Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 9
Biosimilars: potential implications for clinicians
Authors Eleryan M, Akhiyat S, Rengifo-Pardo M, Ehrlich A
Received 13 February 2016
Accepted for publication 30 March 2016
Published 17 June 2016 Volume 2016:9 Pages 135—142
DOI https://doi.org/10.2147/CCID.S91691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Misty G Eleryan,1 Sophia Akhiyat,2 Monica Rengifo-Pardo,1 Alison Ehrlich,1,2
1Department of Dermatology, The George Washington Medical Faculty Associates, 2George Washington University School of Medicine & Health Sciences, Washington, DC, USA
Abstract: With the expiration of patent protection for several biologics looming, the production of highly similar therapeutic agents has begun to emerge on the pharmaceutical market. These alternative drugs are referred to as biosimilars. Many anticipate that the introduction of these agents will result in a reduction in health care costs, which may create a more affordable biopharmaceutical market and also improve patient access. In contrast to generics, which are exact copies of their original products, biosimilars are not identical to their reference products. Due to concern about the safety and efficacy of biosimilars, separate regulatory approval pathways have been developed and implemented by several countries, including the US and Europe. Europe has led the way in acceptance of biosimilars into mainstream clinical practice. Biosimilars are not generic products and require extensive clinical and nonclinical bioequivalence studies before receiving marketing approval. Not only is there a lengthy developmental process, but also they will likely be required to have postmarketing surveillance and ongoing safety monitoring to keep track of issues that may arise, such as immunogenicity. Although US Food and Drug Administration approved the first biosimilar product in March 2015, physicians remain unfamiliar about their indications.
Keywords: biologics, biopharmaceuticals, biomimics, biocopies, interchangeability, immunogenicity
Introduction
The emergence of biologics has been a major advancement in the management of various chronic diseases that were previously unresponsive to conventional therapies. Biologics are large proteins derived from living organisms or cells and manufactured through a highly complex biotechnological process.1 Examples of biological agents are hormones, interferons, monoclonal antibodies, interleukins, and vaccines. Although these drugs are very efficacious, cost is a major factor for patients. Moreover, the growing concern about the cost of drugs, including biologics, has now become a major political issue in many countries including the US. Thus, biosimilars may represent a more economical alternative to the costly biological agents already on the market (Table 1).
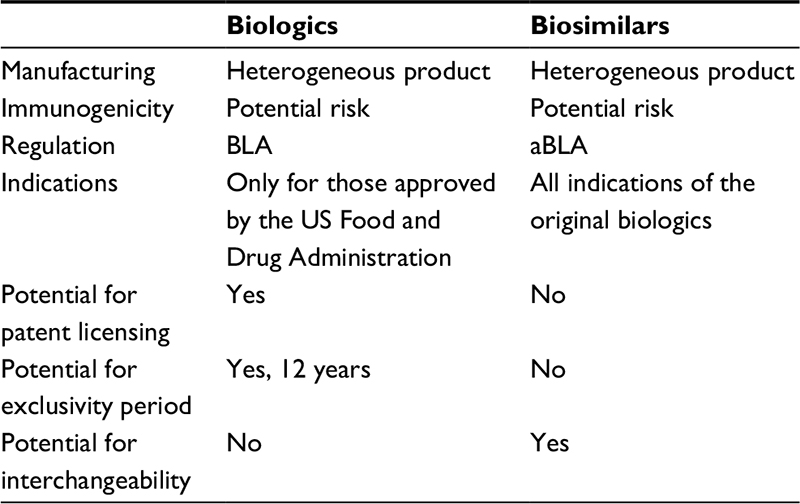 | Table 1 Comparison of biologics and biosimilars Note: Data from Blackstone and Fuhr5 and Camacho et al.9 Abbreviations: BLA, biologics license application; aBLA, abbreviated biologics license application. |
The biologic market is a highly profitable one, with a reported $157 billion in global sales in 2011 with projections to exceed $200 billion by 2016.2,3 Global pharmaceutical sales of biologics are likely to constitute ˜49% of the market by 2018.4 The average daily cost of a biologic drug in the US is $45 when compared to nonbiologic drugs that sell at $2.5 The multifaceted development, manufacturing, and regulation associated with creating a biologic are the factors that cause the drug to have a significant cost.5 According to Silver,3 the development of an innovative biologic drug was estimated to cost at least $1.9 billion in 2012. Similarly, significant investment is required to develop a biosimilar, as its innovation takes an average of 7–8 years.5 However, the cost of this innovation is estimated to range from $100 to $250 million, as compared to the ˜$2 billion required for innovator biologic drugs.5,6 In sharp contrast to biologics and biosimilar development costs, generic drug production costs are estimated to be from $1 to $4 million.5 The production costs for generic drugs are reported to be low once the drug has obtained approval from US Food and Drug Administration (US FDA); in contrast, the production costs of biosimilars may be high, largely due to the unique purification and processing of the drug to protect against immunogenicity and ensure development of a highly similar product.5,7
The European Union (EU) has led the way in the use of biosimilars, with the first approval being epoetin biosimilars (Abseamed®, Binocrit®, epoetin alfa hexal®, Retacrit®, and Silapo®) in 2007 in Europe.8 As of 2014, biosimilars in Europe were priced up to 35% less than the original biologic price.9 The anticipation of these new alternative drugs in the EU resulted in the development of guidelines to assess the similarity and safety between biosimilars and their reference products. The European Medicines Agency (EMA) established a regulatory pathway in 2005.9 Because of their experience with biosimilars, the EMA guidelines are widely accepted as a model for other countries looking to incorporate biosimilars into their pharmaceutical market place, as well as learn from their successes and failures.9
A potential barrier for pharmaceutical companies in biosimilar production is the complexity of their approval requirements. The fact that significant time and monetary investment are required, coupled with the uncertainty of timely approval and the cost of postapproval marketing, encourages pharmaceutical companies to selectively develop biosimilars for licensed biologics that garner higher sales. In this way, extensive investments can be justified by substantial profit.5
In this review, we provide an overview of biosimilars and address important factors for practicing clinicians, including the 1) differences in manufacturing processes, 2) description of the regulatory pathways, and 3) discussion of interchangeability and substitution, immunogenicity, and extrapolation of indications.
Terminology/definitions
The use of unique and new terminology when discussing biopharmaceuticals (biologics, biosimilars, and generics) fuels the misconceptions and uncertainty of clinicians in regard to biosimilar utilization. Here, we define the common terms used when referring to the biopharmaceutical developmental process.
A biologic is a protein created by recombinant DNA expression in live cells that has a therapeutic effect on human diseases.10,11 Examples of biologics include fusion proteins, monoclonal antibodies, hormones, and cytokines.11,12 Indications for use vary widely, including management of psoriasis, chronic urticaria, atopic dermatitis, rheumatoid arthritis, and inflammatory bowel disease.
A biosimilar, also referred to as a “follow-on biologic”, is a biological product that is marketed as an alternative to the original biologic, which shares highly similar, but not identical, safety, and efficacy profiles.8 Biosimilars are highly complex three-dimensional protein structures that are derived from living cell lines and have therapeutic effects.13 They are biological medicine’s analogs to generics for the synthetically derived pharmaceutical market; however, they are not considered to be part of the “generics” category of pharmaceuticals.
Biosimilars are not the same as “biomimics” and “biocopies”, which are intended copies of monoclonal antibodies and fusion proteins, respectively, that have not demonstrated bioequivalence to their reference biologics. These biologic agents are available in a few countries.14 Bolivia, India, People’s Republic of China, and some Latin American countries have approved and licensed several of these drugs, such as Yisaipu® (biocopy of etanercept) and Reditux® (biocopy of rituximab).14–16 Biomimics preceded biosimilars in marketing, including the biomimic Yiasaipu®, which has been on the market in the People’s Republic of China for over a decade.16 Unfortunately, some biomimics have been responsible for more adverse events compared to their reference biologics. For example, Kikuzubam®, a biocopy of rituximab manufactured in Mexico City, was taken off the market in March 2014 due to several reports of anaphylactic reactions occurring when patients switched from the original biologic to the biomimic.14
Generic drugs have small molecular chemical structure and are exact copies of the original nonbiologic drugs. US FDA requires verification of similar pharmacokinetic and pharmacodynamic profiles to the original nonbiologic drug17 and also requires that the ratio of generic drug to the reference product range between 0.80 and 1.25; this verifies adequate bioequivalence to the generic’s reference product.8
In comparison, bioequivalence studies are required to demonstrate that there is no clinically significant difference between biosimilars and the originator reference product in terms of their bioavailability.8
Interchangeability is defined as a high level of biosimilarity when compared to the reference biologic.9 US FDA considers a biosimilar as interchangeable if it is “expected to produce the same clinical result as the reference product in any given patient” and “if administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between use of the biological product and the reference product is not greater than the risk of using the reference product without such (a) switch”.9
Biosimilar development
The notion that biosimilars are “similar but not identical” adds to the concern about the safety and efficacy of biosimilars as compared to biologics. Each product batch of biosimilars is unique from the next in terms of structure, namely due to inevitable posttranslational modifications, referred to as the “microheterogeneity phenomenon”.10 This is not a new phenomenon, as even biologics are subject to microheterogeneity due to biochemical modifications and the complex manufacturing process.18 An example of microheterogeneity in a biosimilar that has been accepted by the EMA is an increased number of phosphorylations on mannose-type structures in epoetin alfa.19 Support for this acceptance is the fact that the different phosphorylated structures are actually common forms of recombinant erythropoietin.19
Generic drugs do not experience microheterogeneity due to the formulaic synthesis of their small chemical structures. In contrast to generic drugs, which are chemically based and have highly predictable developmental processing, biosimilars express an inevitable level of variability in protein folding among preparations even though the primary protein sequences are identical.10 This is due to the unique, organic nature of synthesizing biologics in living organisms. During the intracellular processing of the biosimilar, different modifications (eg, phosphorylations, protein clippings, and glycosylation) are implemented to allow the cell to protect the new protein during transportation into different cellular compartments (Figure 1). Thus, data requirements for the production and marketing of biosimilars are far more than those for generic chemical drugs, which have predictable characteristics. For generic chemical drugs to be approved, demonstration of similar pharmacokinetic profiles (bioequivalence) to the original chemical drug must be established. This proof is typically adequate to conclude therapeutic equivalence between the generic and original chemical drugs. In contrast, biosimilars require meticulous comparison with the reference biologic, which involves a variety of methods including comparability exercises.
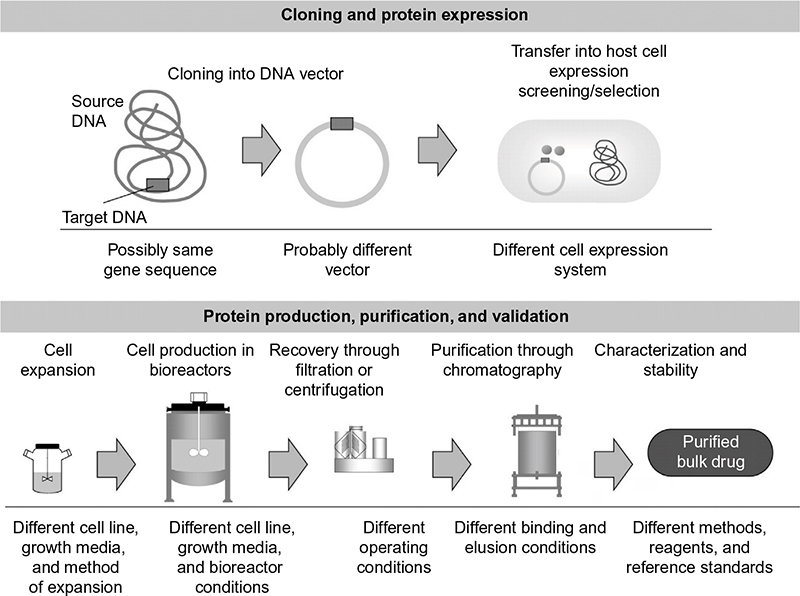 | Figure 1 Variation in manufacturing techniques of biopharmaceuticals. Note: Reproduced from Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–419,31 by permission of Oxford University Press. |
To overcome potential disparity from protein to protein in finished biosimilar batches, a thorough comparability review of structural and functional properties as well as the removal of process-related impurities is required.8 This “comparability exercise” allows regulators to extensively assess the quality, safety, and efficacy of the “newer” version postmanufacturing before final approval is given.18 The analytical assays determine the degree of structural similarity of biosimilars and their reference products with in vitro analysis using protein sequencing, nuclear magnetic resonance spectroscopy, and chromatography to name a few. Because biologic manufacturers realize that their active product undergoes various sequences of modifications from batch to batch, they currently use these well-established methods to determine the comparability of each batch.20 The next course of action to discover variations that may not have been detected during the in vitro molecular testing phase is preclinical testing. This phase focuses on comparing pharmacokinetics and pharmacodynamics of the drug to the reference product in animal model testing.20 Clinical testing on healthy volunteers is the last step in determining noninferiority and similarity between the biosimilar and original product. Phase II clinical trials are omitted because the goal is to establish that there are “no clinically meaningful differences” in the safety and efficacy of biosimilars when compared to the original biologic.10,20
Synthesizing biosimilars under multiple manufacturers with different quality systems presents obstacles, including product drift, evolution, and divergence.
Drift has been defined in a Product Quality Research Institute–US FDA workshop as being “an unintended, unexplained, or unexpected trend of measured process parameter(s) and/or resulting product attribute(s) away from its intended target value in a time-ordered analysis over the lifetime of a process or product”.21 This deviation from the intended biologic product may be a systematic trend or a sudden shift in quality. Product drift may be managed with vigorous quality system monitoring and close evaluation of process parameters.21 Occasionally, biological drugs may drift outside the boundaries of acceptable criteria of similarity due to manufacturing changes, which is a phenomenon described as product evolution.21
Product drift and evolution, over time, may contribute to clinically significant differences between biosimilars and their reference products. This cumulative effect is termed divergence.21 In cases where the biosimilar and reference product are produced by different manufacturers, the two biologics that are initially similar may not remain comparable over time. This loss in similarity may result from unrevised drift by a manufacturer or the evolution of one or both of the agents.21 The clinically meaningful differences of interest include the potency, safety, and immunogenicity profiles. For example, the biosimilar or reference product’s divergence in immunogenicity could affect its safety profile and efficacy of a patient’s therapy.21
Regulatory process
Because biosimilars and generic drugs belong to different drug categories, each is under its own unique regulation. Biosimilars and generic drugs have separate data requirements for licensing in the US. For example, while generic drugs must prove that they are copies of their reference drug, biosimilars must undergo extensive comparison with the reference biologic to confirm close similarity in regard to biological features.10 Generic drugs are monitored by the Federal Food, Drug, and Cosmetic Act, whereas biosimilars are regulated by US FDA under the Public Health Service Act.11 Through an amendment of the Public Health Service Act by the Biologics Price Competition and Innovation Act (BPCIA) of 2009, an approval pathway for biosimilars was established.22
The Affordable Care Act of 2010 included the BPCIA, which aims to provide a solution for protecting against biologic monopoly by allowing competition to enter the market while still encouraging the innovation of new drugs.5 The BPCIA provides a pathway for competition for potential biosimilar entrance into the market once patent protection for biologics has expired.5 Patent protection for originator biologics expires 20 years from the file date of the patent application. Once US FDA issues market approval, the BPCIA grants marketing exclusivity for 12 years and a 4-year data exclusivity for the originator biologic agent. A 6-month exclusivity extension can be given to pediatric applications.5
The BPCIA created an abbreviated application pathway (the 351k path) for biosimilars, which minimizes the testing required in animals and humans, referred to as the abbreviated biologics license application (aBLA).5 The aBLA’s expedited process of biosimilar approval, in comparison with that of biologics, aims to encourage more applicants to reach the market quicker in order to introduce competition, thereby increasing the consumer benefit.5 In order to be approved for the 351k path, products must prove their similarity to the reference in terms of using the same indication, dosing, route of administration, mechanism of action, and potency.11
Although recent legislation has allowed the abbreviated processing of biosimilars, the caveat is that US FDA requires numerous nonclinical studies proving the safety and efficacy, a clinical trial, and an immunogenicity study.9 This may offer less motivation for pharmaceutical companies to invest in biosimilar production through the aBLA. Biologics approved under the biologics license application benefit from an exclusivity period, patent licensing, and a larger sample size. In contrast, biosimilars processed under the aBLA have no rights to exclusivity or patent privileges, which may make investing in biosimilar production a more complex decision for pharmaceutical companies.5
Both domestic and international efforts are underway for promoting the utilization of biosimilars. The World Health Organization composed a guideline for biosimilars in 2009 for the purposes of creating an international resource to guide clinical trials, establish pharmacovigilance, and serve as a reference for countries with less developed pharmacovigilance regulations.23 Since its publication, this universal resource has influenced the construction of many nations’ own approval protocol for biosimilars.
As mentioned previously, the EU leads the nations in pursuing biosimilar utilization. Motivated by the expiration of biologic patents, including epoetin alfa and somatropin, the EMA created the first ever biosimilar regulatory guideline in 2005. The guideline requires proof of biosimilar purity, efficacy, and pharmacovigilance data as validated by clinical studies and extrapolation analysis.9 In 2006, the EMA issued further guidance pertaining to matters concerning biosimilar approval, including quality issues of analytical studies as well as clinical and nonclinical issues. The guidelines sought to ensure that proper reference biologics were chosen for each biosimilar, methods and product indications were clearly stated, biologic activity was sufficiently proven, pharmacophysiologic mechanisms were clear and correct, and postmarketing safety monitoring protocols were in place. With current biosimilar regulatory guidelines in revision, the EMA has also created class-specific biosimilar guidelines that include categories, such as recombinant therapeutic proteins and monoclonal antibodies.9
Although Europe and India have created guidelines to manage biosimilars in 2006 and 2012, respectively, the US is trailing behind having only recently issued the first draft of biosimilar guidelines in 2014.20
US FDA defines biosimilars as being highly similar to its US FDA-approved reference biologic product as far as quality, safety, and potency.11 US FDA has issued draft guidelines to provide a course of action for applicants to demonstrate “biosimilarity”, requiring structural–functional analysis, results of animal studies offering pharmacokinetic and safety data, and extrapolations for indications as supported by evidence-based medicine. The guidelines also request background information of the biosimilars, including information on the manufacturing process, impurities, and drug stability.11 Per guidelines, comparative analytical assay studies are recommended to describe the protein structure, posttranslational modifications, functional activity, and toxicity as compared to reference.9
Following analysis, pharmacophysiologic studies are recommended to demonstrate mechanisms of action. Clinical trials and postmarketing surveillance monitoring are recommended to assess the safety profile of the biosimilar.9 US FDA also addressed the naming of biosimilars. They proposed the use of the reference product’s core name (eg, trastuzumab) with an assigned four-lowercase letter suffix attached with a hyphen (eg, trastuzumab-abcd) to help medical providers differentiate the reference drug from its biosimilar.24
State regulations of US vary in regard to pharmacist substitution for biologics written by the prescribing physician. Whether automatic substitution of a biosimilar for a biologic in written prescriptions is deemed by US FDA to be interchangeable or not depends on each state’s laws as well as judgment from the state pharmacy boards according to the BPCIA of 2009.9 Automatic substitution laws are especially of importance for biologic drugs that are prescribed to patients to be obtained from retail pharmacies for self-administration (eg, supportive care biologics).9 This is in contrast to biologics that must be dispensed in an inpatient or outpatient setting (eg, chemotherapeutic biologics for cancer), where the retail pharmacy substitution process is generally not an issue.5
According to the National Conference of State Legislatures, as of July 2015, 15 states have enacted and signed statutes regarding biosimilar substitutions for biologics, which include Delaware, Florida, Indiana, Massachusetts, North Dakota, Oregon, Utah, Virginia, Washington, Texas, Louisiana, Tennessee, North Carolina, Georgia, and Colorado. Seven states have filed bills that have either failed or were adjourned, and five states have pending legislation. Puerto Rico, New Jersey, and Illinois have passed legislation, but there is no state law. California’s legislation was passed but vetoed in 2013. Typical features of state legislation are listed in Table 2, many of which rely on the interchangeability between biologics and biosimilars.25
 | Table 2 Typical features of state legislation related to biologic medications and substitution of biosimilars according to the NCSL as of 2015 Note: Data from Jelkmann.25 Abbreviations: NCSL, National Conference of State Legislatures; US FDA, US Food and Drug Administration. |
Interchangeability presents a growing concern for clinicians, as they may not be asked for permission to have a biosimilar substituted for the prescribed biologic agent for their patient with automatic substitution. Although generic drugs are routinely interchanged for original therapeutic agents, the risk of immunogenicity from the repeated substitutions is low compared to biosimilars. The EMA allows the individual EU states to decide on this matter.26 US FDA has not yet issued regulatory information about how they plan to determine interchangeability for substitutions. Of note, no biosimilars are currently approved as interchangeable by US FDA.
Safety and efficacy
Repeated switching between biosimilars and their reference products carries a theoretical risk of patients developing immunogenicity; however, this risk is also present when switching occurs between batches of reference products. Immunogenicity is the induction of an antibody response by a drug, a phenomenon unique among biopharmaceuticals.20 Immunogenicity profiles of biologic drugs are of prime interest, as they have the potential to compromise the safety and efficacy of the drug.9 The factors contributing to the immunogenic profile of a biologic include the product characteristics, the underlying disease processes, and the patient’s unique factors. The disease processes and patient factors have already been studied during the development of the associated reference biologic, so the main component of immunogenicity in biosimilars that should be scrutinized is the product characteristics. The inevitable microheterogeneity in biosimilar structures, such as unique posttranslational modifications and protein aggregation, could have a significant impact on immunogenicity.9
A well-known example of immunogenicity was the development of neutralizing antibody-mediated pure red cell aplasia resulting from the substitution of polysorbate 80 and glycine for albumin as a stabilizer in Eprex®. Eprex® was a recombinant human erythropoietin formulation for patients with chronic renal failure in Europe.27 Of note, it was approved as a biologic not as a biosimilar. A second pure red cell aplasia incident occurred with the administration of a subcutaneous epoetin biosimilar HX575® during a clinical trial in Europe. The trial was subsequently terminated after two patients formed neutralizing antibodies while on the medication. Researchers speculated that resultant immunogenicity from HX575® could have been caused by a structural modification during manufacturing.27
Due to the confidentiality of pharmaceutical company trade secrets, such as the details of manufacturing processes for licensed biologics, biosimilar developers must establish their own unique manufacturing process after careful and extensive research of the original biologic.28 Also, having previous knowledge and expertise about the process of developing biologics and the ability to use “reverse engineering” increase the likelihood of biosimilarity.29 However, the inevitable posttranslational modifications that occur during the intracellular processing of the biosimilar causing microheterogeneity from its reference product create a concern about their potential for immunogenicity.
Postmarketing surveillance of the safety of biosimilars is needed for complete reassurance, in addition to premarketing safety analysis and animal studies, in order to ensure ongoing comprehensive monitoring.30 The standard protocol for a new drug application requires the composition of a risk management plan, which discusses potential safety concerns with the drug and submission of the protocol to US FDA for the postmarketing surveillance process. In the case of a biosimilar applicant with a known rare presentation of an immune response that is unlikely to be detected preceding licensing, the biosimilar may undergo expedited approval and require an additional postmarketing study to discover potential incidences.30
US FDA supports the extrapolation of biologic indications to cover their respective biosimilars even if formal investigation of the biosimilar for those additional indications has not been performed.9 A biosimilar used to treat a disease, such as rheumatoid arthritis, may also be approved for another disease, such as psoriasis, without going through a clinical trial since the corresponding reference biologic would have the same indications. For US FDA approval of extrapolations, it is important that the biosimilar applicant clinically supports that it shares the same distribution, mechanism of action, pharmacokinetics, pharmacodynamics, effectiveness, and potential toxicity.9
Conclusion
Biosimilars are at the forefront of medicine changing the landscape and offering an option for helping to alleviate health care’s ever rising costs. By introducing competition, there is potential for reduction in health care costs of associated biopharmaceuticals and the creation of a more affordable biologic market. Newer biosimilars, which are thought of as “biobetters” or “biosuperiors”, are also in development to mimic and improve upon the biologic reference’s administration, mechanism of action, pharmacokinetics, and pharmacodynamics.11
With the influx of new biologic alternatives, there is a critical need for the development of analytical tools and assays with specific end points to better demonstrate similarity of biosimilars in applying for US FDA approval.11 Analytical studies must be enhanced to adequately predict the immunogenicity potential, a required reported component for US FDA approval.9
To date, biosimilar use has been very successful in Europe and other countries, thus serving as a model for the US along with the World Health Organization international guidelines.9 In order to take advantage of the benefits of biosimilars, there is a need for open communication within the medical community to facilitate awareness and knowledge about the safety and efficacy of biosimilars and the appropriate use in patients.
Disclosure
Doctor MGE’s fellowship is funded by Janssen Biotech, Inc. Doctor AE discloses that she is a speaker for Abbvie and Celgene and a principal investigator for Merck, DUSA, Regeneron, Lilly, Leo, and Janssen. The other authors report no conflicts of interest in this work.
References
Kefalas CH, Ciociola AA; FDA-Related Matters Committee of the American College of Gastroenterology. The FDA’s generic-drug approval process: similarities to differences from brand-name drugs. Am J Gastrenterol. 2011;106(6):1018–1021. | ||
Weaver C, Whalen J, Rockoff JD [webpage on the Internet]. Biotech drugs still won’t copy. Wall Street Journal; 2013. Available from: http://online.wsj.com/article/SB10001424127887323864304578318111144984632.html. Accessed April 20, 2016. | ||
Silver S [webpage on the Internet]. Industry Surveys: Biotechnology. Standard & Poor’s; 2013. Available from: https://securingalpha.files.wordpress.com/2014/01/biotechnology-2013.pdf. Accessed April 20, 2016. | ||
Dorey E. How the biologics landscape is evolving. Clin Pharm. 2014;6(9):1–6. | ||
Blackstone E, Fuhr J. The economics of biosimilars. Am Health Drug Benefits. 2013;6(8):469–478. | ||
Citation Machine [webpage on the Internet]. Innovation Over Imitation: High-Cost Biosimilar FOB Development, Slow Uptake. Generics and Biosimilars Initiative. Available from: http://www.citationmachine.net/bibliographies/59804304?new=true. Accessed April 20, 2016. | ||
Winegarden, W [webpage on the Internet]. The Economics of Pharmaceutical Pricing. Pacific Research Institute, June. Available from: http://www.pacificresearch.org/fileadmin/documents/Studies/PDFs/2013-2015/PhamaPricingF.pdf. Accessed April 20, 2016. | ||
Schellekens H. The first biosimilar epoeitin: but how similar is it? Clin J Am Soc Nephrol. 2008;3(1):174–178. | ||
Camacho LH, Frost CP, Abella E, Morrow PK, Whittaker S. Biosimilars 101: considerations for U.S. oncologists in clinical practice. Cancer Med. 2014;3(4):889–899. | ||
U.S. Food and Drug Administration [webpage on the Internet]. Draft Guidance on Biosimilar Product Development. Available from: http://www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsare DevelopedandApproved/ApprovalApplications/TherapeuticBiologic Applications/Biosimilars/default.htm. Accessed April 20, 2016. | ||
Epstein MS, Ehrenpreis ED, Kulkarni PM; FDA-Related Matters Committee of the American College of Gastroenterology. Biosimilars: the need, the challenge, the future: the FDA perspective. Am J Gastroenterol. 2014;109(12):1856–1859. | ||
Hassan I, Aleem S, Sheikh G, Anwar P. Biologics in dermatology: a brief review. BJMP. 2013;6(4):a629. | ||
Kuhlmann M, Covic A. The protein science of biosimilars. Nephrol Dial Transplant. 2006;21(suppl 5):v4–v8. | ||
Dorner T, Kay J. Biosimilars in rheumatology: current perspectives and lessons learnt. Nat Rev Rheumatol. 2015;11(12):1–12. | ||
Mysler E, Scheinberg M. Biosimilars in rheumatology: a view from Latin America. Clin Rheumatol. 2012;31(9):1279–1280. | ||
Castañeda-Hernández G, González-Ramírez R, Kay J, Scheinberg MA. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1(1):e000010. | ||
US Food Administration. Guidance for Industry: Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA. Available from: www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM377465.pdf. Accessed May 12, 2016. | ||
U.S. Food Administration [webpage on the Internet]. Draft Guidelines: Nonproprietary Naming of Biological Products. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM459987.pdf. Accessed April 20, 2016. | ||
Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider C. Biosimilars: the science of extrapolation. Blood. 2014;124(22):3191–3196. | ||
European Medicines Agency [webpage on the Internet]. European Assessment Report on Binocrit. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000725/WC500053615.pdf. Accessed April 20, 2016. | ||
Gupta V, Khaitan BK. Therapeutic potential of biosimilars in dermatology. Indian J Dermatol Venereol Leprol. 2015;81(5):451–456. | ||
Ramanan S, Grampp G. Drift, evolution, and divergence in biologics and biosimilars manufacturing. BioDrugs. 2014;28(4):363–372. | ||
Mayden KD, Larson P, Geiger D, Watson H. Biosimilars in the United States: considerations for oncology advanced practitioners. J Adv Pract Oncol. 2015;6(2):108–116. | ||
WHO [webpage on the Internet]. Guidelines on Evaluation of Similar Biotherapeutic Products (SBPs); 2009. Available from: http://www.who.int/biologicals/areas/biological_therapeutics/BIOTHERAPEUTICS_FOR_WEB_22APRIL2010.pdf. Accessed April 20, 2016. | ||
Jelkmann W. Biosimilar epoetins and other “follow-on” biologics: update on the European experiences. Am J Hematol. 2010;85(10):771–780. | ||
NCSL [webpage on the Internet]. State Laws and Legislation Related to Biologic Medications and Substitution of Biosimilars. National Conference of State Legislature; 2014. Available from: http://www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed April 20, 2016. | ||
Puig L. Biosimilars in dermatology: Starting with infliximab. Actas Dermosifiliogr. 2013;104(3):175–180. | ||
Graser, D [webpage on the Internet]. Industry voices: biosimilars and trade secrets. FierceBiotech; 2012. Available from: www.fiercebiotech.com/story/industry-voices-biosimilars-and-trade-secrets/2012-10-24. Accessed April 20, 2016. | ||
Socinski M, Curigliano G, Jacobs I, Gumbiner B, MacDonald J, Thomas D. Clinical considerations for the development of biosimilars in oncology. MAbs. 2015;7(2):286–293. | ||
Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111–5117. | ||
Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–419. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.