Back to Journals » Drug Design, Development and Therapy » Volume 14
Bisdemethoxycurcumin Inhibits Hepatocellular Carcinoma Proliferation Through Akt Inactivation via CYLD-Mediated Deubiquitination
Authors Qiu C, Liu K, Zhang S, Gao S, Chen W, Li D, Huang Y ![]()
Received 20 September 2019
Accepted for publication 20 February 2020
Published 5 March 2020 Volume 2020:14 Pages 993—1001
DOI https://doi.org/10.2147/DDDT.S231814
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Chengjiang Qiu,1 Kairui Liu,1 Sheng Zhang,1 Simin Gao,2 Weirun Chen,1 Dateng Li,3 Youxing Huang1
1Department of Abdominal Surgery, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, Guangdong Province, People’s Republic of China; 2Department of Breast Surgery, Breast Tumor Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, Guangdong Province, People’s Republic of China; 3Department of Statistical Science, Southern Methodist University, Dallas, TX 75275, USA
Correspondence: Youxing Huang
Department of Abdominal Surgery, The Second Affiliated Hospital of Guangzhou University of Chinese Medicine, No. 111 Dade Road, Guangzhou, Guangdong Province, People’s Republic of China
Tel +86-13632255441
Fax + 86-20-39318790
Email [email protected]
Background: Bisdemethoxycurcumin (BDMC), a stable bioactive ingredient in curcuminoids, is associated with various antitumor functions, such as proliferation inhibition, metastasis suppression and apoptosis induction, in many cancer types. However, the mechanism of BDMC in hepatocellular carcinoma (HCC) remains unclear.
Methods: We assessed the toxicity and the inhibitory effect of BDMC in the HepG2 cell line by using CCK-8 and colony formation assays. The regulatory effects of BDMC on Akt and MAPK signaling were investigated by Western blotting and immunoprecipitation.
Results: We found that the half-maximum inhibitory concentration (IC50) of BDMC after 48 hrs of treatment was 59.13 μM, and BDMC inhibited proliferation in a time- and dose-dependent manner in HepG2 cells. The inhibitory effect was caused by the inactivation of Akt signaling, but not Erk, Jnk or p38 signaling. In addition, the inactivation of Akt signaling was attributed to the inhibition of ubiquitination mediated by K63-Ub but not K48-Ub. Furthermore, we found that BDMC upregulated the expression of CYLD, leading to Akt deubiquitination and inactivation.
Conclusion: BDMC inhibited HCC cell proliferation, and that this effect was induced by Akt inactivation via CYLD-mediated deubiquitination.
Keywords: bisdemethoxycurcumin, hepatocellular carcinoma, proliferation, deubiquitination
Introduction
Hepatocellular carcinoma (HCC), the major histological subtype of liver cancer, is a rapidly progressing, highly chemotherapy-resistant malignancy with frequent recurrence; HCC ranks as the third leading cause of cancer-related deaths worldwide.1,2 Although huge efforts have been made to understand the biological mechanisms that underlie liver cancer development, the morbidity and mortality rates remains dismal.3 In China, where half of liver cancer cases are diagnosed, statistics showed that approximately 466,100 new cases and 422,100 related deaths were estimated in 2015.4 Current treatment algorithm is often futile for patients with advanced HCC because of the loss of surgical opportunity and the very limited effect of chemotherapy.5,6 Therefore, it is of utmost urgency to identify better therapeutic strategies that can complement the present approach.
Tradition Chinese herbal medicine has been used to fight against various cancer types for thousands of years. Curcuminoids, as the main bioactive ingredients of traditional Chinese herbal medicine Curcuma longa L., exert various biological functions, such as anti–inflammation, antioxidation, neuroprotection and anti-carcinogen effects via modulating the expression of molecules involved in cellular signaling pathways.7 Curcuminoids contain curcumin, demethoxycurcumin, and bisdemethoxycurcumin (BDMC); among these BDMC shows increased stability and improved cellular uptake.8 Previous studies revealed that BDMC inhibits proliferation, suppresses migration and invasion, induces apoptosis and generates ROS levels in lung cancer, breast cancer, ovarian cancer and gastric cancer.9–14 Although BDMC inhibits tumorigenesis in various human malignancies, the pharmacological mechanisms regarding its role remain poorly understood, especially in liver cancer.
The serine/threonine protein kinase Akt participates in many aspects of biological functions such as cell proliferation, metabolism, cell cycle and metastasis.15 As such, it is not surprising that deregulated Akt is associated with tumorigenesis and cancer development. The activation of Akt is regulated through Akt phosphorylation at Thr308 and Ser473, which commonly occurs in hepatic oncogenesis and HCC progression.16,17 However, recent studies focused on Akt phosphorylation indicated that ubiquitination and deubiquitination of Akt is also an on-off switch for Akt activity.18,19 For instance, necrosis factor receptor-associated factor 6 (TRAF6) and Skp2-Skp-cullin-F-box-containing (Skp2-SCF) act as E3 ligases to regulate Akt activation through Lys63 (K63)-linked polyubiquitination of Akt in IGF-1 and ErbB receptor signaling, respectively.20,21 Cylindromatosis (CYLD), as a deubiquitinating enzyme (DUB), deubiquitinates and inactivates K63-polyubiquitinated Akt, which suppresses TGF-β signaling.22
In the present study, we found that BDMC attenuated the proliferation of HepG2 cells in a time- and dose-dependent manner via impairing the activation of Akt signaling. In addition, the inactivation of Akt signaling was attributed to the inhibition of ubiquitination mediated by K63-Ub, but not K48-Ub. Furthermore, we demonstrated that BDMC upregulated the expression of CYLD, leading to Akt deubiquitination and inactivation.
Materials and Methods
Cell Culture
The human HCC cell line HepG2 was purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) containing 10% fetal bovine serum (FBS) (Biological Industries, Israel) at 37°C with 5% CO2. Bisdemethoxycurcumin (Selleck, USA) was soluble in dimethyl sulfoxide (DMSO) (Selleck, USA) at the indicated concentrations used for cellular treatment.
Cell Viability Assay
The effects of BDMC on the viability of HepG2 cells were measured by the Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Japan) assay according to the manufacturer’s protocol. Briefly, 1×104 cells in the logarithmic growth phase in a volume of 100 μL DMEM with 10% FBS were plated in each well of a 96-well plate. Cells were treated with 5, 10, 20, 40, 60, 80, 100, 250, 500 and 1000 μM BDMC for the indicated times. Thereafter, 10 μL of CCK-8 solution was added to each well, and the cells were incubated for 2 h at 37°C. The absorbance at 450 nm was measured to evaluate cell viability, with a reference wavelength of 630 nm, using an automated microplate reader (Thermo Fisher Scientific, USA). The half maximum inhibitory concentration (IC50) of BDMC was calculated by using GraphPad Prism 5.01 (GraphPad Software, USA). The experiment was independently repeated three times.
Colony Forming Assay
The HepG2 cells were dissociated into a single cell suspension and diluted to a density of approximately 5×104 cells/mL with DMEM. Then, the HepG2 cells were inoculated in six-well plates at a seeding density of 5×102 cells/well and stimulated with the indicated concentrations of BDMC and DMSO as negative controls. The wells were observed every day using an Olympus IX71 microscope (Olympus, Japan). After 10 days, the cells were fixed in 4% paraformaldehyde for 30 min. The total number of cell colonies was photographed and counted after staining with crystal violet (Solarbio, Beijing, China) for 10 min. The ability to form colonies (groups of 50 or more adhering cells derived from the same mother cell) was assessed. The experiment was independently repeated three times.
Western Blot Analysis
The Western blot protocols were reported previously.23 Briefly, cells were harvested and washed with ice-cold PBS three times and then lysed in RIPA buffer (Beyotime, China) containing protease and phosphatase inhibitors for 30 min on ice. Lysates were obtained via centrifugation at 14,000 rpm at 4°C for 15 mins, after which the supernatants were transferred to new tubes, and their protein concentrations were determined using the BCA protein assay (Thermo Scientific). Equal amounts of each sample diluted in 5x SDS loading buffer were subjected to SDS-polyacrylamide gel electrophoresis and were subsequently transferred to polyvinylidene fluoride (PVDF) membranes (Millipore) for 2 h. The membranes were blocked in 5% nonfat dry milk dissolved in TBST (150 mM NaCl, 50 mM Tris-HCl, pH 7.5, and 0.05% Tween-20) at room temperature for 2 h and then incubated with primary antibodies against GAPDH (1:1000, Cell Signaling Technology, 2118), p-Akt (Ser 473, 1:1000, Cell Signaling Technology, 4060), Akt (1:1000, Cell Signaling Technology, 4691), p-JNK (Thr183/Tyr185, 1:1000, Cell Signaling Technology, 9255), JNK (1:1000, Cell Signaling Technology, 9252), p-Erk (Thr202/Tyr204, 1:1000, Cell Signaling Technology, 4370), Erk (1:1000, Cell Signaling Technology, 4695), p-p38 (Thr180/Tyr182, 1:1000, Cell Signaling Technology, 4511), p38 (1:1000, Cell Signaling Technology, 8690), HA-Tag (1:1000, Cell Signaling Technology, 3724), TTC3 (1:1000, Abcam, ab80061), CYLD (1:1000, Abcam, ab137524), TRAF6 (1:1000, Abcam, ab33915) and Skp2 (1:1000, Abcam, ab68455) overnight at 4°C. The next day, after washing 3 times in TBST, the membranes were incubated with the appropriate HRP-conjugated secondary antibodies (1:3000, Beyotime Institute of Biotechnology, A0216, A0208, A0192) for 1.5 h at room temperature, followed by an additional 3 washes with TBST. The bands were visualized using an ECL Western blot kit (CW0049C, CWBIO). The intensities of the bands were quantified using ImageJ software (NIH, Bethesda, MD, USA, https://imagej.nih.gov/ij/).
Lentivirus Construction and Infection
Protocols were reported previously.23 Briefly, lentiviruses encoding short hairpin RNA (shRNA, GenePharma) targeting CYLD (Sh-CYLD, 5ʹ-GGAAATAAACTCCAGAGTTTC-3ʹ) and the negative control shRNA sequence (Sh-NC, 5ʹ-TTCTCCGAACGTGTCACGTTTC-3ʹ) were constructed. Lentiviruses with constitutively active Akt1 overexpression and their vector controls (CA-akt and Lv-Control) were also purchased from GenePharma. Lentiviruses (109 TU/mL) and polybrene (5 μg/mL, Sigma) were added to the medium and incubated with HepG2 cells for 24 h at a multiplicity of infection (MOI) of 20.
Plasmid Construction and Transfection
Expression plasmid constructs, including full-length pcDNA3.1(+)-HA-Ubiquitin (ubiquitin B, UBB, Homo sapiens), full-length pcDNA3.1(+)-HA-K48-Ubiquitin (ubiquitin B, UBB, Homo sapiens), and pcDNA3.1(+)-HA-K63-Ubiquitin (ubiquitin B, UBB, Homo sapiens) were all constructed by and purchased from Obio Technology Corp Ltd. (Shanghai, China). The Lipofectamine 3000 Transfection Kit (Invitrogen, L3000-015) was used for transfection. Transfection was performed according to the manufacturer’s instructions with minor modifications. Briefly, HepG2 cells were seeded in 6-well plates at a density of 3×105 cells/well. The volume of each plasmid was 2.5 µg/well with 5 µL Lipo3000 and 5 µL P3000, according to the manufacturer’s instructions. The total amount of transfected plasmids in each well was equalized by adding empty pcDNA3.1(+)-vector.
Immunoprecipitation
HepG2 cells were quickly harvested and homogenized in cell lysis buffer for Western blotting and IP (P0013, Beyotime Biotechnology) containing 1% protease and phosphatase inhibitors for 30 min on ice. The cell extracts (approximately 400 μg of total protein) were incubated with an antibody against Akt (1:50, Cell Signaling Technology, 4691) at 4°C overnight with gentle shaking. Then, protein-G agarose beads (40 μL, Beyotime Biotechnology) were added, and the mixture was incubated at 4°C for another 3 h with shaking. The agarose beads were collected, washed, and resuspended in 60 μL of sample buffer containing 50 mM Tris•HCl, pH 7.6, 2% (wt/vol) SDS, 10% (vol/vol) glycerol, 10 mM DTT, and 0.2% bromophenol blue. Afterwards, the samples were boiled for 10 min and analyzed via Western blotting.
Statistical Analysis
All results were determined based on three separate experiments, and each separate experiment in HepG2 cell lines contained three replicates for each condition. All statistical analyses were performed using GraphPad scientific software for Windows (San Diego, CA, USA). Comparisons between two groups were analyzed by independent two-tailed Student’s t tests, and comparisons between more than two groups were analyzed by one-way ANOVA, followed by Tukey’s post hoc test. All data are expressed as the mean±standard deviation. Values of p<0.05 were considered statistically significant.
Result
Bisdemethoxycurcumin (BDMC) Inhibited the Proliferation of Hepatocellular Carcinoma Cells in a Time- and Dose-Dependent Manner
We first detected the toxicity of BDMC at different concentrations for 48 h and calculated the half maximum inhibitory concentration (IC50) using the CCK-8 assay. The results showed that the IC50 value of BDMC after 48 hrs of treatment in HepG2 cells was 59.13 μM (Figure 1A). To avoid the interference of BDMC toxicity, we treated HepG2 cells at concentrations of 5 μM, 10 μM, 20 μM, and 30 μM for 24 h, 48 h and 72 h to measure the inhibitory effect on HepG2 cells through a CCK-8 assay (Figure 1B). At a concentration of 5 μM, the mean absorbance values at 450 nm were 0.571±0.020, 0.725±0.045 and 0.816±0.037 after a 24 h, 48 h and 72 h incubation, respectively. The mean values were 0.537±0.011, 0.645±0.027 and 0.707±0.026 at a concentration of 10 μM, 0.509±0.011, 0.551±0.013 and 0.603±0.018 at a concentration of 20 μM, and 0.455±0.014, 0.457±0.026 and 0.447±0.025 at a concentration of 30 μM. We further confirmed the inhibitory effect via a colony formation assay. As shown in Figure 1C, BDMC significantly suppressed the colony formation of HepG2 cells in a concentration-dependent manner. These results suggested that BDMC inhibited the proliferation of hepatocellular carcinoma cells in a time- and dose-dependent manner.
 |
Figure 1 Effects of BDMC on the viability of the hepatocellular cell line HepG2. (A) The half maximal inhibitory concentrations (IC50) of BDMC were measured by CCK-8 assay. (B) The effects of the indicated concentrations of BDMC on cell viability were measured by CCK-8 assays at 24, 48 and 72 h. (C) The proliferation of HepG2 cell lines at different concentrations of BDMC was detected by a colony formation assay. The data are representative of independent experiments (means ± SD) using one-way analysis of variance (ANOVA) to analyze the differences among groups. *Indicates P<0.05 between groups. |
Bisdemethoxycurcumin (BDMC) Inhibited the Proliferation of HepG2 Cells by Suppressing Akt Signaling Pathway Activation
Previous studies have confirmed that multiple signaling pathways, including the Akt, JNK, Erk and p38 signaling pathways, are involved in the proliferation of hepatocellular carcinoma cells. Thus, we first explored the change in phosphorylation of the abovementioned signaling pathways after treatment with 20 μM BDMC. The results demonstrated that after treatment of HepG2 cell lines with 20 μM BDMC, the activation of the Akt signaling pathway was inhibited significantly, while there were little changes in the activation of the JNK, Erk and p38 signaling pathways after the treatment (Figure 2A). Thus, we further constructed a lentivirus to overexpress constitutively active Akt1 in HepG2 cells. After the overexpression of constitutively active Akt1 in the HepG2 cell line, BDMC lost the ability to inhibit the phosphorylation and activation of Akt (Figure 2B). Finally, we performed the colony formation assay in HepG2 cell lines after transfection of constitutively active Akt1, and the results demonstrated that after overexpression of constitutively active Akt1 in HepG2 cell lines, 20 μM BDMC lost the ability to inhibit the proliferation of HepG2 cells (Figure 2C). In summary, these data indicated that BDMC inhibited the proliferation of hepatocellular carcinoma cells by suppressing Akt signaling pathway phosphorylation and activation.
 |
Figure 2 BDMC inhibited the proliferation of HepG2 cells by suppressing Akt signaling pathway activation. (A) Activation levels of the Akt, JNK, Erk and p38 signaling pathway proteins in each group were detected by Western blotting. The results of Western blotting were quantified as the mean±SD intensity ratio of phosphorylated to nonphosphorylated proteins. (B) Phosphorylation levels of the AKT pathway proteins were assessed by Western blotting after transfection of constitutively active Akt1, which was quantified as the mean±SD intensity ratio of phosphorylated to nonphosphorylated proteins. (C) The proliferation of HepG2 cell lines in each groups was detected by a colony formation assay. The values are presented as the means±SD, using one-way analysis of variance (ANOVA) to analyze the differences among groups. *Indicates P<0.05 between groups. |
Bisdemethoxycurcumin (BDMC) Inhibited the K63-Linked Ubiquitination of Akt
Previous studies have demonstrated that K63-linked ubiquitination of Akt plays an essential role in the phosphorylation and activation of Akt. We first overexpressed HA-Ubiquitin by plasmids in HepG2 cell lines and explored the level of ubiquitination of Akt. The results demonstrated that after treatment of HepG2 cells with 20 μM BDMC, the ubiquitination of Akt was inhibited significantly (Figure 3A). Furthermore, we demonstrated that BDMC inhibited the K63-linked ubiquitination instead of the K48-linked ubiquitination of Akt in HepG2 cells (Figure 3B and C). In conclusion, BDMC inhibited the K63-linked ubiquitination of Akt in HepG2 cell lines.
 |
Figure 3 BDMC inhibited the K63-linked ubiquitination of Akt. (A) The level of ubiquitination of Akt was assayed by Western blotting. (B) The level of K63-linked ubiquitination of Akt was assayed by Western blotting. (C) The level of K48-linked ubiquitination of Akt was assayed by Western blotting. The values are presented as the means±SD, using one-way analysis of variance (ANOVA) to analyze the differences among groups. |
Bisdemethoxycurcumin (BDMC) Inhibited the Activation of the Akt Signaling Pathway and Proliferation of Hepatocellular Carcinoma Cells by Upregulating the Expression of CYLD
Protein ubiquitination is a dynamic process as ubiquitin is attached to substrates by a sophisticated three-step enzymatic cascade, which involves E1 ubiquitin-activating, E2 ubiquitin-conjugating and a variety of E3 ubiquitin-ligating enzymes and several specialized families of proteases, including the deubiquitinases (DUBs), which remove ubiquitin modifications from ubiquitinated proteins. Previous studies have reported several E3 ubiquitin ligases and DUBs that are involved in the ubiquitination of Akt.18 Thus, we first explored the expression of the abovementioned enzymes. The results demonstrated that after treatment of HepG2 cells with 20 μM BDMC, the expression the E3 ubiquitin ligases TTC3, TRAF6 and Skp2 remained stable, while the expression of deubiquitinase CYLD was upregulated significantly after treatment with BDMC (Figure 4A). Thus, we constructed a lentivirus to diminish the expression of CYLD in HepG2 cell lines. After diminishing the expression of CYLD in HepG2 cell lines, BDMC treatment lost the ability to inhibit the K63-linked ubiquitination and phosphorylation of Akt (Figure 4B). Finally, the results of the colony formation assay in HepG2 cell lines demonstrated that CYLD knockdown ablated BDMC-mediated proliferation inhibition in HepG2 cell lines (Figure 4C). In summary, these data demonstrated that BDMC inhibited the K63-linked ubiquitination of Akt, the activation of the Akt signaling pathway and the proliferation of hepatocellular carcinoma cells by upregulating the expression of deubiquitinase CYLD.
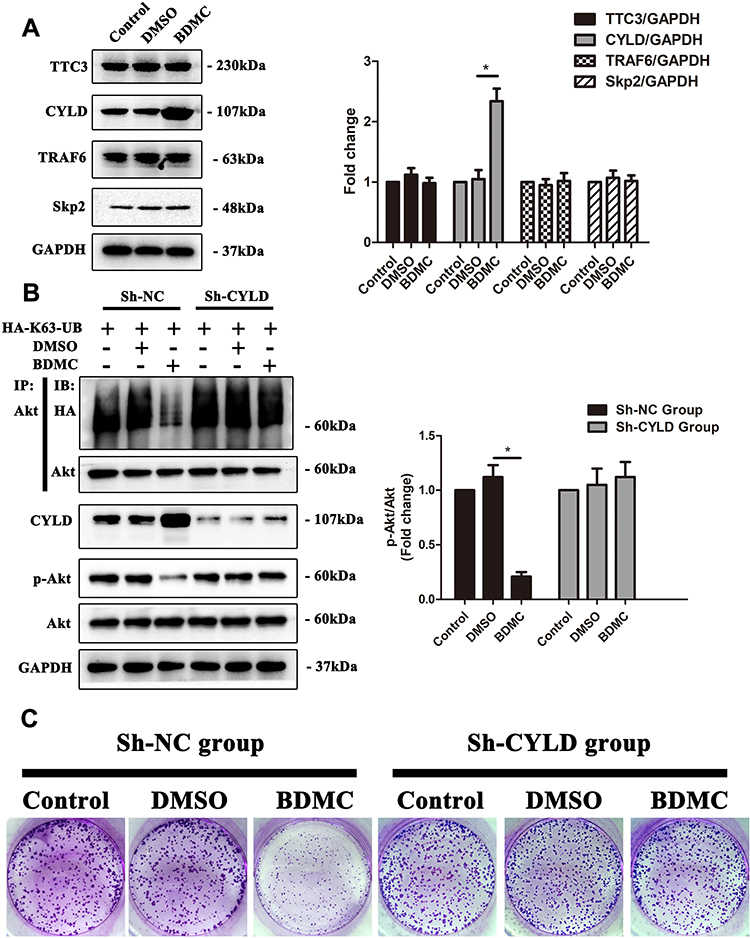 |
Figure 4 BDMC inhibited the activation of the Akt signaling pathway and proliferation of hepatocellular carcinoma cells by upregulating the expression of CYLD. (A) The relevant ubiquitin enzymes TTC3, CYLD, TRAF6 and Skp2 were detected by Western blotting. (B) After transfection with sh-CYLD lentivirus, the levels of K63-linked ubiquitination of Akt, phosphorylation of Akt, and CYLD were assayed by Western blotting. (C) After CYLD knockout, the proliferation of HepG2 cell lines in the control, DMSO and BDMC groups was detected by colony formation assay. The values are presented as the means±SD, using one-way analysis of variance (ANOVA) to analyze the differences among groups. *Indicates P<0.05 between groups. |
Discussion
In this study, we evaluated the effect of BDMC and demonstrated that BDMC inhibited the proliferation of hepatocellular carcinoma cells via upregulating the expression of CYLD to decrease the K63-linked ubiquitination of Akt and the activation of the Akt signaling pathway.
BDMC is one of the most abundant compounds in the dried rhizome, accounting for 10–15% of curcumoids.24 Previous studies revealed that BDMC treatment is related to various antitumor characteristics, such as proliferation inhibition, metastasis suppression and apoptosis induction and that BDMC is engaged in several signaling pathways. For example, BDMC inhibited the viability of MCF-7 cells through regulation of p53/p21 and p16/Rb pathways.9 BDMC regulated E-cadherin and vimentin expression in 95D cells, which suppressed their migration and invasion abilities.14 BDMC also reactivated WIF-1 from a silenced state to induce apoptosis in lung cancer.10 Our results showed that BDMC attenuated the malignant phenotype of HepG2 cells, including proliferation and anchorage-independent growth. The inhibitory effect in the HepG2 cell line is time- and dose-dependent, and the IC50 was 59.13 μM. A similar result has been reported: BDMC inhibited HepG2 cells via G2/M cell arrest and ROS generation.25
During cancerous development, cellular proliferation is an essential process that is dominated by the spatial and temporal activation of signal pathways, including mitogen-activated protein kinase (MAPK) cascades and PI3K/Akt pathways.3,15,16,26 Our study further investigated the potential mechanisms by which BDMC suppressed HCC cell proliferation by detecting the activation of Akt and three clearly characterized MAPK families—JNK, ERK and p38. We found that Akt phosphorylation was decreased following BDMC treatment, as was anchorage-independent growth. Interestingly, BDMC had little effect on JNK, Erk and p38 activity. Although a myriad of crosstalk interactions following pharmaceutical drug treatment affect cell fates, these results led us to speculate that the Akt signaling pathway might be a target of BDMC. We then constructed lentivirus with constitutively activated Akt1 and transduced them into HepG2 cells, and the results showed that the effects of BDMC were abolished. Akt deactivation is one of the potential mechanisms by which BDMC inhibits HCC cell proliferation.
Studies of pharmaceutical drugs detailing the Akt signaling pathway have focused little on its ubiquitination. Ubiquitination is one of the posttranslational modifications triggered by three classes of enzymes, ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligase (E3), through attaching the ubiquitin(s) to lysine residues of proteins; ubiquitination is involved in protein activity and stability and has numerous biological functions. This process can be reversed by deubiquitination enzymes (DUBs). There are seven lysine (K) residues within the ubiquitin, including K6, K11, K27, K29, K33, K48 and K63.19 Lys48-linked chains are the predominant linkage type in cells, while Lys63-linked chains are the second most abundant type. K48-linked ubiquitination often marks proteins for degradation, while K63-linked ubiquitination modification performs various nondegradative roles.27 Previous studies have demonstrated that K63-linked ubiquitination of Akt mediates Akt cell membrane recruitment and activation and leads to tumorigenesis.20,28 In this study, our novel finding is that BDMC treatment not only affected the phosphorylation of Akt but also decreased the K63-linked ubiquitination of Akt. Furthermore, our research also found that BDMC did not influence the K48-linked ubiquitination of Akt, which might indicate that BDMC-inhibited HepG2 cell proliferation did not depend on the ubiquitin-proteasome system. However, how BDMC decreases K63-linked ubiquitination of Akt proliferation remains unknown because multiple molecules, including TRAF6, TTC3 and Skp2, etc., regulate the progression of carcinomas by intervening with this process.20,21,29 In our study, we demonstrated that although BDMC did not affect the expression of TRAF6, TTC3 and Skp2, it upregulated the expression of CYLD to deubiquitinate the K63-linked ubiquitination of Akt and thus inhibited the activation of Akt and proliferation of hepatocellular carcinoma cells. Consistent with our study, previous studies have demonstrated that loss or reduced expression of CYLD is observed in different types of human cancer and that CYLD is a tumor suppressor.30 For example, Yang et al demonstrated that advanced prostate cancer was related to high Akt phosphorylation and low CYLD abundance and that CYLD deficiency promoted prostate cancer cell proliferation, survival, glucose uptake, and xenograft tumor growth.31 Interestingly, CYLD inhibits other proteins and signaling pathways as well.32,33 Therefore, Akt deubiquitination might not be sufficient to illustrate the inhibitory effect of BDMC through upregulating CYLD in our study. Further studies to explore Akt ubiquitination and CYLD may be of great value for identifying therapeutic targets for hepatocellular carcinoma.
In conclusion, in this study, we demonstrated that BDMC inhibited the proliferation of hepatocellular carcinoma cells via upregulating the expression of CYLD to decrease the K63-linked ubiquitination and activation of the Akt signaling pathway. Thus, BDMC may be an effective candidate drug for treating hepatocellular carcinoma. However, there are still some limitations to this study. First, evaluating the effect of BDMC on the proliferation of hepatocellular carcinoma in vivo is of great importance. Second, the mechanism by which BDMC mediates the expression of CYLD remains unclear. These limitations will be the focus of our future study.
Funding
This study was supported by grants from the Science and Technology Planning Project of Guangdong Province (grant no. 2017ZC0180).
Disclosure
The authors report no conflict of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.v68.6
2. Maluccio M, Covey A. Recent progress in understanding, diagnosing, and treating hepatocellular carcinoma. CA Cancer J Clin. 2012;62(6):394–399. doi:10.3322/caac.21161
3. Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106–117. doi:10.1016/j.pharmthera.2017.02.010
4. Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi:10.3322/caac.21338
5. Zhu AX. Systemic therapy of advanced hepatocellular carcinoma: how hopeful should we be? Oncologist. 2006;11(7):790–800. doi:10.1634/theoncologist.11-7-790
6. Carr BI. Hepatocellular carcinoma: current management and future trends. Gastroenterology. 2004;127(5 Suppl 1):S218–24. doi:10.1053/j.gastro.2004.09.036
7. Tomeh MA, Hadianamrei R, Zhao X. A review of curcumin and its derivatives as anticancer agents. Int J Mol Sci. 2019;20(5). doi:10.3390/ijms20051033
8. Basile V, Ferrari E, Lazzari S, Belluti S, Pignedoli F, Imbriano C. Curcumin derivatives: molecular basis of their anti-cancer activity. Biochem Pharmacol. 2009;78(10):1305–1315. doi:10.1016/j.bcp.2009.06.105
9. Li YB, Gao JL, Zhong ZF, Hoi PM, Lee SM, Wang YT. Bisdemethoxycurcumin suppresses MCF-7 cells proliferation by inducing ROS accumulation and modulating senescence-related pathways. Pharmacol Rep. 2013;65(3):700–709. doi:10.1016/S1734-1140(13)71048-X
10. Liu YL, Yang HP, Zhou XD, Gong L, Tang CL, Wang HJ. The hypomethylation agent bisdemethoxycurcumin acts on the WIF-1 promoter, inhibits the canonical Wnt pathway and induces apoptosis in human non-small-cell lung cancer. Curr Cancer Drug Targets. 2011;11(9):1098–1110.
11. Luo C, Du Z, Wei X, Chen G, Fu Z. Bisdemethoxycurcumin attenuates gastric adenocarcinoma growth by inducing mitochondrial dysfunction. Oncol Lett. 2015;9(1):270–274. doi:10.3892/ol.2014.2685
12. Pei H, Yang Y, Cui L, et al. Bisdemethoxycurcumin inhibits ovarian cancer via reducing oxidative stress mediated MMPs expressions. Sci Rep. 2016;6:28773. doi:10.1038/srep28773
13. Liao CL, Chu YL, Lin HY, et al. Bisdemethoxycurcumin suppresses migration and invasion of human cervical cancer HeLa cells via inhibition of NF-kB, MMP-2 and −9 pathways. Anticancer Res. 2018;38(7):3989–3997. doi:10.21873/anticanres.12686
14. Xu J, Yang H, Zhou X, Wang H, Gong L, Tang C. Bisdemethoxycurcumin suppresses migration and invasion of highly metastatic 95D lung cancer cells by regulating E-cadherin and vimentin expression, and inducing autophagy. Mol Med Rep. 2015;12(5):7603–7608. doi:10.3892/mmr.2015.4356
15. Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development. 2016;143(17):3050–3060. doi:10.1242/dev.137075
16. Chung W, Kim M, de la Monte S, et al. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. 2016;370(1):1–9. doi:10.1016/j.canlet.2015.09.016
17. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12(4):487–502. doi:10.1016/j.devcel.2007.03.020
18. Lin K. The Akt DUBbed InAktive. Sci Signal. 2013;6(257):pe1. doi:10.1126/scisignal.2003864
19. Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010;9(3):487–497. doi:10.4161/cc.9.3.10508
20. Yang WL, Wang J, Chan CH, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325(5944):1134–1138. doi:10.1126/science.1175065
21. Chan CH, Li CF, Yang WL, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149(5):1098–1111. doi:10.1016/j.cell.2012.02.065
22. Lim JH, Jono H, Komatsu K, et al. CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat Commun. 2012;3:771. doi:10.1038/ncomms1776
23. Liu K, Wu X, Zang X, et al. TRAF4 regulates migration, invasion, and epithelial-mesenchymal transition via PI3K/AKT signaling in hepatocellular carcinoma. Oncol Res. 2017;25(8):1329–1340. doi:10.3727/096504017X14876227286564
24. Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The essential medicinal chemistry of curcumin. J Med Chem. 2017;60(5):1620–1637. doi:10.1021/acs.jmedchem.6b00975
25. Chen J, Li L, Su J, Chen T. Natural borneol enhances bisdemethoxycurcumin-induced cell cycle arrest in the G2/M phase through up-regulation of intracellular ROS in HepG2 cells. Food Funct. 2015;6(3):740–748. doi:10.1039/C4FO00807C
26. Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18. doi:10.1038/sj.cr.7290105
27. Saeki Y. Ubiquitin recognition by the proteasome. J Biochem. 2017;161(2):113–124. doi:10.1093/jb/mvw091
28. Restuccia DF, Hemmings BA. Cell signaling. Blocking Akt-ivity. Science. 2009;325(5944):1083–1084. doi:10.1126/science.1179972
29. Suizu F, Hiramuki Y, Okumura F, et al. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell. 2009;17(6):800–810. doi:10.1016/j.devcel.2009.09.007
30. Bignell GR, Warren W, Seal S, et al. Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet. 2000;25(2):160–165. doi:10.1038/76006
31. Yang WL, Jin G, Li CF, et al. Cycles of ubiquitination and deubiquitination critically regulate growth factor-mediated activation of Akt signaling. Sci Signal. 2013;6(257):ra3. doi:10.1126/scisignal.2003197
32. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17(1):25–34. doi:10.1038/cdd.2009.43
33. Glittenberg M, Ligoxygakis P. CYLD: a multifunctional deubiquitinase. Fly (Austin). 2007;1(6):330–332. doi:10.4161/fly.5399
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.