Back to Archived Journals » Current Biomarker Findings » Volume 5
Biomarkers for bipolar disorder: current insights
Authors Duong A, Syed B, Scola G ![]()
Received 16 June 2015
Accepted for publication 19 September 2015
Published 3 November 2015 Volume 2015:5 Pages 79—92
DOI https://doi.org/10.2147/CBF.S79138
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Hung Khong
Angela Duong,1 Bushra Syed,1 Gustavo Scola2,3
1Department of Pharmacology and Toxicology, 2Department of Psychiatry, University of Toronto, 3Centre for Addiction and Mental Health, Toronto, ON, Canada
Abstract: Currently, there exists a lack of definitive diagnostic tools for neuropsychiatric disorders, particularly molecular markers that could help assess the illness and develop more personalized treatments for different disorders. Understanding of the neurobiology and potential novel treatments for bipolar disorder (BD), one of the most complex psychiatric illnesses, remains poor. This review aims to compile the most reproducible findings regarding the molecular, genetic, and structural changes that occur in BD. Neuroimaging studies have indicated alterations in neural circuits, disrupted white matter integrity, alterations in reward activation, and decreased gray matter (GM) volume. Genetic studies have identified variations in a number of genes that confer risk for BD development. Studies involving peripheral biomarkers include alterations in the levels of oxidative stress, inflammation, and neurotrophins. These potential molecular markers could be used as tools for diagnosis, to assess illness progression, and to help with the improvement of more specific and personalized treatments for patients with BD. Identification of biologically relevant markers could improve the quality of life of patients with BD and revolutionize public health.
Keywords: biomarkers, neuroimaging, neural activation, gene regulation, microRNAs, oxidative stress, inflammation
Introduction
Bipolar disorder (BD) is a chronic psychiatric illness with partially unknown pathophysiology and symptoms alternating between mania and depression.1 As consistently found in postmortem studies, in BD, morphological alterations are associated with disruption of cerebral functions, leading to impairment in the cellular plasticity and resilience of the brain.2 Currently, many studies have reported alterations in the morphology of brain tissues, brain cells, and in the periphery. These alterations could be directly correlated to dysregulation of the molecular pathways of inflammation and neurotrophins.3,4
The factors that regulate neurological cells are expressed in a region-specific manner in the brain and the peripheral tissues. This suggests the possibility of using peripheral markers to address alterations in the brain that occur during the development of BD and other psychiatric disorders. Biomarkers are essential tools necessary to provide insights into the molecular alterations in BD.2,3 Due to the heterogeneity of this disorder, the possibility of developing specific biomarker is still being explored; however, a set of biomarkers might be available to identify subgroups of patients and also develop new treatments. Our goal is to incorporate and critically review the published literature with regard to the morphological, genetic, and molecular alterations found in BD. The hypothetical use of these findings as potential biomarkers is also examined. Moreover, these markers could be used as tools for diagnosis, to assess illness progression, and to help with the improvement of more specific and personalized treatments for patients with BD. This review is separated into distinct sections that comprise structural, genetic, and peripheral alterations in BD. In addition, all the potential biomarkers discussed in this report are illustrated in Table 1.
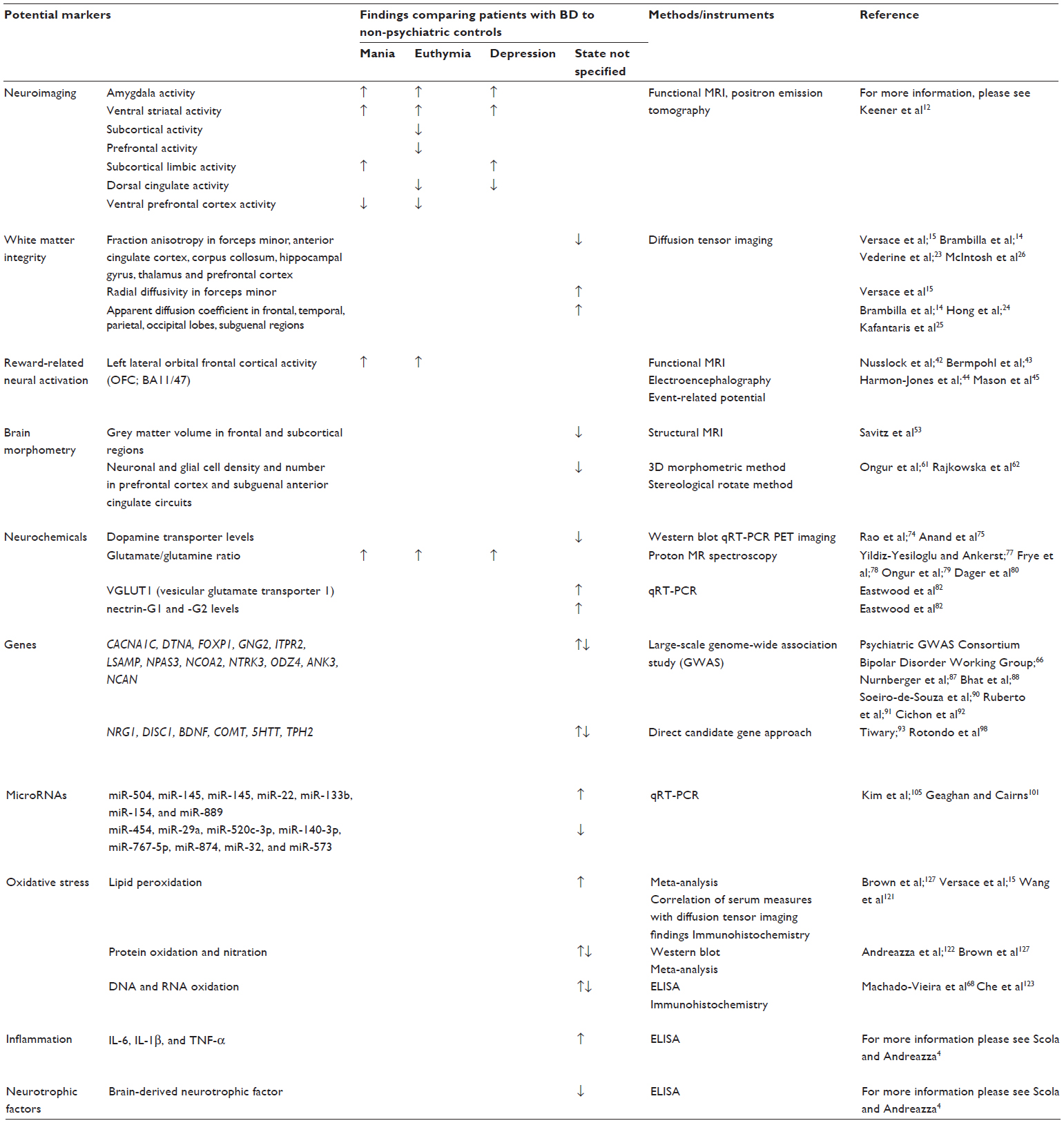 | Table 1 Potential biomarkers for bipolar disorder |
Structural abnormalities and neurochemical alterations
Neuroimaging in BD
BD may present with early episodes of depression, often leading to its misdiagnosis as unipolar depression, which in turn can lead to misdirected treatment.5 Strategies aimed at improving diagnostic accuracy and treatment approaches for BD are therefore crucial. One such approach is using neuroimaging technologies to identify biomarkers. Neuroimaging technologies include structural neuroimaging, functional imaging, diffusion tensor imaging (DTI), and magnetic resonance spectroscopy (MRS).6 Such technologies have allowed a better understanding of the pathophysiology of BD through observations of structural and functional alterations. These alterations may serve as potential biomarkers for BD and have application in diagnostic or prognostic evaluation.
The ventral prefrontal cortex and the amygdala form a corticolimbic network that is involved in emotional regulation.7 Functional deficits such as impaired emotional regulation and attention have been implicated in BD.8 One of the most commonly used tools to characterize such alterations is neuroimaging, which can be divided into either functional or structural studies. Structural neuroimaging showed an increased in volume of the amygdala in patients with BD compared to those with schizophrenia;9 decreased gray matter (GM) volume in dorsal and ventral prefrontal cortices in BD;10 and little or no alteration in hippocampal volume in BD compared to healthy subjects.11 Functional neuroimaging uses functional magnetic resonance imaging (fMRI) and positron emission tomography (PET) to determine alterations in parts of the brain in subjects during the performance of tasks that require emotion processing and executive control.8,12 Several studies have used functional neuroimaging to determine whether a significant difference in emotional processing exists in different states of BD, which can be used as a potential biomarker for diagnostic purposes. Increased amygdala and ventral striatal activity in response to negative stimuli were common in all the three states of BD: euthymia, depression, and mania.12 This increase in the amygdala and ventral striatal activity was also seen in response to positive stimuli in the euthymic and depressive states, but not in mania.12 Decreased subcortical and prefrontal neural activity was seen in the euthymic state, whereas increased subcortical limbic activity was common to the depressive and manic states, and this feature can be used to distinguish BD from unipolar depression.12 As cognitive abilities are also compromised in BD, functional neuroimaging is used to identify changes in brain areas during cognitive functions in BD. Decreased dorsal cingulate activity was common to both depressive and euthymic states, whereas decreased activity in the ventral prefrontal cortex was common to euthymic and manic states.12
It is well established that neural circuit alterations are responsible for the altered emotion and cognition observed in BD. Further discussion of neuroimaging findings that demonstrates altered neural circuits involved in emotion processing and regulation in BD is presented in a review by Phillips and Swartz.13 Although neuroimaging studies have provided the pathophysiological basis for BD, their use in diagnosis, early detection, and treatment of patients with BD still lacks significant impact. It is important to consider that one of the challenges in using neuroimaging to identify biomarkers is the cost involved. Neuroimaging techniques such as fMRI, to analyze response to different attentional or emotional paradigms, and DTI are expensive and are done only in research settings, which limits their feasibility of application in psychiatric assessment. Another challenge is that neuroimaging measures, at present, do not offer adequate specificity and sensitivity to accurately characterize alterations in BD. However, if more specific neural alterations are found in BD and can be differentiated among other psychiatric conditions, then their applications do hold promise in identifying reliable valid biomarkers. This warrants more established findings to generate a strong consensus on alterations in activation patterns in different brain regions in BD.
In the following sections, we review some studies on neuroimaging to address functional, morphometric, and neurochemical alterations in BD. We aim to largely evaluate differences between patients with BD and nonpsychiatric controls. Although this may provide insights into potential biomarkers that may not be conclusive, they will guide the initial step toward biomarker development that could reflect pathophysiological processes in BD.
Disruption of white matter integrity in BD
White matter integrity in BD can be assessed by neuroimaging. White matter mainly consists of myelinated axons and is made up of glial cells and oligodendrocytes.14,15 Abnormalities in white matter integrity are most commonly measured using DTI or diffusion-weighted imaging (DWI) that measure fraction anisotropy (FA), radial diffusivity (RD), and apparent diffusion coefficient (ADC).14,15 FA and ADC measurements indicate white matter consistency and its ability to prevent diffusion of water into the tissues, whereas RD value indicates myelin abnormalities and axonal damage.14,15 Abnormal values of these measures indicate demyelination and abnormalities in white matter integrity.14,15
Since white matter hyperintensities, detected by structural magnetic resonance imaging (MRI), were first reported in patients with BD,16 they have become one of the most consistent findings replicated in neuroimaging studies of BD.17,18 White matter hyperintensities were most frequently observed in patients with at least one manic episode (BD-I) with poor treatment response. In addition, patients with at least one hypomanic episode and one depressive episode, also known as BD-II, were characterized by elevated number of white matter hyperintensities in comparison to nonpsychiatric controls.19 These white matter hyperintensities are thought to impair connectivity involved in control of mood, and thus may lead to underlying mood dysregulation associated with BD.20 However, these findings are relatively nonspecific to BD, since they are involved in the pathophysiology of other diseases and healthy aging.21
Although structural MRI offers an incomplete understanding of the brain’s white matter integrity, one neuroimaging approach that is sensitive in detecting white matter microstructure and integrity is DTI.22 Several studies have examined the FA and ADC from different areas of the brain in patients with BD. Reduced FA and higher RD values were observed in patients with BD, compared to healthy controls, in the forceps minor, indicating myelin abnormalities.15 In patients with BD, reduced FA was also seen in the anterior cingulate cortex and corpus collosum, while significantly increased ADC was found in the frontal, temporal, parietal, and occipital lobes, indicating demyelination, axonal loss, and disruption of microstructure organization of the white matter.14 A meta-analysis conducted has reported decreased FA near the hippocampal gyrus and the right anterior and subgenual cingulate cortices.23 The right anterior and subgenual cingulate cortices are involved in emotion processing; a decrease in ADC in the subgenual regions was also observed.24,25 Decreased FA in tracts between the thalamus and prefrontal cortex was also observed.26 White matter abnormalities in these regions, especially in the anterior cingulate cortex, may explain the characteristic alterations in emotional processes seen in BD.27 Lower FA and increased ADC may indicate a loss of organization and connectivity in the white matter, which could explain the compromise of white matter integrity seen in patients with BD.
Although interest in reduced white matter connectivity is emerging, majority of DTI results across studies have been controversial and inconsistent when compared to those in other disorders, such as schizophrenia. Some studies have reported decreased,15,28 increased,29–33 as well as both increased and decreased integrity in distinct white matter tracts in the same patients with BD.34 Such differences in DTI findings may suggest the complexity of pathology in different cohorts of BD, differences on how the DTI data were acquired, and the effects of medications, which were not accounted for in the majority of the studies analyses.
DTI could be a useful candidate to assess white matter integrity and facilitate diagnosis and assessment of illness progression in the early stages of BD.14,23,24,27 Indeed, low FA was found to correlate with genetic vulnerability to BD as well as with lipid peroxidation markers, suggesting the potential role of oxidative stress in altering white matter microstructure.15,35 However, white matter findings remain inconsistent, suggesting that further studies are needed to validate its potential use as a biomarker for BD.
Reward-related neural activation as biological marker in BD
Imaging studies also provide meaningful insights into how the brain’s reward circuitry regulates reward experiences and affections that guide complex behavior in BD.36,37 The reward hypersensitivity theory proposes that a factor that confers a risk to bipolar spectrum disorders involves increased motivation to expend effort toward attaining rewards and goals following reward-related events.37,38 This hypersensitivity is thought to precipitate the hypomanic/manic phase of BD.38,39 In contrast, reward hypersensitivity can also trigger the depressive phase when goals and rewards are not achieved.38 This model suggests differential alterations in neural activation during reward processing in BD and depression. Due to frequent misdiagnosis between BD and unipolar depression,40 it is important to consider the degree to which reward-related neural activation reflects a valuable biomarker that is specific for assessing the traits of BD.
The frontal-striatal neural circuit forms a complex system involved in regulating various aspects of reward processing.41 Excessive activation of this neural circuit has been implicated in BD.42 Using fMRI, one study reported elevated activation of the left lateral orbital frontal cortex (OFC; BA11/47) in patients with BD assessed during the manic phase while anticipating for increasing gains, and decreased left OFC activation during anticipation of increasing loss.43 Increased OFC and ventral striatal responses during reward anticipation were also found in a subset of euthymic patients compared with non-psychiatric controls, suggesting that reward activation is not state-dependent.42 Increased left frontal cortical activity is supported by electroencephalographic (EEG) assessment – a neurophysiological index of reward sensitivity – in individuals susceptible to bipolar spectrum diagnosis.44 Another neurophysiological index is event-related potential (ERP), which is used to examine neuronal responses to feedback (feedback-related negativity [FRN]).45 Indeed, individuals at risk for hypomania displayed lower FRN during a monetary reward task, suggesting greater reward sensitivity and decreased response to negative feedback.45 This suggests that patients with BD may display extremely high motivational drives during reward striving, leading to decreased propensity to negatively regulate their elevated reward response. Thus, it is likely that hypersensitivity of the frontal striatal network is a risk factor that predisposes to altered episodes in patients with BD during reward processing.
It is important, however, to have a measureable reward-related biomarker that is consistent throughout all phases of BD, including depression. As depressive patients with BD usually seek treatments and may present this episode initially, measureable biomarkers are needed to facilitate assessment in order to ensure that BD is not misdiagnosed as unipolar depression.5 Although dysfunction of the reward system is thought to play an important role in both unipolar depression and BD, it can be used for differential diagnosis and assessment.42,46 EEG and ERP studies have identified diminished left frontal cortical activity47 and decreased FRN in patients with unipolar depression,48 suggesting deficits in reward processing and responses. Similarly, diminished activity of the frontostriatal neural circuitry was also seen in other psychiatric illnesses, such as schizophrenia.49 Depressive patients with BD, on the other hand, maintained increased activity of the left frontal cortex,42 suggesting that reward processing in this brain region is not dependent on mood state. As an elevated frontal-striatal neural circuit is maintained throughout all phases of BD, it may be a potential biomarker for differential diagnosis and assessment of disease progression in patients with BD versus those with unipolar depression.
Further investigations are needed to unravel different reward-related mechanisms and targets of depression. This would improve our ability to differentiate between bipolar depression and unipolar depression, without relying on the main feature underlying BD – mania.
Brain morphometric and neurotransmitter biomarkers in BD
Brain morphometric studies have become increasingly significant since the introduction of noninvasive MRI and other neuroimaging technologies. These advancements allow for the quantification of brain abnormalities based on shape and size over time, providing a whole paradigm of novel diagnostic approaches. In BD, morphometric alterations of certain brain regions might offer insights into potential biological markers and pathologies that are not fully understood at present. These morphometric changes in the brain may dysregulate neuronal circuitry by modifying neurotransmitter release, and thus could impair synaptic plasticity, connectivity, and cellular resilience.50 Thus, considering neurotransmitter systems as potential biomarkers is important as they are widely distributed throughout the neuronal circuit and are believed to reflect motivational, cognitive, and behavioral manifestations of BD.51,52
Structural MRI in patients with BD have indicated decreased GM volume of both frontal and subcortical regions of the brain.53 Volumetric reduction of GM in these brain areas is considered to be attributable to the ventricular enlargement in BD.54–57 Interestingly, mood stabilizers such as lithium were able to significantly increase GM volume in previously nonmedicated patients with BD compared to healthy controls.58,59 Following lithium treatment, elevated GM density was demonstrated in diffuse cortical areas independent of mood states, particularly with greatest significance in regions involved in motivational and emotional drives.60 These findings suggest that patients with BD may have distinct and region-specific morphological profiles of the brain that might be involved in regulating mood and visceral manifestations. Thus, structural MRI could be a potential biomarker for assessing the degree of GM reduction in patients with BD.29 However, further studies are needed to verify which specific regions are altered in BD and whether GM volume is a marker that reflects clinical outcome.
There is also growing evidence that the reduction of GM volume may reflect morphometric abnormalities of the neurons and glial cells. Several postmortem studies reported a significant reduction of neuronal cell density in the prefrontal cortex and subguenal anterior cingulate circuits (ACC) in individuals with BD.61–63 The reduction in neuronal density and number appear to be region-, layer-, and cell type-specific.63–66 Furthermore, reduction in glial cell size might also be implicated in the pathophysiology of BD.62 Subjects with BD who were not medicated with lithium or valproate had significantly reduced glial density in the amygdala, suggesting that mood stabilizers might be involved in protecting the integrity of glial cytoarchitecture.67 Additionally, elevated levels of the glial marker protein, S100B, was found in the serum of patients with BD, reflecting potential glial cell injury and malfunction.68 Thus, S100B levels could be regarded as a potential indicator of glial alteration and status.69–71 As glial cells provide many important functions to the neurons, loss of glial cells in BD can directly affect the number of neurons. Thus, neuronal and glial cell loss may provide important indications of alterations in neuronal circuits involved in normal mood functioning.
Furthermore, it is important to consider how the observed morphological alterations in the brain could translate into abnormal neurotransmission in BD. First, dysregulation of the dopaminergic system is postulated to underlie extreme moods associated with BD.72 It was proposed that increased dopaminergic activity during the manic phase results in reduced sensitivity of dopaminergic receptors over time, which in turn may initiate a conversion into the depressive phase.72 Increased dopaminergic activity is thought to result from alterations in dopamine reuptake mechanisms.73 Significantly reduced mRNA and protein expression levels of dopamine transporter (DAT) in frontal cortex in subjects with BD compared to healthy controls supports this hypothesis.74 PET imaging also revealed reduced DAT availability in the bilateral dorsal caudate areas of patients with BD.75 Thus, altered DAT expression may lead to excessive dopamine transmission underlying the mania of BD. It is important to consider that the dopamine dysregulation in BD is not completely understood and is presently speculative. Further studies are necessary to better understand the mechanisms that underlie dopamine dysregulation in BD. This would help identify important targets related to dopamine neurotransmission that could be used as potential biomarkers to guide diagnosis and assessment of illness progression for patients with BD.
In contrast, glutamatergic dysregulation was also proposed to be involved in the pathophysiology of BD. Proton MR spectroscopy (H-MRS) is a noninvasive brain imaging technology that can quantify metabolites such as glutamate and glutamine in the brain.76 H-MRS revealed increased levels of combined glutamate/glutamine (Glx) in various cortical regions of patients with BD, independent of mood state.77 Some studies have shown that levels of glutamate differ between patients with BD and those with unipolar depression. Elevated glutamate levels were consistently observed in ACC of depressive patients with BD,78–80 whereas decreased glutamate levels were found in the same brain region in patients with unipolar depression.81 Furthermore, gene expression of VGLUT1 and nectrin-G1 and -G2 were found to be increased in ACC of patients with BD.82 As VGLUT1 and nectrin-G proteins are known to be involved in glutamate transmission and formation of synaptic connectivity of excitatory pathways,82 this suggests that their elevated expression may result in increased glutamate transmission and potential excitotoxic processes in the brains of patients with BD. Furthermore, H-MRS represents a promising biomarker because of its ability to reflect changes in Glx levels in response to treatment. For instance, lithium was able to reduce Glx levels,83 suggesting its neuroprotective effects during states of increased glutamatergic signaling. As Glx levels in BD were increased independent of mood state, differed from unipolar depression, and decreased in response to lithium, H-MRS findings of glutamate are a potential biomarker for the diagnosis and assessment of BD.
Genetic alterations and gene regulation
Genetic alterations in BD
Biomarkers for BD are not only limited to specific structural and neurochemical alterations. Compelling genetic findings in patients with BD have well replicated a number of significant susceptibility genes that may hold clinical promises and allow for the identification of specific biological pathways of interest that underlie the pathophysiology of BD. Currently, no single causative risk gene has been identified as being central to predicting the risk of BD development. However, evidence from molecular genetics strongly suggests polygenic effects where many genetic polymorphisms of small additive effect sizes contribute to the risk of BD.84
A large-scale genome-wide association study (GWAS) has examined multitudes of candidate genes with intriguing variable findings, reflective of high heterogeneity in BD.85,86 Recently, a study that compiled multiple GWAS found altered expression of genes that regulate biological pathways such as hormones, calcium channels, second messenger systems, and glutamate receptor signaling in BD.87 Particularly, CACNA1C, DTNA, FOXP1, GNG2, ITPR2, LSAMP, NPAS3, NCOA2, and NTRK3 were found to be differentially expressed in the dorsolateral prefrontal cortex in patients with BD, and are hypothesized to be involved in the predisposition to BD.87 Among these genes, CACNA1C has been one of the most consistent replicable findings in BD.88 CACNA1C is a gene that encodes for the alpha 1C subunit of the L-type voltage-gated calcium channel and plays an important role in regulating calcium processes involved in neuronal signaling.89 A polymorphism in the CACNA1C gene is thought to influence executive and cognitive functioning, both of which are strongly implicated in BD.90 Such endophenotypes have been associated with dysfunction in various neural circuits such as the hippocampus, amygdala, and the mesolimbic reward system.88 Other genetic risk variants for BD identified by GWAS that are involved in brain-specific functions include: odd Oz/ten-m (ODZ4), which encodes for cell surface proteins important for neuronal signaling;66 ankyrin 3 (ANK3), a gene involved in the regulation of action potentials via sodium channels assembly is thought to influence cognitive processes in BD;91 and neurocan (NCAN), a gene that codes for extracellular matrix glycoproteins is theorized to be involved in cognitive and emotional regulation in BD.92
In contrast, direct candidate gene approach has also identified various risk genes for BD. Such genes include NRG1, DISC1, BDNF, and COMT.93 These risk genes are known to interact and appear to confer risks to not only BD but also depression and schizophrenia.93 Interestingly, a nonconservative polymorphism in the BDNF gene that swaps valine for methionine at codon 66 was found to increase mature-BDNF/pro-BDNF levels in the serum of patients with BD, suggesting aberrations in the conversion process and trafficking of BDNF.94 The variant BDNF was associated with vulnerability toward rapid cycling, impairment in CNS function, and risk of suicidal behavior in BD.95–97 Moreover, other gene variants involved in neurotransmission include 5HTT and TPH2, which have been linked to BD.98 Particularly, a polymorphism in the 5HTT promoter region was found to affect lithium response and increase susceptibility to manic episodes associated with antidepressant use in patients with BD.99,100
Therefore, it is plausible that genetic alterations may underpin endophenotypes of BD characterized by specific structural and peripheral alterations, drug responses, and CNS functions. Further studies will need to investigate the link between these genetic polymorphisms and structural and biological pathways observed in BD.
Posttranscriptional dysregulation and microRNAs in BD
Despite the identification of a number of susceptibility genes of relevance to BD, only a few studies have explored how these genes are regulated and why such gene expressions are differentially altered in BD. Exploring regulation of expression patterns of potential susceptibility genes at the posttranscriptional level could provide clues on genetic markers for BD. One level of posttranscriptional gene regulatory mechanism associated with BD involves microRNAs (miRNAs).101 miRNAs are small noncoding RNA molecules that play an important role in regulating gene expression via complementary binding to the 3′-untranslated region of a specific mRNA molecule.102 Genetic variations in miRNA binding sites could alter gene expression, which could modulate neuronal development and plasticity.103 This could explain the heterogeneity of BD risk among individuals.104
There is emerging evidence from expression studies, suggesting a link between dysregulation of miRNA and BD. A recent review that assessed a wide variety of differentially regulated miRNA reported, on postmortem examination, an upregulation of seven miRNAs and a downregulation of eight miRNAs in the dorsolateral prefrontal cortex of patients with BD in comparison to controls (Table 1).101,105 Another study also validated gene expression using quantitative real-time polymerase chain reaction (qRT-PCR) and reported that patients with BD presented a trend toward decreased miRNA expression levels, specifically in 19% of miRNAs that were analyzed in the prefrontal cortex (Table 1).106 More recently, a negative correlation was found between miRNA expression and their gene targets,105 suggesting that miRNA may act to negatively regulate gene expression.107 Notably, microarray studies emphasized that individual miRNAs can target multiple genes regulating diverse biological processes, indicating that miRNA can have pleiotropic effects on gene regulation.108 This supports a key regulatory role of these miRNAs on modulating gene expression of susceptibility genes associated with BD, and suggests that posttranscriptional dysregulation may be involved in BD.
Posttranscriptional dysregulation in BD is also supported by genetic and molecular studies on a number of miRNAs and their target genes. A recent study has shown that polymorphisms in the miRNA-206 gene (rs16882131) and BDNF gene (rs6255) interact to modulate the expression of BDNF, affect treatment response to lithium and valproate, and influence genetic susceptibility to BD-I.109 Some miRNAs have been described as important regulators of neurogenesis and formation of synaptic plasticity,110–112 where a majority of their targeted genes are involved in regulation of ion channels and neuronal processes – both of which have been previously implicated in BD.113 This supports the potential role of miRNAs in the regulation of brain function and thus, may be involved in the pathophysiology of BD.113
Another important fact that must be discussed in this section is the effect of treating patients with BD with mood stabilizers. A number of miRNAs have been found to be upregulated after treatment with both lithium and valproate.114 However, this finding is being questioned due to a recent study which reports that valproate causes degradation of an important protein involved in the production of miRNA, leading to a global downregulation of miRNA levels.115 For example, plasma levels of miR-134 were found reduced in non-medicated and medicated patients at the manic stage compared to controls, increased on following treatment with mood stabilizers, and also correlated with illness severity. These findings suggests that plasma miR-134 levels may be considered a potential peripheral biomarker reflecting the manic symptoms of BD and effective treatment response to mood stabilizers.
Therefore, a group of differentially expressed miRNA has been detected in patients with BD, suggesting its potential as a promising novel biomarker. However, these results should be interpreted in the light of the study’s limitations and should be confirmed to check the potential of these molecules as possible biomarkers.
Peripheral alterations
Mitochondrial dysfunction and oxidative stress in BD
While structural and genetic biomarkers appear to hold clinical utility in diagnosis, a majority of studies to date focus on biochemical markers that are measureable in the peripheral blood of patients with BD. Particularly, mitochondrial alterations and oxidative stress are important contributors to the pathophysiology of BD.2 Mitochondria are known to play a critical role in many fundamental processes such as energy production, apoptosis, and reactive oxygen species (ROS) formation under physiological conditions.117 Dysfunction of the mitochondrial electron transport chain is a consistent reporting in many diseases, including BD.118 Alterations in mitochondrial functionality are associated with increased production of ROS that can induce oxidative stress and lead to redox modulations of macromolecules.119,120 Alterations to proteins, lipids, and DNA induced by mitochondrial dysfunction were found in postmortem brain samples,121–124 as well as in peripheral blood cells,68,122,125,126 from patients with BD.
Changes in mitochondrial function and redox modulation patterns were strongly associated with lipid peroxidation and DNA aberrations, which include changes in methylated and oxidized DNA.3 Changes in protein conformation due to carbonylation and nitrosylation were also found; however, these alterations do not represent a strong correlation with the pathophysiology of BD.127 Importantly, our group and others are already investigating studies with regard to how the lipid peroxidation process and DNA aberrations caused by mitochondrial dysfunction occur in BD. These findings will be very important in guiding us to develop new tools for the measurement of illness progression and might also be helpful in the development of more specific treatments for this disorder.
Inflammation and neurotrophic factors
Mitochondrial dysfunction was also suggested to trigger activation of the inflammatory pathway.128 Inflammation plays an important role in the pathophysiology of BD. A recent review emphasized the potential role of nod-like receptor pyrin domain containing 3 (NLRP3) as being closely linked to inflammation and mitochondrial dysfunction in BD.128 In this review, the authors explored a very interesting mechanism of action with regard to the assembly of the NLRP3 inflammasome and initiation of the inflammatory cascade upon mitochondrial dysfunction-induced ROS release.128 There has been a lack of agreement across a number of studies regarding alterations in expression levels of various inflammatory cytokines.129 So far, it is well described that interleukin-6 (IL-6), IL-1β, and tumor necrosis factor alpha (TNF-α) are inflammatory markers consistently increased in BD, supporting their potential use as a biomarker.130–132 More information regarding inflammatory biomarkers can be found in Scola and Andreazza.3
Neurotrophic factors, such as BDNF, are strongly implicated in BD. BDNF plays an important role in supporting the growth, survival, and differentiation of neurons.133 More information on their general description, mechanisms of action, and their effects on BD is presented in Scola and Andreazza.4 Currently, decreased peripheral levels of BDNF represent the most consistent finding replicated in studies of BD and is also considered as a potential peripheral biomarker for assessing disease progression.2 In addition, more studies are needed to better understand how these interleukins and trophic factors exhibit their activities in a multifaceted environment such as BD.
Future directions
This review focuses mainly on comparing BD with nonpsychiatric controls by evaluating the differences in their pathophysiology. Simply, this is the first step towards biomarker development as it only provides insights into potential biomarkers for diagnosis, but does not provide definitive biomarkers. In order to identify biomarkers that can assess the progression of BD, longitudinal studies are needed. Moreover, it is important to consider whether the biomarker is specific to BD. By assessing whether biomarkers of unipolar depression will uncover the depressive episode in BD or can help differentiate BD from other psychiatric illnesses such as schizophrenia would be clinically relevant. As studies on biomarker development are still very preliminary, some of the findings described in this review are not specific for BD. This suggests that these outcomes must be replicated in larger cohorts to assess the reliability and validity of these potential biomarkers.
Although there is a clear need for specific and clinically relevant biomarkers for BD, these biomarkers will likely be limited to a particular group of patients or the type of treatment these patients are taking. Therefore, identification of a subset of biomarkers to stratify specific patients within a population is needed in order to tailor treatments to the specific diagnosis of a patient. This would give information on how patients respond to a particular treatment. Moreover, although great improvement has been made in relating alterations to BD, further studies are needed to characterize how these markers addressed are altered in response to treatment with mood stabilizers and severity of the illness. Determining how genetic markers are associated with structural and peripheral alterations in BD would be meaningful as well. Finally, in order for biomarkers for BD to become a practicable goal, the field must progress beyond its current dependence on diagnosis. This involves integrating biomarkers into large-scale prospective cohorts and randomized controlled trials to validate the clinical utility of these biomarkers in determining the risk of BD for it to aid in the development of novel therapies.2
Conclusion
Recent advances in BD have improved our understanding of its pathophysiology, leading to the search of relevant biomarkers. Indeed, the most promising biomarker for BD would be a marker that shows altered levels with disease development, varies according to the severity of disease, and normalizes following treatment with mood stabilizers. Such a marker would be a valuable tool that can be used for diagnosis, prognosis, and monitoring of treatment responses in BD. However, the field of biomarker development in BD is still in an early stage of research. Neuroimaging studies suggest a number of structural changes that could be regarded as potential biomarkers, such as alterations in neural circuits, disrupted white matter integrity, activation of reward-related frontal-striatal neural circuitry, and decreased GM volume. These structural alterations could affect neurotransmission that can drive oscillation between mood states. Studies evaluating the genetic basis of BD highlighted polymorphisms in a number of susceptibility genes, such as those involved in calcium processes and second messenger systems. These genetic insights paved the way for understanding the role of miRNA-mediated posttranscriptional dysregulation in BD. Finally, studies involving peripheral biomarkers include increased oxidative stress, increased proinflammatory markers, and decreased BDNF levels. Taken together, BD is a very complex illness that encompasses structural, peripheral, and genetic alterations. These alterations are potential biomarkers that could provide insights into treatment approaches and development of novel therapies.
Acknowledgment
The authors thank the CAMH Foundation for funding this work.
Disclosure
The authors report no conflicts of interest in this work.
References
Malhi GS, Fritz K, Allwang C, et al. Agitation for recognition by DSM-5 mixed features specifier signals fatigue? Aust N Z J Psychiatry. 2015;49(6):499–501. | |
Frey BN, Andreazza AC, Houenou J, et al. Biomarkers in bipolar disorder: a positional paper from the international society for bipolar disorders biomarkers task force. Aust N Z J Psychiatry. 2013;47(4):321–332. | |
Scola G, Andreazza AC. Current state of biomarkers in bipolar disorder. Curr Psychiatry Rep. 2014;16(12):514. doi: 10.1007/s11920-014-0514-4. | |
Scola G, Andreazza AC. The role of neurotrophins in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2015;56:122–128. | |
Keck PE Jr, Kessler RC, Ross R. Clinical and economic effects of unrecognized or inadequately treated bipolar disorder. J Psychiatr Pract. 2008;14 Suppl 2:31–38. | |
Bandettini PA. What’s new in neuroimaging methods? Ann N Y Acad Sci. 2009;1156:260–293. | |
Hariri AR, Bookheimer SY, Mazziotta JC. Modulating emotional responses: effects of a neocortical network on the limbic system. Neuroreport. 2000;11(1):43–48. | |
Brooks JO 3rd, Vizueta N. Diagnostic and clinical implications of functional neuroimaging in bipolar disorder. J Psychiatr Res. 2014;57:12–25. | |
Altshuler LL, Bartzokis G, Grieder T, et al. An MRI study of temporal lobe structures in men with bipolar disorder or schizophrenia. Biol Psychiatry. 2000;48(2):147–162. | |
Lopez-Larson MP, DelBello MP, Zimmerman ME, Schwiers ML, Strakowski SM. Regional prefrontal gray and white matter abnormalities in bipolar disorder. Biol Psychiatry. 2002;52(2):93–100. | |
Blumberg HP, Kaufman J, Martin A, et al. Amygdala and hippocampal volumes in adolescents and adults with bipolar disorder. Arch Gen Psychiatry. 2003;60(12):1201–1208. | |
Keener MT, Phillips ML. Neuroimaging in bipolar disorder: a critical review of current findings. Curr Psychiatry Rep. 2007;9(6):512–520. | |
Phillips ML, Swartz HA. A critical appraisal of neuroimaging studies of bipolar disorder: toward a new conceptualization of underlying neural circuitry and a road map for future research. Am J Psychiatry. 2014;171(8):829–843. | |
Brambilla P, Bellani M, Yeh PH, Soares JC, Tansella M. White matter connectivity in bipolar disorder. Int Rev Psychiatry. 2009;21(4):380–386. | |
Versace A, Andreazza AC, Young LT, et al. Elevated serum measures of lipid peroxidation and abnormal prefrontal white matter in euthymic bipolar adults: toward peripheral biomarkers of bipolar disorder. Mol Psychiatry. 2014;19(2):200–208. | |
Dupont RM, Jernigan TL, Gillin JC, Butters N, Delis DC, Hesselink JR. Subcortical signal hyperintensities in bipolar patients detected by MRI. Psychiatry Res. 1987;21(4):357–358. | |
Stoll AL, Renshaw PF, Yurgelun-Todd DA, Cohen BM. Neuroimaging in bipolar disorder: what have we learned? Biol Psychiatry. 2000;48(6):505–517. | |
Kempton MJ, Geddes JR, Ettinger U, Williams SC, Grasby PM. Meta-analysis, database, and meta-regression of 98 structural imaging studies in bipolar disorder. Arch Gen Psychiatry. 2008;65(9):1017–1032. | |
Altshuler LL, Curran JG, Hauser P, Mintz J, Denicoff K, Post R. T2 hyperintensities in bipolar disorder: magnetic resonance imaging comparison and literature meta-analysis. Am J Psychiatry. 1995;152(8):1139–1144. | |
Vataja R, Pohjasvaara T, Leppavuori A, et al. Magnetic resonance imaging correlates of depression after ischemic stroke. Arch Gen Psychiatry. 2001;58(10):925–931. | |
Gootjes L, Teipel SJ, Zebuhr Y, et al. Regional distribution of white matter hyperintensities in vascular dementia, alzheimer’s disease and healthy aging. Dement Geriatr Cogn Disord. 2004;18(2):180–188. | |
Beaulieu C. The basis of anisotropic water diffusion in the nervous system – a technical review. NMR Biomed. 2002;15(7–8):435–455. | |
Vederine FE, Wessa M, Leboyer M, Houenou J. A meta-analysis of whole-brain diffusion tensor imaging studies in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35(8):1820–1826. | |
Heng S, Song AW, Sim K. White matter abnormalities in bipolar disorder: Insights from diffusion tensor imaging studies. J Neural Transm. 2010;117(5):639–654. | |
Kafantaris V, Kingsley P, Ardekani B, et al. Lower orbital frontal white matter integrity in adolescents with bipolar I disorder. J Am Acad Child Adolesc Psychiatry. 2009;48(1):79–86. | |
McIntosh AM, Munoz Maniega S, Lymer GK, et al. White matter tractography in bipolar disorder and schizophrenia. Biol Psychiatry. 2008;64(12):1088–1092. | |
Mahon K, Burdick KE, Szeszko PR. A role for white matter abnormalities in the pathophysiology of bipolar disorder. Neurosci Biobehav Rev. 2010;34(4):533–554. | |
Macritchie KA, Lloyd AJ, Bastin ME, et al. White matter microstructural abnormalities in euthymic bipolar disorder. Br J Psychiatry. 2010; 196(1):52–58. | |
Haznedar MM, Roversi F, Pallanti S, et al. Fronto-thalamo-striatal gray and white matter volumes and anisotropy of their connections in bipolar spectrum illnesses. Biol Psychiatry. 2005;57(7):733–742. doi: 10.1016/j.biopsych.2005.01.002. | |
Houenou J, Wessa M, Douaud G, et al. Increased white matter connectivity in euthymic bipolar patients: diffusion tensor tractography between the subgenual cingulate and the amygdalo-hippocampal complex. Mol Psychiatry. 2007;12(11):1001–1010. | |
Versace A, Almeida JR, Hassel S, et al. Elevated left and reduced right orbitomedial prefrontal fractional anisotropy in adults with bipolar disorder revealed by tract-based spatial statistics. Arch Gen Psychiatry. 2008;65(9):1041–1052. | |
Wessa M, Houenou J, Leboyer M, et al. Microstructural white matter changes in euthymic bipolar patients: a whole-brain diffusion tensor imaging study. Bipolar Disord. 2009;11(5):504–514. | |
Yurgelun-Todd DA, Silveri MM, Gruber SA, Rohan ML, Pimentel PJ. White matter abnormalities observed in bipolar disorder: a diffusion tensor imaging study. Bipolar Disord. 2007;9(5):504–512. | |
Torgerson CM, Irimia A, Leow AD, et al. DTI tractography and white matter fiber tract characteristics in euthymic bipolar I patients and healthy control subjects. Brain Imaging Behav. 2013;7(2):129–139. | |
Chaddock CA, Barker GJ, Marshall N, et al. White matter microstructural impairments and genetic liability to familial bipolar I disorder. Br J Psychiatry. 2009;194(6):527–534. | |
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14(9):609–625. doi: 10.1038/nrn3381; 10.1038/nrn3381. | |
Urosevic S, Abramson LY, Harmon-Jones E, Alloy LB. Dysregulation of the behavioral approach system (BAS) in bipolar spectrum disorders: review of theory and evidence. Clin Psychol Rev. 2008;28(7):1188–1205. doi: 10.1016/j.cpr.2008.04.004; 10.1016/j.cpr.2008.04.004. | |
Alloy LB, Abramson LY. The role of the behavioral approach system (BAS) in bipolar spectrum disorders. Curr Dir Psychol Sci. 2010;19(3):189–194. doi: 10.1177/0963721410370292. | |
Johnson SL. Mania and dysregulation in goal pursuit: a review. Clin Psychol Rev. 2005;25(2):241–262. doi: 10.1016/j.cpr.2004.11.002. | |
Singh T, Rajput M. Misdiagnosis of bipolar disorder. Psychiatry (Edgmont). 2006;3(10):57–63. | |
Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010;35(1):4–26. | |
Nusslock R, Young CB, Damme KS. Elevated reward-related neural activation as a unique biological marker of bipolar disorder: assessment and treatment implications. Behav Res Ther. 2014;62:74–87. doi: 10.1016/j.brat.2014.08.011; 10.1016/j.brat.2014.08.011. | |
Bermpohl F, Kahnt T, Dalanay U, et al. Altered representation of expected value in the orbitofrontal cortex in mania. Hum Brain Mapp. 2010;31(7):958–969. doi: 10.1002/hbm.20909; 10.1002/hbm.20909. | |
Harmon-Jones E, Abramson LY, Nusslock R, et al. Effect of bipolar disorder on left frontal cortical responses to goals differing in valence and task difficulty. Biol Psychiatry. 2008;63(7):693–698. doi: 10.1016/j.biopsych.2007.08.004. | |
Mason L, O’Sullivan N, Bentall RP, El-Deredy W. Better than I thought: positive evaluation bias in hypomania. PLoS One. 2012;7(10):e47754. doi: 10.1371/journal.pone.0047754; 10.1371/journal.pone.0047754. | |
Satterthwaite TD, Kable JW, Vandekar L, et al. Common and dissociable dysfunction of the reward system in bipolar and unipolar depression. Neuropsychopharmacology. 2015;40(9):2258–2268. | |
Thibodeau R, Jorgensen RS, Kim S. Depression, anxiety, and resting frontal EEG asymmetry: a meta-analytic review. J Abnorm Psychol. 2006;115(4):715–729. doi: 10.1037/0021-843X.115.4.715. | |
Foti D, Hajcak G. Depression and reduced sensitivity to non-rewards versus rewards: evidence from event-related potentials. Biol Psychol. 2009;81(1):1–8. doi: 10.1016/j.biopsycho.2008.12.004; 10.1016/j.biopsycho.2008.12.004. | |
Grimm O, Heinz A, Walter H, et al. Striatal response to reward anticipation: evidence for a systems-level intermediate phenotype for schizophrenia. JAMA Psychiatry. 2014;71(5):531–539. | |
Schloesser RJ, Huang J, Klein PS, Manji HK. Cellular plasticity cascades in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology. 2008;33(1):110–133. doi: 10.1038/sj.npp.1301575. | |
Drevets WC. Neuroimaging studies of mood disorders. Biol Psychiatry. 2000;48(8):813–829. | |
Post RM. Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev. 2007;31(6):858–873. doi: 10.1016/j.neubiorev.2007.04.003. | |
Savitz JB, Price JL, Drevets WC. Neuropathological and neuromorphometric abnormalities in bipolar disorder: view from the medial prefrontal cortical network. Neurosci Biobehav Rev. 2014;42:132–147. doi: 10.1016/j.neubiorev.2014.02.008; 10.1016/j.neubiorev.2014.02.008. | |
Kato T, Shioiri T, Murashita J, Hamakawa H, Inubushi T, Takahashi S. Phosphorus-31 magnetic resonance spectroscopy and ventricular enlargement in bipolar disorder. Psychiatry Res. 1994;55(1):41–50. | |
Nasrallah HA, McCalley-Whitters M, Jacoby CG. Cortical atrophy in schizophrenia and mania: a comparative CT study. J Clin Psychiatry. 1982;43(11):439–441. | |
Pearlson GD, Garbacz DJ, Tompkins RH, et al. Clinical correlates of lateral ventricular enlargement in bipolar affective disorder. Am J Psychiatry. 1984;141(2):253–256. | |
Strakowski SM, DelBello MP, Zimmerman ME, et al. Ventricular and periventricular structural volumes in first- versus multiple-episode bipolar disorder. Am J Psychiatry. 2002;159(11):1841–1847. | |
Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK. Lithium-induced increase in human brain grey matter. Lancet. 2000;356(9237):1241–1242. | |
Sassi RB, Nicoletti M, Brambilla P, et al. Increased gray matter volume in lithium-treated bipolar disorder patients. Neurosci Lett. 2002;329(2):243–245. | |
Bearden CE, Thompson PM, Dalwani M, et al. Greater cortical gray matter density in lithium-treated patients with bipolar disorder. Biol Psychiatry. 2007;62(1):7–16. doi: 10.1016/j.biopsych.2006.10.027. | |
Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc Natl Acad Sci U S A. 1998;95(22):13290–13295. | |
Rajkowska G, Halaris A, Selemon LD. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry. 2001;49(9):741–752. | |
Benes FM, Vincent SL, Todtenkopf M. The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry. 2001;50(6):395–406. | |
Rajkowska G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry. 2000;48(8):766–777. | |
Cotter D, Mackay D, Chana G, Beasley C, Landau S, Everall IP. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb Cortex. 2002;12(4):386–394. | |
Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43(10):977–983. | |
Bowley MP, Drevets WC, Ongur D, Price JL. Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry. 2002;52(5):404–412. | |
Machado-Vieira R, Andreazza AC, Viale CI, et al. Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci Lett. 2007;421(1):33–36. | |
Dean B, Gray L, Scarr E. Regionally specific changes in levels of cortical S100beta in bipolar 1 disorder but not schizophrenia. Aust N Z J Psychiatry. 2006;40(3):217–224. doi: 10.1111/j.1440-1614.2006.01777.x. | |
Schroeter ML, Sacher J, Steiner J, Schoenknecht P, Mueller K. Serum S100B represents a new biomarker for mood disorders. Curr Drug Targets. 2013;14(11):1237–1248. | |
Schroeter ML, Abdul-Khaliq H, Sacher J, Steiner J, Blasig IE, Mueller K. Mood disorders are glial disorders: evidence from in vivo studies. Cardiovasc Psychiatry Neurol. 2010;2010:780645. doi: 10.1155/2010/780645; 10.1155/2010/780645. | |
Berk M, Dodd S, Kauer-Sant’anna M, et al. Dopamine dysregulation syndrome: implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr Scand Suppl. 2007;(434)(434):41–49. doi: 10.1111/j.1600-0447.2007.01058.x. | |
de Bartolomeis A, Buonaguro EF, Iasevoli F, Tomasetti C. The emerging role of dopamine-glutamate interaction and of the postsynaptic density in bipolar disorder pathophysiology: implications for treatment. J Psychopharmacol. 2014;28(6):505–526. doi: 10.1177/0269881114523864. | |
Rao JS, Kellom M, Reese EA, Rapoport SI, Kim HW. Dysregulated glutamate and dopamine transporters in postmortem frontal cortex from bipolar and schizophrenic patients. J Affect Disord. 2012;136(1–2):63–71. doi: 10.1016/j.jad.2011.08.017; 10.1016/j.jad.2011.08.017. | |
Anand A, Barkay G, Dzemidzic M, et al. Striatal dopamine transporter availability in unmedicated bipolar disorder. Bipolar Disord. 2011;13(4):406–413. doi: 10.1111/j.1399-5618.2011.00936.x; 10.1111/j.1399-5618.2011.00936.x. | |
Stork C, Renshaw PF. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psychiatry. 2005;10(10):900–919. | |
Yildiz-Yesiloglu A, Ankerst DP. Neurochemical alterations of the brain in bipolar disorder and their implications for pathophysiology: a systematic review of the in vivo proton magnetic resonance spectroscopy findings. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30(6):969–995. doi: 10.1016/j.pnpbp.2006.03.012. | |
Frye MA, Watzl J, Banakar S, et al. Increased anterior cingulate/medial prefrontal cortical glutamate and creatine in bipolar depression. Neuropsychopharmacology. 2007;32(12):2490–2499. | |
Ongur D, Jensen JE, Prescot AP, et al. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry. 2008;64(8):718–726. | |
Dager SR, Friedman SD, Parow A, et al. Brain metabolic alterations in medication-free patients with bipolar disorder. Arch Gen Psychiatry. 2004;61(5):450–458. | |
Auer DP, Putz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biol Psychiatry. 2000;47(4):305–313. | |
Eastwood SL, Harrison PJ. Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder. Biol Psychiatry. 2010;67(11):1010–1016. doi: 10.1016/j.biopsych.2009.12.004; 10.1016/j.biopsych.2009.12.004. | |
Shibuya-Tayoshi S, Tayoshi S, Sumitani S, Ueno S, Harada M, Ohmori T. Lithium effects on brain glutamatergic and GABAergic systems of healthy volunteers as measured by proton magnetic resonance spectroscopy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(1):249–256. doi: 10.1016/j.pnpbp.2007.08.015. | |
International Schizophrenia Consortium, Purcell SM, Wray NR, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185; 10.1038/nature08185. | |
Craddock N, Sklar P. Genetics of bipolar disorder. Lancet. 2013; 381(9878):1654–1662. doi: 10.1016/S0140-6736(13)60855-7; 10.1016/S0140-6736(13)60855-7. | |
Maletic V, Raison C. Integrated neurobiology of bipolar disorder. Front Psychiatry. 2014;5:98. doi: 10.3389/fpsyt.2014.00098; 10.3389/fpsyt.2014.00098. | |
Nurnberger JI Jr, Koller DL, Jung J, et al. Identification of pathways for bipolar disorder: a meta-analysis. JAMA Psychiatry. 2014;71(6):657–664. doi: 10.1001/jamapsychiatry.2014.176; 10.1001/jamapsychiatry.2014.176. | |
Bhat S, Dao DT, Terrillion CE, et al. CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol. 2012;99(1):1–14. doi: 10.1016/j.pneurobio.2012.06.001; 10.1016/j.pneurobio. 2012.06.001. | |
Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev. 2008;88(4):1407–1447. doi: 10.1152/physrev.00002.2008; 10.1152/physrev.00002.2008. | |
Soeiro-de-Souza MG, Bio DS, Dias VV, Vieta E, Machado-Vieira R, Moreno RA. The CACNA1C risk allele selectively impacts on executive function in bipolar type I disorder. Acta Psychiatr Scand. 2013;128(5):362–369. doi: 10.1111/acps.12073; 10.1111/acps. 12073. | |
Ruberto G, Vassos E, Lewis CM, et al. The cognitive impact of the ANK3 risk variant for bipolar disorder: initial evidence of selectivity to signal detection during sustained attention. PLoS One. 2011;6(1):e16671. doi: 10.1371/journal.pone.0016671; 10.1371/journal.pone. 0016671. | |
Cichon S, Muhleisen TW, Degenhardt FA, et al. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. 2011;88(3):372–381. doi: 10.1016/j.ajhg.2011.01.017; 10.1016/j.ajhg.2011.01.017. | |
Tiwary BK. The severity of mental disorders is linked to interaction among candidate genes. Integr Biol (Camb). 2012;4(9):1096–1101. doi: 10.1039/c2ib20066j; 10.1039/c2ib20066j. | |
Sodersten K, Palsson E, Ishima T, et al. Abnormality in serum levels of mature brain-derived neurotrophic factor (BDNF) and its precursor proBDNF in mood-stabilized patients with bipolar disorder: a study of two independent cohorts. J Affect Disord. 2014;160:1–9. doi: 10.1016/j.jad.2014.01.009; 10.1016/j.jad.2014.01.009. | |
Green EK, Raybould R, Macgregor S, et al. Genetic variation of brain-derived neurotrophic factor (BDNF) in bipolar disorder: case-control study of over 3000 individuals from the UK. Br J Psychiatry. 2006;188:21–25. doi: 10.1192/bjp.bp.105.009969. | |
Rybakowski JK, Borkowska A, Skibinska M, et al. Prefrontal cognition in schizophrenia and bipolar illness in relation to Val66Met polymorphism of the brain-derived neurotrophic factor gene. Psychiatry Clin Neurosci. 2006;60(1):70–76. doi: 10.1111/j.1440-1819. 2006.01462.x. | |
Vincze I, Perroud N, Buresi C, et al. Association between brain-derived neurotrophic factor gene and a severe form of bipolar disorder, but no interaction with the serotonin transporter gene. Bipolar Disord. 2008;10(5):580–587. doi: 10.1111/j.1399-5618.2008.00603.x; 10.1111/j.1399-5618.2008.00603.x. | |
Rotondo A, Mazzanti C, Dell’Osso L, et al. Catechol o-methyltransferase, serotonin transporter, and tryptophan hydroxylase gene polymorphisms in bipolar disorder patients with and without comorbid panic disorder. Am J Psychiatry. 2002;159(1):23–29. | |
Rybakowski JK, Suwalska A, Czerski PM, Dmitrzak-Weglarz M, Leszczynska-Rodziewicz A, Hauser J. Prophylactic effect of lithium in bipolar affective illness may be related to serotonin transporter genotype. Pharmacol Rep. 2005;57(1):124–127. | |
Ferreira Ade A, Neves FS, da Rocha FF, et al. The role of 5-HTTLPR polymorphism in antidepressant-associated mania in bipolar disorder. J Affect Disord. 2009;112(1–3):267–272. doi: 10.1016/j.jad.2008.04.012; 10.1016/j.jad.2008.04.012. | |
Geaghan M, Cairns MJ. MicroRNA and posttranscriptional dysregulation in psychiatry. Biol Psychiatry. 2015;78(4):231–239. | |
Sun E, Shi Y. MicroRNAs: small molecules with big roles in neurodevelopment and diseases. Exp Neurol. 2015;268:46–53. | |
Forero DA, van der Ven K, Callaerts P, Del-Favero J. miRNA genes and the brain: implications for psychiatric disorders. Hum Mutat. 2010; 31(11):1195–1204. | |
Kim J, Bartel DP. Allelic imbalance sequencing reveals that single-nucleotide polymorphisms frequently alter microRNA-directed repression. Nat Biotechnol. 2009;27(5):472–477. | |
Kim AH, Reimers M, Maher B, et al. MicroRNA expression profiling in the prefrontal cortex of individuals affected with schizophrenia and bipolar disorders. Schizophr Res. 2010;124(1–3):183–191. | |
Moreau MP, Bruse SE, David-Rus R, Buyske S, Brzustowicz LM. Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol Psychiatry. 2011;69(2):188–193. | |
Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34(5):1646–1652. | |
Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. | |
Wang Z, Zhang C, Huang J, et al. MiRNA-206 and BDNF genes interacted in bipolar I disorder. J Affect Disord. 2014;162:116–119. | |
Cheng LC, Pastrana E, Tavazoie M, Doetsch F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat Neurosci. 2009;12(4):399–408. | |
Yoo AS, Staahl BT, Chen L, Crabtree GR. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature. 2009;460(7255):642–646. | |
Rajasethupathy P, Fiumara F, Sheridan R, et al. Characterization of small RNAs in aplysia reveals a role for miR-124 in constraining synaptic plasticity through CREB. Neuron. 2009;63(6):803–817. | |
Shih WL, Kao CF, Chuang LC, Kuo PH. Incorporating information of microRNAs into pathway analysis in a genome-wide association study of bipolar disorder. Front Genet. 2012;3:293. | |
Zhou R, Yuan P, Wang Y, et al. Evidence for selective microRNAs and their effectors as common long-term targets for the actions of mood stabilizers. Neuropsychopharmacology. 2009;34(6):1395–1405. | |
Zhang Z, Convertini P, Shen M, et al. Valproic acid causes proteasomal degradation of DICER and influences miRNA expression. PLoS One. 2013;8(12):e82895. | |
Rong H, Liu TB, Yang KJ, et al. MicroRNA-134 plasma levels before and after treatment for bipolar mania. J Psychiatr Res. 2011;45(1):92–95. | |
Ben-Shachar D. The interplay between mitochondrial complex I, dopamine and Sp1 in schizophrenia. J Neural Transm. 2009;116(11):1383–1396. | |
Scola G, Kim HK, Young LT, Andreazza AC. A fresh look at complex I in microarray data: clues to understanding disease-specific mitochondrial alterations in bipolar disorder. Biol Psychiatry. 2013;73(2):e4–e5. | |
Hovatta I, Juhila J, Donner J. Oxidative stress in anxiety and comorbid disorders. Neurosci Res. 2010;68(4):261–275. | |
Xing M. Oxidative stress: A new risk factor for thyroid cancer. Endocr Relat Cancer. 2012;19(1):C7–C11. | |
Wang JF, Shao L, Sun X, Young LT. Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord. 2009;11(5):523–529. | |
Andreazza AC, Shao L, Wang JF, Young LT. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch Gen Psychiatry. 2010;67(4):360–368. | |
Che Y, Wang JF, Shao L, Young T. Oxidative damage to RNA but not DNA in the hippocampus of patients with major mental illness. J Psychiatry Neurosci. 2010;35(5):296–302. | |
Andreazza AC, Wang JF, Salmasi F, Shao L, Young LT. Specific subcellular changes in oxidative stress in prefrontal cortex from patients with bipolar disorder. J Neurochem. 2013;127(4):552–561. | |
Andreazza AC, Kauer-Sant’anna M, Frey BN, et al. Oxidative stress markers in bipolar disorder: a meta-analysis. J Affect Disord. 2008; 111(2–3):135–144. | |
Andreazza AC, Kapczinski F, Kauer-Sant’Anna M, et al. 3-nitrotyrosine and glutathione antioxidant system in patients in the early and late stages of bipolar disorder. J Psychiatry Neurosci. 2009;34(4):263–271. | |
Brown NC, Andreazza AC, Young LT. An updated meta-analysis of oxidative stress markers in bipolar disorder. Psychiatry Res. 2014; 218(1–2):61–68. | |
Kim HK, Chen W, Andreazza AC. The potential role of the NLRP3 inflammasome as a link between mitochondrial complex I dysfunction and inflammation in bipolar disorder. Neural Plast. 2015; 2015:408136. | |
Munkholm K, Brauner JV, Kessing LV, Vinberg M. Cytokines in bipolar disorder vs healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res. 2013;47(9):1119–1133. | |
Goldstein BI, Fagiolini A, Houck P, Kupfer DJ. Cardiovascular disease and hypertension among adults with bipolar I disorder in the united states. Bipolar Disord. 2009;11(6):657–662. | |
Drexhage RC, Knijff EM, Padmos RC, et al. The mononuclear phagocyte system and its cytokine inflammatory networks in schizophrenia and bipolar disorder. Expert Rev Neurother. 2010;10(1):59–76. | |
Hope S, Dieset I, Agartz I, et al. Affective symptoms are associated with markers of inflammation and immune activation in bipolar disorders but not in schizophrenia. J Psychiatr Res. 2011;45(12):1608–1616. | |
Acheson A, Conover JC, Fandl JP, et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374(6521):450–453. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.