Back to Journals » International Journal of Nanomedicine » Volume 15
Biomimetic Synthesis and Evaluation of Interconnected Bimodal Mesostructured MSF@Poly(Ethyleneimine)s for Improved Drug Loading and Oral Adsorption of the Poorly Water-Soluble Drug, Ibuprofen
Authors Xin W, Wang Y, Guo X, Gou K, Li J, Li S, Zhao L, Li H
Received 22 July 2020
Accepted for publication 1 September 2020
Published 5 October 2020 Volume 2020:15 Pages 7451—7468
DOI https://doi.org/10.2147/IJN.S272796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Wei Xin,1,2 Yumei Wang,3 Xianmou Guo,3 Kaijun Gou,3 Jing Li,3 Sanming Li,3 Lin Zhao,1 Heran Li1
1School of Pharmacy, China Medical University, Shenyang 110122, People’s Republic of China; 2The First Affiliated Hospital of China Medical University, Shenyang 110001, People’s Republic of China; 3School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, People’s Republic of China
Correspondence: Heran Li; Lin Zhao
School of Pharmacy, China Medical University, 77 Puhe Road, Shenyang North New Area, Shenyang 110122, People’s Republic of China
Tel +8613897945866
; +8618900911856
Email [email protected]; [email protected]
Purpose: The aim of this study was to improve the oral bioavailability and anti-inflammatory activity of the poorly soluble drug ibuprofen (IBU) by employing a new kind of poly(ethyleneimine)s (PEIs)-based mesocellular siliceous foam (MSF) called B-BMSF@PEI as drug carrier.
Methods: B-BMSF@PEI was biomimetically synthesized by using PEIs as templates, catalysts and scaffolds under ambient conditions, and the structural characteristics, including size, morphology, mesoscopic structure and pore properties, were estimated by TEM, SEM, FTIR and N2 desorption/adsorption measurement. Then, IBU was incorporated into B-BMSF@PEI at the drug:carrier weight ratio of 1:1. The structural features of IBU before and after drug loading were systemically characterized. IBU and B-BMSF@PEI were then subject to in vitro drug release study and wettability analysis. Finally, in vivo pharmacokinetics and anti-inflammatory pharmacodynamics studies were carried out to evaluate the efficacy of B-BMSF@PEI on improving the oral adsorption of IBU.
Results: The results demonstrated that B-BMSF@PEI was a meso–meso porous silica material with foam appearance. It consisted of uniform spherical cells (40 nm) with interconnected pore networks. IBU can be successfully loaded into B-BMSF@PEI with high efficiency (as high as 39.53%), and crystal IBU was effectively converted to an amorphous state during this process. Benefiting from the great architectures of B-BMSF@PEI, IBU/B-BMSF@PEI performed good wetting property and significantly improved the dissolution rate in both simulated gastric fluid (SGF) and simulated intestinal fluid (SIF). Notably, IBU exhibited very satisfactory relative bioavailability (681.4%) and anti-inflammatory effects (the inhibition rates were between the ranges of 113.5% to 1504.3%).
Conclusion: B-BMSF@PEI with bimodal mesoporous system and interconnected nanopores was obtained owing to the dynamic self-assembly functions of PEIs. It had superiority in drug loading and could improve the oral adsorption of ibuprofen to a satisfactory level.
Keywords: biomimetic synthesis, MSF, bimodal mesostructured, bioavailability, ibuprofen
Introduction
In modern drug discovery, one of the main limitations to the development of highly potent pharmaceuticals is the poor solubility of drugs, since approximately 40% of the marketed oral drugs are poorly water soluble and about 90% of new innovative drug candidates are practically insoluble.1–3 The low solubility of the drugs ultimately results in a series of problems in the clinical work, such as poor absorption, low bioavailability, dose escalation and unsatisfactory clinical outcome, especially in oral drug administration which is still the most common and preferable delivery route for most various therapeutic agents with the best patient compliance and fewest safety concerns.4–8 For example, ibuprofen is a well-documented long-term nonsteroidal anti-inflammatory drug, mainly functions to reduce fever, inflammation and pain. Due to its safety and effectiveness, it is widely used in both adults and children, and is popular as an OTC analgesic-antipyretic drug.7–9 Like many traditional drugs classified as BCS II, IBU is poorly water soluble, and the dissolution process is the rate-limiting step for the absorption of IBU.7,10
The basic two principles for improving the oral bioavailability of poorly water-soluble drugs are increasing the solubility to enhance the drug release and controlling the drug delivery to maintain stable blood concentrations.1,2 Several effective strategies (such as co-solvents, micronizations, nanosizing, salifications and complexations) and formulations (such as aqueous/organic gels, nanoemulsions, polymeric nanoparticles, liposomes, vesicles and micelles) have been developed to overcome these obstacles.1,5,6,11–16 Different from the traditional “soft” organic drug delivery systems mentioned above, silica-containing materials with inorganic “rigid” matrices possess thermal/chemical stability, mechanical strength, physiologically inertness and are a prime asset to protect the guest molecules inside.17,18 Furthermore, mesoporous silica nanoparticles (MSNs, the pore size range of 2 nm-50 nm) with large surface area and pore volume have integrated the advantages of nanostructured materials and silica-based fabrications, and proved to be excellent candidates to serve as hosting systems in the field of drug delivery.17–21 A substantial number of drug delivery systems based on MSNs have been put forward since 2001 due to the unique structural and textural features. The large diversity and tunability in particle morphology, particle size, pore structures and surface chemistry not only provide better control of drug loading and release performance, but also favored the biological behavior with tissues and cells. Compared with the most common MSNs such as MCM41 and SBA15, mesocellular siliceous foams (MSF) composed of uniformlysized, nanoparticle spherical cells are interconnected to create a 3D pore network.22 The interconnected nature of the new mesostructured silicas are favorable for a wide range of applications and are promising candidates to be used in the field of drug delivery.23
How to precisely control the structures of silica nanoparticles is crucial to the practical applications and often achieved by a top-down synthetic approach including chemical vapor deposition and lithography/etching processes which require harsh conditions (such as extreme pH, temperature and pressure).24 Inspired by the biominerals existing widely in nature with complex nanostructures or nanopatterns, such as diatoms, radiolarians and sponges, novel biological silica formation synthetic routes called biosilicification have been established to fabricate inorganic materials, which is a nontrivial bottom-up synthetic procedure.24,25 Particularly, biosilicification usually occurs at ambient temperature and pressure in aqueous solution as opposed to the traditional “heat and beat” synthesis of silica-based materials. The formation of biominerals is genetically controlled by specialized biomacromolecules such as polypeptides, polysaccharides, amino acid derivatives and proteins that catalyze the polymerization and hydrolysis through a sol-gel process.23–27 Recently, biomimetic synthesis of silica fabrication has attracted increasing attention due to the cost-effective manufacture of nanopatterns and the precise control over shape. A great number of amine-containing biomacromolecules are easily available and in turn facilitate the versatility of silica structure carriers, thus resulting in multiple morphogenesis of biomimetic synthesized silicas including nanospheres, nanoflakes, nanoribbons, nanosheets, twisted nanorods, helical nanofibers, curved nanofilms and large aggregates.28–34
Poly(ethyleneimine)s (PEIs) are weak polyelectrolytes which are extensively used as flocculating agents, adhesives and thickeners, especially in the paper industry and biomedical applications.27,35 PEIs are able to assemble to form superhydrophobic multilayered surfaces and are used to formulate hybrid inorganic materials.24,35,36 After adsorbing PEI molecules onto the surface of materials, they distribute perpendicularly and can significantly increase the number of surface/segment contacts.35 In these applications, the dynamic and static interfacial properties of PEIs are of paramount importance. To the best of our knowledge, PEIs can be architecturally divided into two kinds: linear PEIs which are prepared from the hydrolysis of linear polyoxazoline, and branched PEIs that are formed from the ring opening polymerization of cyclic ethyleneimine.28,35–37 Notably, branched chain PEIs repress the crystallization, and, as a result, are amorphous in the bulk state and freely soluble in water, whereas linear architecture of PEIs can exhibit four crystalline phases including anhydrate, hemihydrate, sesquihydrate and dihydrate per EI unit, depending on the humidity of the atmosphere. Double-stranded helical conformation chains are adopted by the parallel array of linear PEIs in the anhydrate state, while all-trans planar-zigzag conformations can be constituted by the fully extended form for the hemihydrate, dihydrate, and sesquihydrate.36 Meanwhile, as a kind of polyamine, the obvious highlight and advantage of PEIs is their nontoxic nature at low molecular weight, which minimizes the safety concerns even if some or all the template remains in the synthesized silica products.37 Ford and Yang covalently grafted PEIs onto hydrogel films to direct the silica deposition through a sol-gel reaction and discovered that film thickness and silica morphology could be impacted by the molecular weight of PEIs.24 Recently, Li et al, successfully fabricated silica xerogel by PEIs with low molecular weight (<25 kDa) through three formation stages, which were sol, semi-solid gels and dry gels.37 During the synthetic process, methanol effectively regulated the morphology of the xerogel, in which compactness of product increased with decreasing methanol volume ratio. Meanwhile, the authors further explored its potential to be a drug carrier and revealed that such PEIs-based silica xerogel showed superior performance in delivering both water-soluble drugs (including propranolol hydrochloride, diltiazem hydrochloride, and levofloxacin hydrochloride) and a poorly water-soluble drug (indomethacin).37–39
In view of the aforementioned, in this work we successfully designed and biomimetically fabricated mesoporous silica by using linear PEIs. Novel biomimetic mesoporous silica named B-BMSF@PEI was synthesized and employed for the delivery of a poorly water-soluble drug. The obvious advantages of as-synthesized B-BMSF@PEI were listed as follows. (1) B-BMSF@PEI can be obtained through a biomimetic synthesis route at ambient conditions (normal pressure, normal temperature and neutral pH) and the synthetic process was easy and low cost; (2) by using PEIs with a non-toxic nature as scaffold, template and catalyst, silica condensation occurred quickly; (3) the attractive MSF type morphology, with interconnected bimodal mesostructures, was successfully obtained. Afterwards, the structure of B-BMSF@PEI was systemically characterized using a transmission electron microscope (TEM), scanning electron microscopy (SEM), and Fourier transform infrared spectroscopy- (FTIR). Then ibuprofen was selected as a model drug, and was loaded into B-BMSF@PEI at the drug:carrier ratio of 1:1 (w/w) according to the solvent dispersion method. Apart from its poor water-soluble nature, IBU was also chosen as a model drug because of its suitable molecule size of about 1.0 nm, which warranted the drug loading into the mesoporous ranged internal channel of B-BMSF@PEI. The drug loading amount and drug release behaviors of B-BMSF@PEI were deeply studied to explore the storage capacity and controlled release functions of B-BMSF@PEI. Meanwhile, X-ray diffraction (XRD), differential scanning calorimetry (DSC), FTIR and N2 desorption/adsorption studies were carried out to confirm the conversion of drug crystalline state during drug loading and the host-guest interactions between drug and carriers. Then wetting ability and the release behaviors of drug loaded samples were investigated to evaluate the controlled release properties of B-BMSF@PEI. Finally, pharmacokinetics and pharmacodynamics studies were performed to analyze the effect of B-BMSF@PEI on improving the oral adsorption of IBU.
Materials and Methods
Chemicals and Reagents
Tetraethoxysilane (TEOS) was purchased from Aladdin (Shanghai, China). Linear PEIs (MW = 20 kDa) were obtained from Chengdu Xiya Chemical Technology Co., Ltd. (Chengdu, China). IBU was provided by Wuhan Dinghui Chemical Co., Ltd. (Wuhan, China). Double deionized water was prepared by ion exchange. All other chemicals were of reagent grade and used without any further purification.
Synthesis of B-BMSF@PEI
A novel silica-based material denoted as B-BMSF@PEI was synthesized by employing Linear PEIs as templates, scaffolds and catalysts. In a typical run, PEIs (0.316 g) were dissolved in 1 mL deionized water under stirring condition at room temperature and kept for 4 h. After that, the resulting solution system was transferred to the ice-water bath to induce the crystallization of PEIs and left for 20 min. The white solid species was then added into 30 mL deionized water, and 6 mL TEOS was introduced to the mixture under vigorous stirring for 24 h. The mixture was aged for 72 h statically and collected by centrifugation, washed multiple times by deionized water and ethanol, dried in an oven overnight, and finally removed the template through thermal treatment (calcined at 550°C for 6 h).
Morphology and Structure Characterization of B-BMSF@PEI
FTIR analyses of PEIs and of B-BMSF@PEI samples before and after calcine were taken using a FTIR spectrometer (Spectrum 1000, Perkin Elmer, USA) at room temperature from the spectral region 400–4000 cm−1. Samples were thoroughly dried, crushed, and mixed with KBr. A Tecnai G2-F30 TEM instrument (FEI, The Netherlands) was used for evaluation of the shape and detailed morphology of B-BMSF@PEI. A JSM-6510A scanning electronic microscope (SEM) instrument (JEOL, Japan) was also employed to study the morphology and size (calculated from 200 nanoparticles). Meanwhile, the size distribution of B-BMSF@PEI was also evaluated using a Zeta-Potential/Particle Sizer (3000 HAS, UK) and the value was the average of three consecutive measurements. The porous structure and texture properties of B-BMSF@PEI were characterized by N2 desorption/adsorption tests using an adsorption analyzer (V-Sorb 2800P, Gold APP, China). Before analysis, sample was degassed at 120°C for 6 h. The specific surface area (SBET) of the prepared sample was analyzed by the Brunauer–Emmett–Teller (BET) method. The pore size distribution (WBJH) and pore volume (Vt) were derived from the adsorption branches of the isotherms using the Barrett-Joyner-Halenda (BJH) method.
Drug Loading Procedure
Ibuprofen was selected as a model drug, and was incorporated into B-BMSF@PEI by the base of solvent deposition method, which was proved to be an effective method for the preparation of drug loaded systems of poorly water-soluble drugs. The high-concentration hexane solution of IBU was prepared by dissolving 20 mg of IBU in 2 mL hexane. Then B-BMSF@PEI was soaked in hexane solution of IBU at the drug:carrier ratio of 1:1 (w/w) under magnetic stirring for 24 h in an attempt to achieve maximum drug loading in the internal channels. The drug loaded mixture named as IBU/B-BMSF@PEI was recovered by drying at 40°C under vacuum to remove the solvent.
The IBU loading content was measured by suspending 4 mg IBU/B-BMSF@PEI in 100 mL methanol under ultrasound at room temperature for 30 min. After passing through the 0.22 μm polytetrafluoroethylene (PTFE) membrane filter, the IBU concentration was determined by high performance liquid chromatography (HPLC) method. A Kromasil ODS-2 C18 column (250 mm × 4.6 mm) was used for separation. A mixture of acetonitrile and the pH 2.5 sodium acetate buffer (50:50; v/v) was used as the mobile phase at a flow rate of 1.5 mL/min. Quantitative analysis of IBU was achieved by using a UV-vis detector at the wavelength of 363 nm and was calculated by using a calibration curve. The drug loading capacity was calculated by weight loss according to the following equation:

Besides, the physical mixture of IBU and B-BMSF@PEI (1:1, w/w) was prepared by simple mixing.
Characteristics of IBU/B-BMSF@PEI
N2 desorption/adsorption test of IBU/B-BMSF@PEI was carried out on an adsorption analyzer (V-Sorb 2800P, Gold APP, China). The sample was degassed at 55°C for 12 h to remove physically adsorbed water before the examination. Besides, the BET method and BJH method were applied to investigate the pore characteristics. FTIR spectra of samples before and after drug loading were taken from 400 to 4000 cm−1 wavenumber range using a pressed KBr tablet method. DSC measurement of IBU, B-BMSF@PEI and IBU/B-BMSF@PEI was conducted from 25 to 200°C on a thermal analyzer (Q1000, TA Instrument, USA) by heating samples at steps of 10°C/min under a N2 flow. The XRD test of samples was carried out on an X’pert PRO x-ray diffractometer (PANalytical, Netherlands). Data was collected at the diffraction angle range of 5° to 40° (2θ) at a scanning rate of 0.05°.
Wettability Measurement
To investigate the interfacial properties of B-BMSF@PEI, the contact angles were determined using a model JCY series drop angle apparatus (Shanghai Fangrui Instrument Co., Ltd., Shanghai, China) through sessile drop method. In details, 200 mg B-BMSF@PEI was compressed into a tablet. A droplet of water was carefully exposed to the tablet, and the wettability procedure was imaged by a high-resolution CCD digital camera. Photos were then transferred to a computer, and the contact angle was measured using the tangent method. Both the initial contact angle and the equilibrium contact angle were investigated. The wettability measurements of IBU and IBU/B-BMSF@PEI were also conducted to evaluate the functionality of the carrier. The measurements were performed in triplicate.
In vitro Release Study
In vitro drug release was performed on a dissolution apparatus (ZRS-8G, Huanghai Medicament Test Instrument, China) using the USP II paddle method. IBU (5 mg) and IBU/B-BMSF@PEI (at a dose equivalent to 5 mg IBU) were suspended in 250 mL of release medium. To further assess the influence of pH on the release behaviors of IBU preparations, simulated gastric fluid (SGF, pH 1.0) and simulated intestinal fluid (SIF, pH 6.8) were respectively served as release medium. The dissolution cup with release medium were incubated at 37°C with continuous agitation at 50 rpm for 24 h. 5 mL of aliquots were withdrawn and replaced by an equivalent amount of fresh isothermal supplement at the predetermined time intervals. After the filter procedure (through 0.22 μm PTFE membrane filter), the drug release amounts were quantified by HPLC, and the cumulative release rate of IBU was calculated by the following expression. All in vitro release studies were repeated three times.
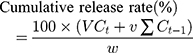
where Ct is the concentration of IBU determined at time t, Ct-1 is the concentration of IBU measured at time (t-1), v the sampled volume of release medium taken at specific time interval, and V is the total volume of release medium, w is the content of IBU in the composite.
The Similarity factor (F2) was employed to compare the dissolution profile of IBU/B-BMSF@PEI.40,41 The obtained release data was then analyzed with the zero order, Higuchi, Ritger-Peppas and Hixson-Crowel equations. The computational formula's mathematical models were according to the previous papers.32,42,43 The equation that best fitted with the release data was evaluated based on the correlation coefficient (R2).
In vivo Pharmacokinetics Study
All the experiments involving animals were approved by the Institutional Animal Care and Use Committee at China Medical University (protocol number # CMU2019194). The study complied with guidelines for the Care and Use of Laboratory Animals.
8 male Sprague-Dawley rats (SD rats, 200 ± 20 g body weight) were randomly divided into 2 groups (n=4 for each studied group): IBU group and IBU/B-BMSF@PEI group. Prior to dosing, animals were fasted overnight with free access to water. Animals were orally administered the water suspensions of IBU and IBU/B-BMSF@PEI with a drug dose of 25 mg/kg, respectively. At predetermined time intervals, an approximately 0.5 mL aliquot blood sample was collected via the retro-orbital venous sinus, immediately centrifuged for 10 min at 8000 rpm to separate the plasma, and stored at −20°C for the further HPLC analysis.
In the pretreatment process, 40 μL of indomethacin methanol solution (30 μg/mL) as an internal standard was sufficiently mixed with 200 μL plasma sample by a vortex mixer for 1 min. Then 100 μL NaOH (0.01 mol/L) was added into the system and vortexed for another 1 min. 1.5 mL diethyl ether was introduced into the above mixture and centrifuged for 12 min at 6000 rpm to separate the organic layer (2 mL). After evaporating at room temperature under a slight stream of N2, the residuals were reconstituted with 100 μL mobile phase, vortexed, centrifuged (8000 rpm for 5 min), and injected for HPLC analysis. The separation process of plasma samples was conducted on a Kromasil ODS-2 C18 column (250 mm × 4.6 mm) equipped with a JanuSep C18 pre-column. The column temperature was maintained at 30°C. The UV detector was set at 263 nm. The mixture of pH 2.5 sodium acetate buffer and acetonitrile (65:35, v/v) was selected as the optimized mobile phase with a flow rate of 0.5 mL/min. The IBU content in plasma was calculated by the internal standard method, and the main pharmacokinetic parameters were calculated using the software of DAS 2.0.
In vivo Pharmacodynamics Study
As is known to all, acute inflammation can always be related to the symptoms of swelling, pain, redness, and dysfunction in a short-term process. As a kind of acidic non-steroidal anti-inflammatory drug, the pharmacodynamic evaluations of the IBU preparations were probed by mouse ankle swelling test (MAST), mouse ear swelling test (MEST) and mouse writhing test (MWT).
Mouse Ankle Swelling Test
20 SD male rats (180–220 g body weight) were separated into 5 groups (n=4) at random: Normal Saline group (serving as negative control), IBU group (considered as positive control), IBU/B-BMSF@PEI (low dose) group, IBU/B-BMSF@PEI (middle dose) group and IBU/B-BMSF@PEI (high dose) group. Before dosing, animals were fasted overnight, and the right ankle initial circumferences of each rat were measured. The Normal Saline group, IBU group and IBU/B-BMSF@PEI (middle dose) group were orally administered 2 mL normal saline, 2 mL normal saline suspension of IBU and IBU/B-BMSF@PEI at a dose of 25 mg/kg IBU, respectively. Animals in IBU/B-BMSF@PEI (low dose) group and IBU/B-BMSF@PEI (high dose) group were orally administered half and twice of the dose in IBU/B-BMSF@PEI middle dose group which equaled to 12.5 mg/kg and 50 mg/kg of IBU, respectively. At 30 min post administration, 1% carrageenan (w/v) was injected into the right ankle area of each rat to induce the acute swelling. The circumferences of the right ankles after injection were measured at desired time intervals (0.5 h, 1 h, 1.5 h, 2 h, 3 h, 4 h and 6 h). The repression rate and the swelling rate were calculated by the following equation:

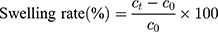
Where c0 is the initial circumference of ankle, and 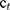 stands for the circumference measured at time t.
stands for the circumference measured at time t.
Mouse Ear Swelling Test
20 Kunming male mice (weighing 20 ± 2 g) were divided into 5 groups (Normal Saline group, IBU group, IBU/B-BMSF@PEI low dose group, IBU/B-BMSF@PEI middle dose group and IBU/B-BMSF@PEI high dose group, n=4) randomly. Prior to oral administration, animals were fasted overnight. Mice in Normal Saline group and IBU group were orally administered with normal saline and IBU (0.6 mg), and served as negative control and positive control, respectively. Animals in IBU/B-BMSF@PEI (low dose) group, IBU/B-BMSF@PEI (middle dose) group and IBU/B-BMSF@PEI (high dose) group were respectively orally administered IBU/B-BMSF@PEI at a dose of 0.5 mg IBU, 1 mg IBU and 2 mg IBU. At 30 min post administration, xylene (0.1 Ml) was exposed onto the left ear of each mouse. 50 min later, mice were sacrificed, and the left and right ears were carefully cut to circular pieces with a diameter of 0.6 cm by hole punch. The weights of the left and right ears were measured to quantify the ear swelling rate and the repression rate.

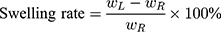
Where, 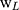 denotes the weight of the tissue cut from left ear;
denotes the weight of the tissue cut from left ear; 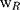 stands for the weight measured from right ear tissue of the same mice.
stands for the weight measured from right ear tissue of the same mice.
Mouse Writhing Test
Mouse writhing tests were carried out to investigate the antinociceptive effects of IBU preparations. 20 SPF-grade Kunming male mice weighing 20 ± 2 g were enrolled in the study and were divided into 5 groups (each containing 4 animals): Normal Saline group (negative control), IBU group (positive control), IBU/B-BMSF@PEI (low dose) group, IBU/B-BMSF@PEI (middle dose) group and IBU/B-BMSF@PEI (high dose) group. In IBU/B-BMSF@PEI (middle dose) group, IBU preparations with the drug dose of 1 mg were suspended in 0.5 mL normal saline and oral administered to mice, while animals in IBU/B-BMSF@PEI (low dose) group and IBU/B-BMSF@PEI (high dose) group received oral treatment of 0.5 mg and 2 mg IBU. At 60 min post administration, each animal was intraperitoneally injected with 0.4 mL acetic acid 0.4% (v/v) to induce pain, and was placed in a separate mouse cage. The numbers of mice writhing, which was considered as an abdominal stretching or constriction of at least one hind limb, was counted by the same experimenter who was blinded to interventions for 20 min. Then the repression rate was calculated as:
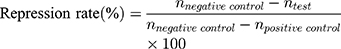
Where n is the writhing numbers.
Statistical Analysis
Data were expressed as the mean ± SD. One-way ANOVA was employed to compare the control and test values. Student’s t-test was carried out to determine identification levels of significance between the groups. Significant difference was considered as p < 0.05.
Results and Discussion
Formation Mechanism of B-BMSF@PEI
PEIs consist of a large amount of basic amine groups on their surface, and are regarded as a kind of soft template owing to the solution state. It was reported that linear PEIs possessed a strong ability to self-assemble into crystalline units in water or water-based media.36–38 In the synthesized program of B-BMSF@PEI, the template solution was alkaline after the addition of PEIs. By cooling down the PEIs solution, soft nanocrystalline aggregates could be afforded, which enabled them to function as templates for structure-direction (spatial control).37,38 Then the silica condensation was facilely accomplished by dispersing the PEIs crystalline aggregate into the silica source solutions which consisted of a certain amount of water/TEOS (5:1 v/v) without any additional catalysts. Along with the addition of TEOS, PEIs actively participated in the forming of hydrogen bonding with Si-OH and could highly concentrate the silica source to exert the functionality of a scaffold.37,38 Meanwhile, the alternating presences of nonprotonated and protonated amine groups in PEIs chains allowed the formation of a hydrogen bond with the oxygen adjacent to silica in the precursor and thus catalyzing –Si-O-Si- bond formation (temporal control). The nanostructured silica product with controlled shape denoted as B-BMSF@PEI was successfully obtained in such a very simple, low cost and low impact synthetic route.
It should be noted that the synthesis could be ascribed to biomimetic synthesis, because amines catalyzed the condensation of silica precursors, and the synthesis occurred under neutral pH and ambient conditions, which all coincide with the principle of silicification.27,28
Figure 1A shows the FTIR spectra of PEIs and B-BMSF@PEI before and after calcine. In the FTIR spectra of PEIs, characteristic absorptions identified as N-H stretching vibrations at 2930.7 cm−1 and 2831.9 cm−1, as well as the bending vibration of N-H at 1630.1 cm−1 could be observed. After silica condensation, the B-BMSF@PEI was successfully yielded, as evidenced by the disappearance of amine bands and the existence of the silica material characteristic bands, including bands at 3427.1 cm−1 relative to the antisymmetric stretching of Si-OH cm−1, and bands at 1091.2 cm−1 and 466.4 cm−1 respectively attributed to the antisymmetric stretching vibration and bending vibration of Si–O–Si.44
 |
Figure 1 FTIR spectra of samples. (A) FTIR spectra of (a) PEIs, (b) B-BMSF@PEI before calcine, and (c) B-BMSF@PEI after calcine; (B) FTIR spectra of (a) IBU, (b) the physical mixture of IBU and B-BMSF@PEI, (c) IBU/B-BMSF@PEI, and (d) B-BMSF@PEI. |
Characteristics of B-BMSF@PEI
The morphology and pore structure of B-BMSF@PEI was mainly analyzed by TEM and SEM. As shown in Figure 2, B-BMSF@PEI pieces of foam consisted of close-connected particles (cells, Figure 2A–D) which possessed spherical shapes with a uniform diameter of about 40 nm (35–40 nm accounted for the maximum percentage, Figure 2E). Mesoporous sized inter-particle spaces (windows) existed on the boundary of nanoparticles. Since the cell's nanoparticle was extremely small, it could easily form siloxane bridges and become highly agglomerated as a consequence of the higher interface energy.45 As shown in Figure 2F, The particle size distribution of B-BMSF@PEI was also analyzed by a Zeta-Potential/Particle Sizer. The particle size distribution curve showed a main peak at 43.22 nm ascribed to the small nanoparticle cells and a second peak at 820.3 nm belonging to the foam appearance. Due to the hydration effect, the measured value is slightly larger than the actual size of B-BMSF@PEI. However, although the nanoparticle cells were uniform, the PDI value was 0.440±0.112, because the small nanoparticles were highly aggregated to form the mesocellular siliceous foam. Meanwhile, well-developed mesopores were homogeneously distributed on the surface of the B-BMSF@PEI, which showed significant difference with the traditional MSNs with well-ordered lattice fringes (such as MCM41 and SAB15). The mesostructure was highly interconnected (Figure 2A, pointed out at red arrows) with short and twisty pore networks. These findings unambiguously confirmed the successful control of morphology at structural level programed by the dynamic self-assembly function of PEIs through the bio-silification process.
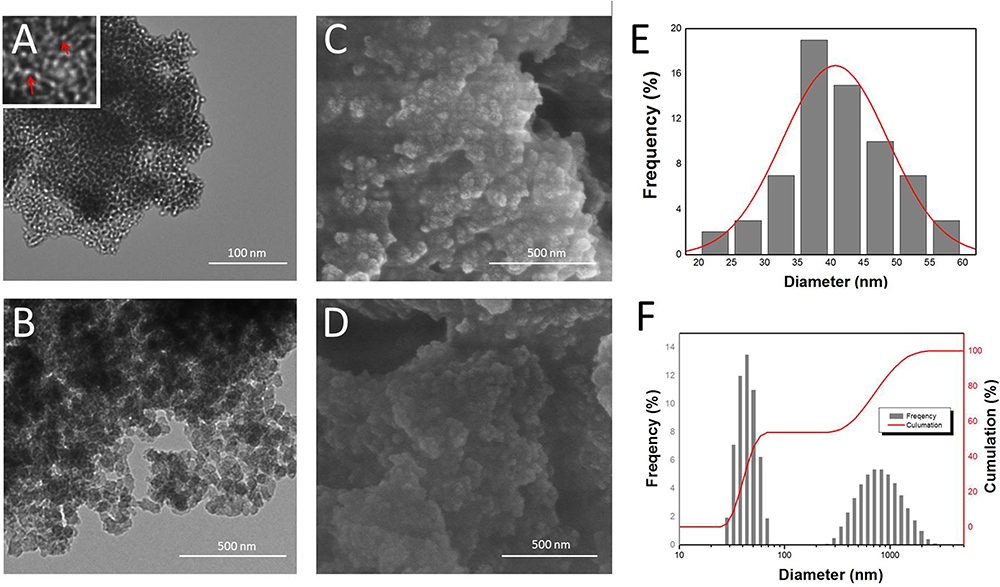 |
Figure 2 Structural characteristics of B-BMSF@PEI. (A) and (B) TEM images of B-BMSF@PEI; (C) and (D) SEM images of B-BMSF@PEI; (E) particle size distribution of B-BMSF@PEI calculated from the SEM images; (F) particle size distribution of B-BMSF@PEI obtained from the Zeta-Potential/Particle Sizer. |
The porous properties and texture characteristics of B-BMSF@PEI were provided by N2 adsorption/desorption measurements. As collected in Figure 3A, the N2 adsorption–desorption isotherms of B-BMSF@PEI were type IV isotherm with two pronounced hysteresis loops respectively at the relative pressure of p/p0 0.2–0.6 and 0.7–1.0, confirming the presence of a bimodal mesoscopic structure. The BJH pore size distribution curve (Figure 3B) of B-BMSF@PEI suggested these nanoparticles possessed a meso–meso structure. The first peak, corresponding to nitrogen capillary condensation at the relative pressure of 0.2–0.6 was observed at less than 2.23 nm, and the second broad peak which then resulted to an adsorption step at the relative pressure of 0.7–1.0 was centered at about 12.65 nm which was obvious due to the inter-particle aggregations. The BET surface area and the total pore volume of B-BMSF@PEI were found to be 503.21 m2/g and 0.98 cm3/g, respectively, suggesting effective active sites for potential application as a host platform in a drug delivery system. The new mesostructured silica material B-BMSF@PEI was a promising candidate for the host and release of drugs.
 |
Figure 3 (A) N2 adsorption/desorption isotherm of B-BMSF@PEI; (B) pore size distribution of B-BMSF@PEI; (C) N2 adsorption/desorption isotherm of IBU/B-BMSF@PEI; (D) pore size distribution of IBU/B-BMSF@PEI. |
Characteristics of IBU/B-BMSF@PEI
Drug Loading
In the present study, IBU was selected as the model drug, and was loaded into B-BMSF@PEI by using the solvent deposition method. The drug loading process involved two steps, including the initial adsorption equilibrium stage and subsequent solvent evaporation stage.21 According to the N2 adsorption/desorption results, after drug loading, all the texture parameters (including SBET, Vt and WBJH) were decreased sharply to very low values (Figure 3C and D and Table 1). It provided sufficient evidence that IBU was effectually loaded into B-BMSF@PEI, thus taking up a certain amount of space. Furthermore, both the first pronounced hysteresis loop at the relative pressure of p/p0 0.2–0.6 and the second one at relative pressure of 0.7–1.0 showed a decrease in intensity in Figure 3C. It seemed that, IBU occupied both small mesopores and inter-particle spaces.
 |
Table 1 BET Surface Area, Total Pore Volume and Average Pore Diameter of B-BMSF@PEI Before and After Drug Loading |
The drug loading amount of IBU/B-BMSF@PEI (at the drug:carrier ratio of 1:1, w/w) ensured by the HPLC analysis was 39.53%±1.27% w/w. It was worth mentioning that the IBU loading capacity of B-BMSF@PEI was higher compared with the normal mesoporous silica carriers by using the same drug loading procedure. As reported in previous studies, the IBU loading content for MCM41 was around 25% w/w, the value increased to 30–36% w/w by using amino modified MCM41 as drug carrier, for SBA15 it was 18–30% w/w, for the organo-functionalized and sulfonic acid functionalized SBA15 it was 28% and 35% w/w, for a new kind of chiral mesoporous silica it was 36% w/w, and for mesoporous silica nanorod and amino functionalized mesoporous silica nanorod it was 23% w/w and 29% w/w.46–51 It should be noted that most of these aforementioned carriers, as well as other MSNs that are widely used for biomedical applications, possess large particle sizes (200–500 nm) and long straight lattice fringed pore networks with poor connectivity.21,31,45 As demonstrated by Perez et al, the structural properties of nanoparticles strongly influence the uptake of cargo molecules.52 The superiority of B-BMSF@PEI in drug loading was due to the unique meso-meso structure and the resulting large pore volume.48 Firstly, the outstanding drug loading capacity of B-BMSF@PEI could be ascribed to its pore morphology.21,31,52,53 Compared to the aforementioned carriers such as MCM41-typed and SAB15-typed MSNs with unconnected nanopores, the short and highly interconnected nanopores within the cells (as revealed by TEM images) meant shorter transmission distance and smaller steric hindrance. Furthermore, the mesopore diameter was one of the key factors to influence the drug loading behaviors, because the pore size determined the maximum size of the guest molecules that could be incorporated and the steric hindrance they would encounter during the loading process.52,53 In this case, the larger mesoporous sized inter-particle spaces of B-BMSF@PEI indicated small steric hindrance and extra storage space for drug loading, and further facilitated the mass transport during drug incorporation.
Interactions Between Drug and Carrier
FTIR was employed to identify the successful loading of IBU and the interactions between the IBU and silica carrier. As displayed in Figure 1B, IBU presented an absorbance at 1720.0 cm−1 belonging to the stretching vibration of the carboxyl group, and absorbance assigned to the quaternary carbon atom of benzene ring at 1508.1 cm−1 and 1460.7 cm−1.7,35 The FTIR spectra of the physical mixture of IBU and B-BMSF@PEI was the overlap of the spectra of IBU and B-BMSF@PEI with the maintained carbonyl band at 1719.9 cm−1. After drug loading, most of the characteristic absorption bands of IBU became undetectable, suggesting it was effectively incorporated into B-BMSF@PEI. Meanwhile, the carboxyl group of IBU was involved in the forming of hydrogen bonding with the silanol groups on the silica surface thus resulting in the disappearance of the carbonyl band.
Drug Crystalline State
To assess the physical state of the drug before and after loading into B-BMSF@PEI, DSC and XRD studies were performed. Figure 4A depicted the XRD diffraction pattern of samples. IBU was highly crystalline in nature as indicated by the numerous peaks.51 Contrastingly, the absence of distinctive peaks suggested that B-BMSF@PEI was a kind of amorphous material. After drug loading, IBU existed in an amorphous state, evidenced by the broad bands without any obvious diffraction peaks.48,51,54. The uniform mesopores of B-BMSF@PEI could produce space confinement, which prevented the crystallization of IBU due to the finite-size effect.55 DSC analysis was also carried out to investigate the conversion of the crystalline state. As shown in Figure 4B, the endothermic event of pure IBU comprised of a single endothermic procedure at 77°C, which was consistent with its melting points.51,55 Whereas IBU/B-BMSF@PEI exhibited the amorphous broad peak without any glass transitions or bulk phase transition thermograms.51,53 After being incorporated into B-BMSF@PEI, the DSC thermogram was almost a smooth line without melting point depressions, demonstrating the amorphous state of IBU/B-BMSF@PEI. The DSC results were in accordance with the XRD findings, and further illustrated the transformation of the drug's crystalline form, caused by the limited space of the mesoporous material. Compared to the drug in its crystalline state, the nanosized drug with B-BMSF@PEI as delivery platform had reduced the lattice energy and might subsequently lead to improved dissolution rate and enhanced bioavailability.55
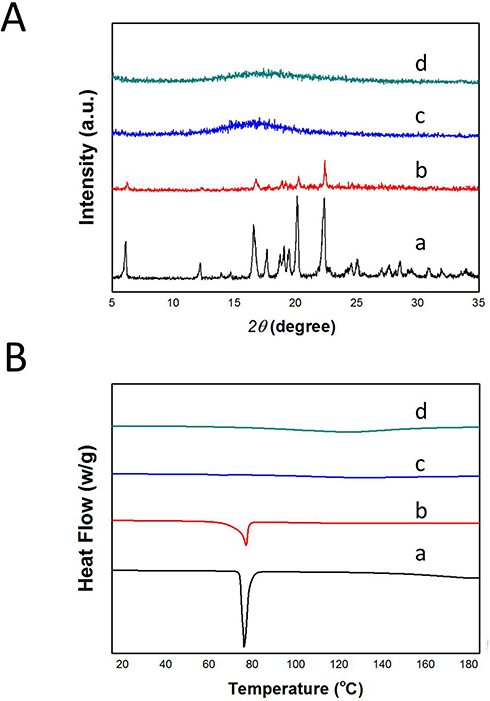 |
Figure 4 (A) XRD diffractograms of (a) IBU, (b) the physical mixture of IBU and B-BMSF@PEI, (c) IBU/B-BMSF@PEI, and (d) B-BMSF@PEI; (B) DSC thermograms of (a) IBU, (b) the physical mixture of IBU and B-BMSF@PEI, (c) IBU/B-BMSF@PEI, and (d) B-BMSF@PEI. |
Wettability Measurement
As an extremely critical interfacial property, the wettability of drug carriers and drug delivery systems was proved to be vital for their functionality and bioactivity.56,57 In the present study, the contact angles of B-BMSF@PEI, IBU, the physical mixture and IBU/B-BMSF@PEI, which formed between the liquid/vapor interface and the liquid/solid interface, were measured to investigate their interfacial behavior. As seen in Figure 5A and Table 2, the initial contact angle of B-BMSF@PEI was 22.33±2.17°, demonstrating the hydrophilic nature. Furthermore, as a kind of mesoporous silica carrier, the contact angle of B-BMSF@PEI reduced with time and the equilibrium contact angle was 9.48±1.88°. B-BMSF@PEI with enriched pore structure adsorbed water from the humid surroundings due to the strong capillary condensation happening in the mesopores (as indicated by the TEM and nitrogen adsorption studies). On the other hand, the silanol groups on the internal and external surface could actively participate in the forming of hydrogen bonds with H2O, thus benefiting the wetting process.57 Therefore, the dissolution medium could easily enter into the nanopores of B-BMSF@PEI to sufficiently contact and dissolve out their encapsulated cargo while performing the delivery tasks.
 |
Table 2 The Contact Angles (°) of Samples Measured at Different Times |
 |
Figure 5 Wettability test of samples. (A) Initial contact angles of (a) IBU, (b) the physical mixture of IBU and B-BMSF@PEI, (c) IBU/B-BMSF@PEI, and (d) B-BMSF@PEI; (B) contact angles measured at different times and the equilibrium contact angles of (a) IBU, (b) the physical mixture of IBU and B-BMSF@PEI, (c) IBU/B-BMSF@PEI, and (d) B-BMSF@PEI. |
As a kind of hydrophilic excipient, the potential of B-BMSF@PEI to improve the wettability of a poorly soluble drug was also explored by using IBU as a model drug, and the results were collected in Figure 5B. The initial contact angle of IBU and IBU/B-BMSF@PEI were 73.82±3.47°and 45.05±3.12°, and the equilibrium contact angle of IBU and IBU/B-BMSF@PEI were respectively 68.41±4.67° and 20.64±2.16°. The results suggested that insoluble drug, IBU, had poor water wetting property as evidenced by the high contact angles. After loading into B-BMSF@PEI, the contact angle of IBU decreased sharply, because B-BMSF@PEI can improve wetting and led to supersaturation in the diffusion layer. The huge hydrophilic surface area of B-BMSF@PEI facilitated the wetting and dispersion of the stored drug, which might result in fast dissolution. Besides, B-BMSF@PEI converted crystal IBU into an amorphous form to increase the solubility. IBU/B-BMSF@PEI performed better wetting property compared to pure IBU and the physical mixture, which helped to dissolve the drug and would subsequently suggest an increase in absorption and bioavailability of the poorly soluble drug.
In vitro Drug Release Study
Inspired by the special structural features of B-BMSF@PEI, an in vitro drug release study was then carried out. Ibuprofen was a weak acid mainly absorbed in the stomach and proximal intestine during the oral delivery.7,10 Since the pH of physiological conditions varied from 1–2 in the stomach to 5–7 in the antrum, and to 7–8 in the proximal intestine, the drug release study was conducted at SGF and SIF, respectively. As can be observed in Figure 6A, very incomplete dissolution was observed in SGF for IBU, in which only 31.46±1.34% was released within 24 h. At this condition (pH 1, pH < pKa), IBU molecules existed in the stationary phase without ionization, owing to the abundance of protons under acid environment.49,51 However, IBU molecules showed faster release rate in SIF (pH 6.8, pH > pKa), in which most of the IBU was ionized and lessremained in forms of stable prototype compounds.48,51 Importantly, in all release mediums, the dissolution rate of IBU was significantly improved after loading into B-BMSF@PEI. In SGF, IBU/B-BMSF@PEI exhibited an increased release amount from 31.46±1.34% to 73.84±1.81% within 24 h. Meanwhile, IBU/B-BMSF@PEI could almost fully release the loaded IBU (the cumulative release amount was larger than 95%, Figure 6B) in SIF. The dissolution profile of IBU and IBU/B-BMSF@PEI were compared by similarity factor (F2, Table 3). The F2 value of IBU/B-BMSF@PEI in SGF and SIF were 27 and 22, respectively, which are both smaller than the critical value of 49.5.40,41 So that, the dissolution profiles of IBU and IBU/B-BMSF@PEI were identified as “unsimilar”, which confirmed the efficacy of IBU/B-BMSF@PEI on improving the release behaviour of IBU.
 |
Table 3 Mathematic Models and Similarity Factor of Samples Released in SGF and SIF |
 |
Figure 6 (A) In vitro drug release profiles of IBU and IBU/B-BMSF@PEI in SGF; (B) In vitro drug release profiles of IBU and IBU/B-BMSF@PEI in SIF. |
The dissolution-promoting effect of B-BMSF@PEI for in vitro release of IBU was obvious, on account of the change in crystalline state and the reduction of drug particle size. The amorphization of IBU (as confirmed by the DSC and XRD results) in the spatially limited mesoporous material increased the solubility of poorly water-soluble drugs as a result of the higher energy state.55 Compared with the crystalline drug, the amorphous drug with mesoporous silica as carriers had reduced lattice energy. The high internal energy and specific volume of the amorphous state relative to the crystalline state could lead to enhanced dissolution and bioavailability.55 Moreover, the nanoscale mesoporous channels of B-BMSF@PEI (as indicated by the TEM and nitrogen adsorption studies) could effectively reduce the particle size of IBU from the micron range to the lower nanometer range thus increasing the effective surface area in diffusion layer, enhancing the apparent solubility of the drug (Ostwald–Freundlich equation), and further increasing the drug dissolution rate according to Noyes-Whitney equation.58,59 Overall, B-BMSF@PEI could afford attractive drug release performance.
Kinetics of Release
To evaluate the kinetics of drug release from B-BMSF@PEI, the obtained dissolution data were fitted to mathematical models and the results are displayed in Table 3. The model that best fitted with the obtained data was determined according to the correlation coefficient (R2). For IBU in both SGF and SIF, The R2 values obtained for the zero order model, Higuchi model and Ritger-Peppas model were within the range of 0.398–0.832. However, the release behaviors of IBU exhibited a good fitness with the Hixson-Crowel model (R2 were within the range of 0.982–0.983). For IBU/B-BMSF@PEI, the zero order and Higuchi models fail to explain the IBU release, in which the R2 values for zero order equations and Higuchi model were within the range of 0.542–0.865. While, the R2 values acquired for the Ritger-Peppas model were within the range of 0.995–0.998, for the Hixson-Crowel model they were within the range of 0.986–0.991. It can be concluded that the release of IBU from B-BMSF@PEI followed the Ritger-Peppas model better than the other equations. It is worth mentioning that the exponent n in the Ritger-Peppas model confirmed the release mechanism: if n < 0.5, the release mechanism followed the Fickian diffusion model.32,42 Herein the release exponent n for IBU/B-BMSF@PEI was respectively valued at 0.179 and 0.229, and was relative to the Fickian diffusion transport mechanism. This finding was in good agreement with the previous studies that drug dissolution out from MSNs was a mainly diffusion-controlled process.
It was generally accepted that drug release from mesoporous silica could be regarded as a diffusion-controlled process, governed by the mesostructure of the carrier.52,53 When the release started, the dissolution medium began to migrate to the nanopores of the carrier to dissolute drug molecules.20 Then the drug solution escaped from the pores, along with the pore channels filled with the refresh solvent. In detail, IBU/B-BMSF@PEI showed two-stage dissolution processes in SGF and SBF including a faster release stage in the first 5 min and then a prolonged release stage. In the first step, a burst release phenomenon was observed, because IBU diffused out from the inter-particle spaces of B-BMSF@PEI. 24.01±0.69% and 30.27±0.66% of IBU was able to release in SGF and SIF within 5 min, which was 2.25 and 1.72-fold higher than that of IBU, respectively. The enlarged mesoporous space in B-BMSF@PEI provided more chance for the stayed drug molecules to escape from carriers. In the second step, the solvent started to diffuse into the small pores, and the release profile became gentler. In other words, owing to the geometry facilitation, the small channels in the bimodal meso-meso system contributed to the amorphization of IBU, while the larger spacious inter-particle spaces and the interconnected structure meant less diffusion resistance which helped the loaded guest molecules move more easily into the dissolution medium.
In vivo Pharmacokinetics Study
To estimate the capability of B-BMSF@PEI for improving drug absorption in vivo, pharmacokinetic studies of IBU/B-BMSF@PEI were conducted with pure IBU as a reference. The mean plasma concentration time profiles of IBU formulations after oral administration were shown in Figure 7A. The pharmacokinetic parameters were summarized in Table 4. For pure IBU, very poor and incomplete adsorption was observed, and the plasma level peaked at 2 h (Tmax) with the maximum concentration (Cmax) of 0.89±0.11 μg/mL. It was obvious that, B-BMSF@PEI could effectively improve the oral adsorption of drug, and the Tmax was effectively prolonged to 6 h post administration, which was favored for its clinically therapeutical effect. Moreover, IBU/B-BMSF@PEI could achieve higher relative bioavailability of 681.4% and higher Cmax of 6.51±0.41 μg/mL, which was approximately 7.3-fold higher than pure IBU. The result of the pharmacodynamics study revealed that the bioavailability of IBU was significantly improved after loading into B-BMSF@PEI, which was in accord with the in vitro release behaviors. Apart from the transformation of the crystalline state, the dramatic higher oral adsorption was also due to the beneficial architectures of B-BMSF@PEI with smaller cell nanoparticles (40 nm). Decreasing the size of MSNs always led to an increase in cell uptake and diffusion coefficient.58 According to the Stokes−Einstein equation, a reduction in the diameter of nanoparticle could improve the diffusion coefficient, making the small particles easier to penetrate through the mucus.58,59 In other words, the significantly enhanced oral adsorption of IBU ensured the potential of B-BMSF@PEI as the next-generation platform for the oral delivery of various poorly soluble drugs.
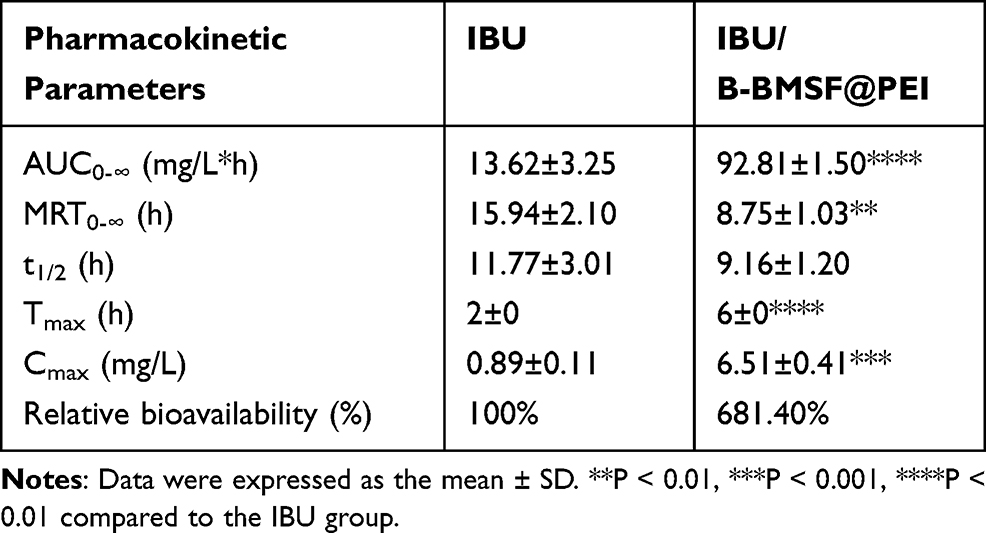 |
Table 4 Pharmacokinetic Parameters Obtained After Oral Administration of IBU and IBU/B-BMSF@PEI |
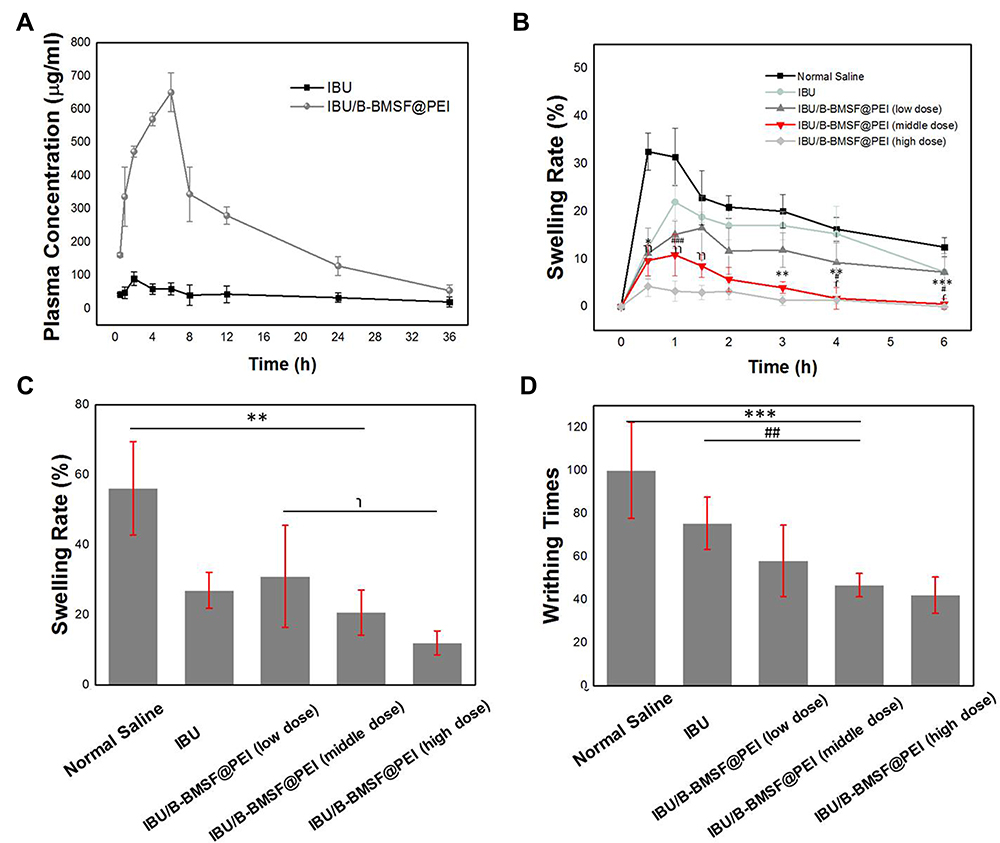 |
Figure 7 Pharmacokinetics and pharmacodynamics evaluations of samples. (A) Plasma concentration–time profiles of IBU and IBU/B-BMSF@PEI; (B) the anti-inflammatory effects of IBU and different dosed IBU/B-BMSF@PEI in MAST; (C) the anti-inflammatory effects of IBU and different dosed IBU/B-BMSF@PEI in MEST; (D) the analgesic effects of IBU and different dosed IBU/B-BMSF@PEI in MWT. *P < 0.05, **P < 0.01, ***P < 0.001 compared to the negative control group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared to the positive control group; ɾP < 0.05 compared to the low dose group; ɿP < 0.05, ɿɿP < 0.01 compared to the high dose group. |
In vivo Pharmacodynamics Study
The pharmacodynamics study was carried out by evaluating the anti-inflammatory effects of IBU and IBU/B-BMSF@PEI through mouse ankle swelling test, mouse ear swelling test and mouse writhing test. It can be seen from Figure 7B, in the Normal Saline group, the maximum swelling degree was found at 0.5 h after the injection of λ-carrageenan, indicating the establishment of the ankle swelling model. Besides, IBU was orally administrated to animals and served as the positive control. In the IBU group, the maximum swelling degree underwent a reduction from 32.3±3.89% to 19.3±2.93% due to the functions of IBU as a non-selective inhibitor of the cyclo-oxygenase (COX 1 and 2) enzymes in relieving inflammation and swelling.7 After loading into B-BMSF@PEI, the maximum swelling degree ratio was only 10.8±3.24% in the middle dose group, as a result of the remarkably improved bioavailability. The inhibition rates of IBU/B-BMSF@PEI (middle dose) were within the range between 113.5% and 1504.3% (Table 5). Furthermore, almost no swelling was observed in the IBU/B-BMSF@PEI (high dose) group.
 |
Table 5 Inhibition Rate (%) of IBU/B-BMSF@PEI Samples at Different Dose in MAST |
As shown in Figure 7C, in the mouse ear swelling test, the swelling rate of animals in the Normal Saline group, IBU group and IBU/B-BMSF@PEI (middle dose) group was respectively 42.81±13.38%, 21.75±5.17% and 16.86±6.24% at the same dose. IBU/B-BMSF@PEI was found to be more powerful in suppressing ear swell in mice compared to IBU. IBU/B-BMSF@PEI significantly and dose dependently suppressed the swelling induced by xylene, and very mild swelling happened in the IBU/B-BMSF@PEI (high dose) group (the inhibition rate was as high as 159.6%, Table 6). All above results indicated that the anti-inflammation effect of IBU was effectively enhanced after being loaded into B-BMSF@PEI.
 |
Table 6 Inhibition Rate (%) of IBU/B-BMSF@PEI Samples at Different Dose in MEST and MWT |
The mouse writhing test was also carried out to further evaluate the analgesic effect of IBU/B-BMSF@PEI, and the results are shown in Figure 7D. In the Normal Saline group, acetic acid caused significant pain, and the average writhing number was as high as 100.0±22.3 times. By oral administration of IBU, which was regarded as the positive control, an attenuation of pain was observed, and the writhing number was 75.4±12.2 times. The application B-BMSF@PEI as drug carrier (in a middle dose equivalent to 1 mg IBU) contributed to the inhibition of pain, and the writhing number reduced to 50.7±5.42 times (the inhibition rate was as high as 180.6%). Additionally, the inhibition rate on MWT followed the sequence: B-BMSF@PEI (low dose) < B-BMSF@PEI (middle dose) < B-BMSF@PEI (high dose). Notably, B-BMSF@PEI showed dose related inhibitory effect on ankle writhing of mouse.
These pharmacodynamics findings of the pharmacodynamics study also provided convincing evidence for the improvement of drug oral absorption by loading into B-BMSF@PEI.
Conclusion
In the present study, a novel drug delivery system established by B-BMSF@PEI with a foam appearance was successfully prepared through a biomimetic method by using PEI as templates, catalysts and scaffolds. Structure characteristics of B-BMSF@PEI and its application as a drug carrier were intensively investigated. As a kind of meso–meso material, B-BMSF@PEI consisted of uniform spherical cells (40 nm) with a bimodal mesoporous system and interconnected pore morphology. B-BMSF@PEI was then employed to provide an effective strategy for increasing the bioavailability of the poorly water-soluble drug ibuprofen. Benefiting from the pore architectures, B-BMSF@PEI was able to host IBU with high capacity (as high as 39.53%). During the process, crystal IBU was effectively converted into an amorphous state with the forming of hydrogen bonds between IBU and B-BMSF@PEI. As a result the wetting property and dissolution of insoluble drug IBU was significantly improved. Particularly, IBU exhibited very satisfactory relative bioavailability (681.4%) and anti-inflammatory effects. We believe that the development of a B-BMSF@PEI-based drug delivery system was effective in improving the oral bioavailability of poorly water-soluble drugs and would contribute to the rational design of bio-materials.
Funding
This work was supported by the National Natural Science Foundation of China (No. 81773672; No. 81903550) and National Science and Technology Major Project [No. 2017ZX09101001-006-012].
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Ren XN, Cheng S, Liang Y, et al. Mesoporous silica nanospheres as nanocarriers for poorly soluble drug itraconazole with high loading capacity and enhanced bioavailability. Micro Meso Mat. 2020;110389.
2. Peltonen L, Hirvonen J. Drug nanocrystals-versatile option for formulation of poorly soluble materials. Int J Pharm. 2018;537(1–2):73–83. doi:10.1016/j.ijpharm.2017.12.005
3. Dolinina ES, Vorobyeva EV, Parfenyuk EV. Development of novel delivery system for warfarin based on mesoporous silica: adsorption characteristics of silica materials for the anticoagulant: pharmaceutical development and technology. Pharm Dev Technol. 2015;21:1546.
4. Nagai N, Ogata F, Otake H, Kawasaki N. Oral administration system based on meloxicam nanocrystals: decreased dose due to high bioavailability attenuates risk of gastrointestinal side effects. Pharmaceutics. 2020;12(4):313. doi:10.3390/pharmaceutics12040313
5. Xie XM, Li Y, Zhao DZ, et al. Oral administration of natural polyphenol-loaded natural polysaccharide-cloaked lipidic nanocarriers to improve efficacy against small-cell lung cancer. Nanomed-Nanotechnol. 2020;29:102261. doi:10.1016/j.nano.2020.102261
6. Upponi JR, Jerajani K, Nagesha DK, et al. Polymeric micelles: theranostic co-delivery system for poorly water-soluble drugs and contrast agents. Biomaterials. 2018;170:26–36.
7. Russell J, Fletcher EM, Michalek JE. Treatment of primary fibrositis/fibromyalgia syndrome with ibuprofen and alprazolam, A double-blind, placebo-controlled study. Arthritis Rheumatol. 2014;34(5):552–560. doi:10.1002/art.1780340507
8. Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–182. doi:10.1146/annurev.biochem.69.1.145
9. Warn DE, Thompson SG, Spiegelhalter DJ. Bayesian random effects meta-analysis of trials with binary outcomes: methods for the absolute risk difference and relative risk scales. Stat Med. 2010;21(11):1601–1623. doi:10.1002/sim.1189
10. Searle S, Muse D, Paluch E, Leyva R, Depadova E, Cruz-Rivera M. Efficacy and safety of single and multiple doses of a fixed-dose combination of ibuprofen and acetaminophen in the treatment of postsurgical dental pain: results from 2 Phase 3, randomized, parallel-group, double-blind, placebo-controlled studies. Clin J Pain. 2020;36:495–504.
11. Ma Z, Wang N, He H, Tang X. Pharmaceutical strategies of improving oral systemic bioavailability of curcumin for clinical application. J Control Release. 2019;316:359–380. doi:10.1016/j.jconrel.2019.10.053
12. Yen -C-C, Yi-Chen C, Wu M-T, et al. Nanoemulsion as a strategy for improving the oral bioavailability and anti-inflammatory activity of andrographolide. Int J Nanomed. 2018;13:669–680. doi:10.2147/IJN.S154824
13. Rayamajhi S, Marchitto J, Nguyen TDT, Marasini R, Aryal S. Ph-responsive cationic liposome for endosomal escape mediated drug delivery. Colloids Surf B Biointerfaces. 2020;188:110804. doi:10.1016/j.colsurfb.2020.110804
14. Jafar M, Salahuddin M, Bolla SR. Gastric floating in-situ gel as a strategy for improving anti-inflammatory activity of meloxicam. J Appl Pharm Sci. 2018;8(11):95–102.
15. Weyna DR, Cheney ML, Shan N, Hanna M, Zaworotko MJ, Sava V. Improving solubility and pharmacokinetics of meloxicam via multiple-component crystal formation. Mol Pharm. 2012;9(7):2094–2102. doi:10.1021/mp300169c
16. Khurana S, Jain NK, Bedi PMS. Development and characterization of a novel controlled release drug delivery system based on nanostructured lipid carriers gel for meloxicam. Life Sci. 2013;93(21):763–772. doi:10.1016/j.lfs.2013.09.027
17. Hu BB, Wang JX, Li J, Li SM, Li HR. Superiority of L-tartaric acid modified chiral mesoporous silica nanoparticle as a drug carrier: structure, wettability, degradation, bio-adhesion and biocompatibity. Int J Nanomed. 2020;15:601–618. doi:10.2147/IJN.S233740
18. Xu C, Cao Y, Lei C, Li Z, Ye Q. Polymer-mesoporous silica nanoparticle core-shell nanofibers as a dual drug delivery system for guided tissue regeneration. ACS Appl Nano Mater. 2020;3(2):1457–1467. doi:10.1021/acsanm.9b02298
19. Ibrahim AH, Smtt JH, Govardhanam NP, Ibrahim HM, Rosenholm JM. Formulation and optimization of drug-loaded mesoporous silica nanoparticle-based tablets to improve the dissolution rate of the poorly water-soluble drug silymarin. Eur J Pharm Sci. 2019;142:105103. doi:10.1016/j.ejps.2019.105103
20. Bharti C, Nagaich U, Pal AK, Gulati N. Mesoporous silica nanoparticles in target drug delivery system: a review. J Pharm Investig. 2015;5(3):124–133. doi:10.4103/2230-973X.160844
21. Zhou Y, Quan G, Wu Q, Zhang X, Niu B, Wu B. Mesoporous silica nanoparticles for drug and gene delivery. Acta Pharm Sin B. 2018;8(2):165–177. doi:10.1016/j.apsb.2018.01.007
22. Li WK, Lin P, Dai S, Sun XQ, Shen YL. Preparation of a mesocellular siliceous foam supported lanthanide-sensitive polymer for the selective adsorption of lanthanides. Dalton Trans. 2018;47(14):4840–4846. doi:10.1039/C7DT04255H
23. Hu N, Sridhar M, Gunugunuri K. Preparation, characterization and lysozyme immobilization studies on siliceous mesocellular foams: effect of precursor chemistry on pore size, wall thickness and interpore spacing. Micro Meso Mat. 2014;190:215–226. doi:10.1016/j.micromeso.2014.02.004
24. Ford J, Yang S. Directed synthesis of silica nanoparticles on micropatterned hydrogel templates tethered with poly(ethyleneimine). Chem Mater. 2007;19(23):5570–5575. doi:10.1021/cm071566q
25. Wang F, Jiang F, Li Y, Wang Q, Zhang X. Formation of new biosilica-like structures by flow-induced forces. RSC Adv. 2012;2(13):5738–5747. doi:10.1039/c2ra20127e
26. Zhao R, Su BL. Self-assembly of phosphorylated poly(ethyleneimine) for use as biomimetic templates in the formation of hybrid hollow silica spheres. Mater Lett. 2012;74:163–166. doi:10.1016/j.matlet.2012.01.023
27. Jin RH, Yuan JJ. Shaped silicas transcribed from aggregates of four-armed star polyethyleneimine with a benzene core. Chem Mater. 2006;18:3390–3396. doi:10.1021/cm060533s
28. Jin RH, Yuan JJ. Simple synthesis of hierarchically structured silicas by poly(ethyleneimine) aggregates pre-organized by media modulation. Macromol. Chem Phys. 2005;206:2160–2170.
29. Naik RR, Whitlock PW, Rodriguez F, et al. Controlled formation of biosilica structures in vitro. Chem Comm. 2003;238–239. doi:10.1039/b210635c
30. Jackson E, Ferrari M, Cuestas-Ayllon C, et al. Protein-templated biomimetic silica nanoparticles. Langmuir. 2015;31(12):3687–3695. doi:10.1021/la504978r
31. Wang YM, Li W, Liu T, et al. Design and preparation of mesoporous silica carriers with chiral structures for drug release differentiation. Mat Sci Eng C. 2019;103:109737. doi:10.1016/j.msec.2019.109737
32. Li HR, Ke J, Li H, et al. Mesoporous silicas templated by heterocyclic amino acid derivatives: biomimetic synthesis and drug release application. Mat Sci Eng C. 2018;94:407–418. doi:10.1016/j.msec.2018.07.081
33. Atluri R, Iqbal MN, Bacsik Z, Hedin N, L A V, Garcia-Bennett AE. Self-assembly mechanism of folate-templated mesoporous silica. Langmuir. 2013;29:12003–12012. doi:10.1021/la401532j
34. Hao N, Nie Y, Zhang JXJ. Biomimetic hierarchical walnut kernel-like and erythrocyte-like mesoporous silica nanomaterials: controllable synthesis and versatile applications. Micro Meso Mat. 2018;261:144–149. doi:10.1016/j.micromeso.2017.11.003
35. Deng Y, Sun J, Ni X, Xiong D. Multilayers of poly(ethyleneimine)/poly(acrylic acid) coatings on ti6al4v acting as lubricated polymer-bearing interface. J Biomed Mater Res B. 2020;108(5):2141–2152. doi:10.1002/jbm.b.34553
36. Mészáros R, Varga I, Gilányi T. Adsorption of poly(ethyleneimine) on silica surfaces: effect of ph on the reversibility of adsorption. Langmuir. 2004;20(12):5026–5029. doi:10.1021/la049611l
37. Li J, Xu L, Liu H, Wang Y, Wang Q, Chen H. Biomimetic synthesized nanoporous silica@poly(ethyleneimine)s xerogel as drug carrier: characteristics and controlled release effect. Int J Pharm. 2014;467(1–2):9–18. doi:10.1016/j.ijpharm.2014.03.045
38. Li J, Xu L, Wang H, Yang B, Liu H, Pan W. Comparison of bare and amino modified mesoporous silica@poly(ethyleneimine)s xerogel as indomethacin carrier: superiority of amino modification. Mat Sci Eng C. 2015;S0928493115305038.
39. Li J, Wang H, Li H, Xu L, Guo Y, Lu F. Mutual interaction between guest drug molecules and host nanoporous silica xerogel studied using central composite design. Int J Pharm. 2016;498(1–2):32–39. doi:10.1016/j.ijpharm.2015.12.005
40. Shah VP, Tsong Y, Sathe P, Liu JP. In vitro dissolution profile comparison—statistics and analysis of the similarity factor, f2. Pharm Res. 1998;15(6):889–896. doi:10.1023/A:1011976615750
41. Costa P. An alternative method to the evaluation of similarity factor in dissolution testing. Int J Pharm. 2001;220(1–2):77–83. doi:10.1016/S0378-5173(01)00651-2
42. Jangra S, Girotra P, Chhokar V, Tomer VK, Sharma AK, Duhan S. In-vitro drug release kinetics studies of mesoporous SBA-15-azathioprine composite. J Porous Mat. 2016;23(3):679–688. doi:10.1007/s10934-016-0123-1
43. Dolinina ES, Vlasenkova MI, Parfenyuk EV. Effect of trehalose on structural state of bovine serum albumin adsorbed onto mesoporous silica and the protein release kinetics in vitro. Colloid Surface A. 2017;527:101–108. doi:10.1016/j.colsurfa.2017.05.014
44. Irani M, Jacobson AT, Gasem KAM, Fan M. Facilely synthesized porous polymer as support of poly(ethyleneimine) for effective co 2 capture. Energy. 2018;157(15):1–9. doi:10.1016/j.energy.2018.05.141
45. Maleki A, Hamidi M. Dissolution enhancement of a model poorly water-soluble drug, atorvastatin, with ordered mesoporous silica: comparison of MSF with SBA-15 as drug carriers. Expert Opin Drug Deliv. 2016;13(2):171–181. doi:10.1517/17425247.2015.1111335
46. Skorupska E, Jeziorna A, Paluch P, Potrzebowski MJ. Ibuprofen in mesopores of mobil crystalline material 41 (mcm-41): a deeper understanding. Mol Pharm. 2014;1(5):1512–1519. doi:10.1021/mp400670f
47. Ghedini E, Signoretto M, Pinna F, Guarascio D, Cerrato G. Ibuprofen delivery behaviour on mcm-41: influence of organic groups amount. Stud Surf Sci Catal. 2008;174(08):429–432.
48. Li J, Xu L, Nan Z, Lu FZ, Wang H, Li SM. Biomimetic synthesized bimodal nanoporous silica: bimodal mesostructure formation and application for ibuprofen delivery. Mat Sci Eng C. 2016;58:1105–1111.
49. Hwang DH, Lee D, Lee H, Choe D, Lee SH, Lee K. Surface functionalization of SBA-15 particles for ibuprofen delivery. Korean J Chem Eng. 2010;27(4):1087–1092. doi:10.1007/s11814-010-0225-4
50. Rehman F, Volpe PLO, Airoldi C. The applicability of ordered mesoporous SBA-15 and its hydrophobic glutaraldehyde-bridge derivative to improve ibuprofen-loading in releasing system. Colloids Surf B Biointerfaces. 2014;119:82–89. doi:10.1016/j.colsurfb.2014.03.043
51. Wang Y, Ke J, Gou KJ, et al. Amino functionalized mesoporous silica with twisted rod-like shapes: synthetic design, in vitro and in vivo evaluation for ibuprofen delivery. Micro Meso Mat. 2020;294:109896. doi:10.1016/j.micromeso.2019.109896
52. Perez RA, Singh RK, Kim TH, Kim HW. Silica-based multifunctional nanodelivery systems toward regenerative medicine. Mater Horiz. 2017;4:772–799.
53. Li H, Li H, Wei C, Ke J, Li J, Xu L. Biomimetic synthesis and evaluation of histidine-derivative templated chiral mesoporous silica for improved oral delivery of the poorly water-soluble drug, nimodipine. Eur J Pharm Sci. 2018;117:321–330. doi:10.1016/j.ejps.2018.03.013
54. Newa M, Bhandari KH, Kim JO, Im JS, Kim JA. Enhancement of solubility, dissolution and bioavailability of ibuprofen in solid dispersion systems. Chem Pharm Bull. 2008;56(4):569–574. doi:10.1248/cpb.56.569
55. Hancock BC, Zografi G. Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci. 1997;86:1–12. doi:10.1021/js9601896
56. Cheng HC, Jiang ZX, Chang TL, Chen PH. Roughness and wettability properties of plain and silica-coated copper surfaces textured with picosecond laser. Appl Surf Sci. 2020;514:145918. doi:10.1016/j.apsusc.2020.145918
57. Li H, Wu X, Yang B, et al. Evaluation of biomimetically synthesized mesoporous silica nanoparticles as drug carriers: structure, wettability, degradation, biocompatibility and brain distribution. Mat Sci Eng C. 2019;94:453–464. doi:10.1016/j.msec.2018.09.053
58. Yu MR, Xu L, Tian FL, Su Q, Zheng N, Yang YW. Rapid transport of deformation-tuned nanoparticles across biological hydrogels and cellular barriers. Nat Commun. 2018;9(1).
59. Ran F, Lei W, Cui Y, Jiao J, Wang S. Size effect on oral absorption in polymer-functionalized mesoporous carbon nanoparticles. J Colloid Inter Sci. 2018;511:57–66. doi:10.1016/j.jcis.2017.09.088
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.