, Barton LS, et al. Potent, selective, orally active 3-Oxo-1,4-benzodiazepine GPIIb/IIIa integrin antagonists. J Med Chem. 1996;39(25):4867–4870. | ||
Liu F, Craft RM, Morris SA, Carroll RC. Lotrafiban: an oral platelet glycoprotein IIb/IIIa blocker. Expert Opin Investig Drugs. 2000;9(11):2673–2687. | ||
Yu SJ, Kang MW, Chang HC, Chen KM, Yu YC. Bright fluorescent nanodiamonds: no photobleaching and low cytotoxicity. J Am Chem Soc. 2005;127(50):17604–17605. | ||
Leclerc JR. Platelet glycoprotein IIb/IIIa antagonists: lessons learned from clinical trials and future directions. Crit Care Med. 2002;30(5):S332–S340. | ||
Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Curr Opin Chem Biol. 2010;14(1):71–79. |
Supplementary materials
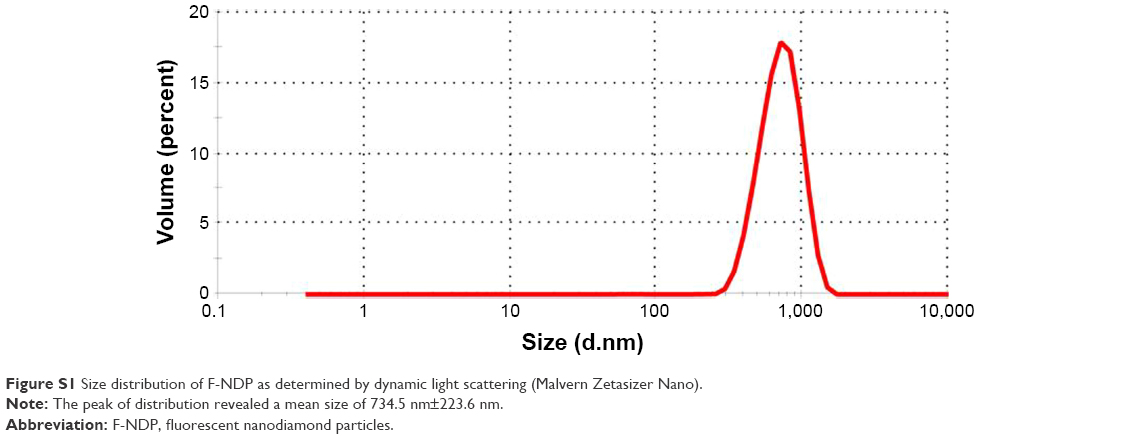 | Figure S1 Size distribution of F-NDP as determined by dynamic light scattering (Malvern Zetasizer Nano). |
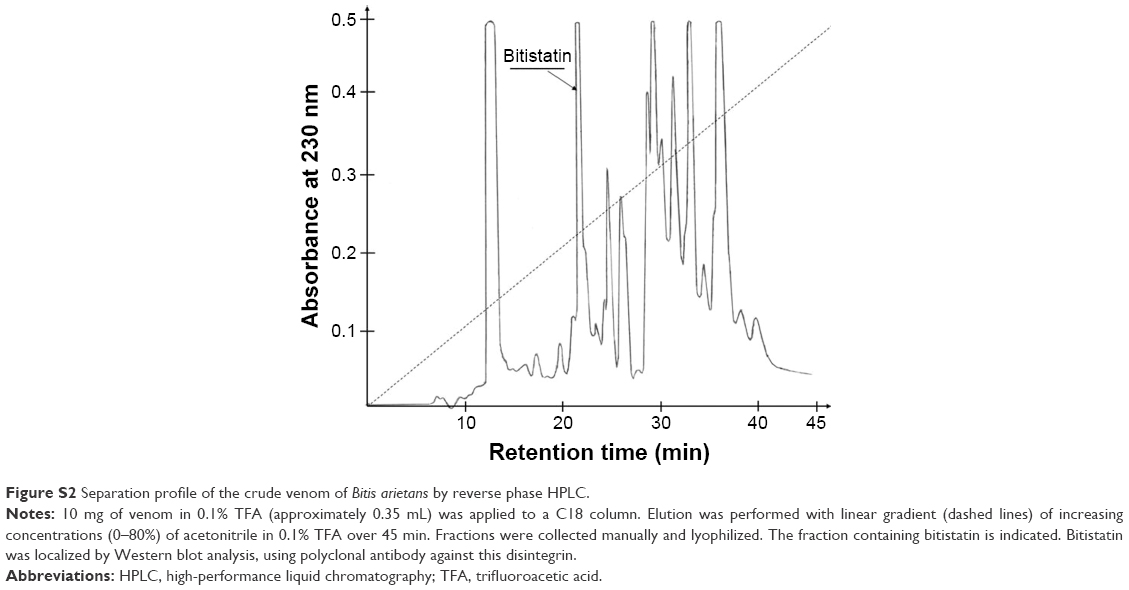 | Figure S2 Separation profile of the crude venom of Bitis arietans by reverse phase HPLC. |
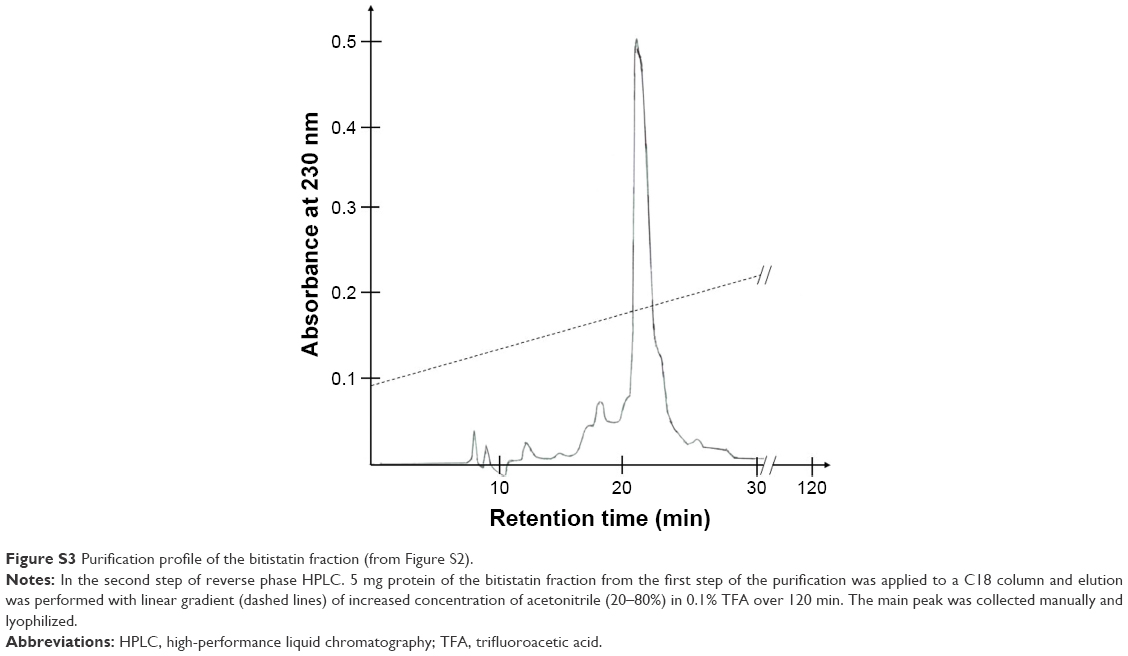 | Figure S3 Purification profile of the bitistatin fraction (from Figure S2). |
 | Figure S4 Verification of the purity of native bitistatin by SDS-PAGE. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.