Back to Journals » Clinical and Experimental Gastroenterology » Volume 11
Bioavailability of dexlansoprazole delayed-release capsule granules when administered via nasogastric tube or orally via syringe
Authors Kukulka M, Nudurupati S, Perez MC
Received 1 April 2017
Accepted for publication 28 July 2018
Published 5 October 2018 Volume 2018:11 Pages 381—389
DOI https://doi.org/10.2147/CEG.S138580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Andreas M. Kaiser
Michael Kukulka,1 Sai Nudurupati,2 Maria Claudia Perez3
1Department of Clinical Pharmacology, Takeda Development Center Americas, Inc., Deerfield, IL, USA; 2Department of Analytical Sciences, Takeda Development Center Americas, Inc., Deerfield, IL, USA; 3Department of Clinical Science, Takeda Development Center Americas, Inc., Deerfield, IL, USA
Objective: To assess the effect of route of administration on the bioavailability of dexlansoprazole 60 mg delayed-release capsule granules.
Methods: One open-label, Phase I, single-dose, 3-period crossover study was conducted in healthy adults. The bioavailability of Dexilant® (dexlansoprazole) after dexlansoprazole capsule granules were mixed with water and administered via 16 French nasogastric tube or orally via syringe was compared to administration of the intact capsule in the fasted state, swallowed with water. Blood samples were collected before and after dosing to determine dexlansoprazole pharmacokinetic parameter estimates and plasma concentrations.
Results: Similar values for area under the plasma concentration–time curve and observed maximum plasma concentration were achieved when the dexlansoprazole 60 mg capsule was administered as the intact capsule or when the granules were mixed with water and administered via nasogastric tube or orally via syringe. The primary endpoints of maximum plasma concentration and area under the plasma concentration–time curve demonstrated bioequivalence when assessing these alternative routes of administration. Most adverse events were rated as mild and were comparable irrespective of administration route.
Conclusion: Systemic exposure to dexlansoprazole was equivalent regardless of administration route. The dexlansoprazole capsule was well tolerated.
Keywords: TAK-390MR, dexlansoprazole, dual delayed release, pharmacokinetics, proton pump inhibitor
Introduction
Gastroesophageal reflux disease (GERD) is a common digestive disorder characterized by the reflux of stomach contents into or beyond the esophagus most notably associated with symptoms of heartburn and acid regurgitation.1 A systematic review of the epidemiology of GERD reports a prevalence rate of up to 20% in North American adults.2 Drug therapy options include acid-suppressing agents including antacids, histamine 2-receptor antagonists, and proton pump inhibitors (PPIs). The treatment of choice for GERD is PPIs for relieving symptoms and healing of esophageal erosions.1
Suppression of acid secretion by PPIs occurs via covalent binding to the hydrogen–potassium adenosine triphosphatase pump in the gastric parietal cell, resulting in inhibition of the final step of acid production.3 Most PPIs are acid-labile and are formulated as delayed-release products to protect the active moiety from degradation by exposure to stomach acid.4 These delayed-release formulations are designed to provide a single release of drug in the proximal small intestine,5 where absorption of the drug occurs.6
Dexlansoprazole is a PPI that is approved for use in adult patients and in pediatric patients 12–17 years old.7 Unlike other delayed-release PPI formulations, the dexlansoprazole capsule has a pH-dependent dual delayed-release mechanism that releases drug in the proximal and distal portions of the small intestine.6,8 After dosing with the dual delayed-release formulation, the first release of dexlansoprazole takes place within 1–2 hours, with a second release occurring within 4–5 hours.9 This approach allows prolonged drug exposure and an extended duration of acid suppression with once-daily dosing.8,10 Dexlansoprazole 60 mg delayed-release capsules once daily are approved for healing of erosive esophagitis (EE), and dexlansoprazole 30 mg delayed-release capsules are indicated for treatment of symptomatic nonerosive GERD and maintenance of healed EE and relief of heartburn for up to 6 months.7 The pharmacokinetic, pharmacodynamic, efficacy, and safety profiles of dexlansoprazole capsules following administration of doses of 30, 60, and 90 mg have been extensively studied in more than 5,000 adults in completed, randomized, double-blind, controlled clinical studies.6,8,11–14
Most PPIs in capsule or tablet form are not ideal for patients with difficulty swallowing (dysphagia) since they are intended to be swallowed intact.15,16 The prevalence of dysphagia is substantially higher in patients with GERD and increases with severity of EE.16–18 Dysphagia is common in patients with neurological or muscular disorders, as well as other conditions that might impede esophageal movement.19–23 Short-term use of a nasogastric (NG) tube is indicated when dysphagia leads to insufficient oral intake and malnutrition. Conditions that may require NG intubation include multiple sclerosis, liver disease, inflammatory bowel disease, cystic fibrosis, and renal disease.24,25
Although the dexlansoprazole capsule can be opened and the granules sprinkled on a tablespoon of applesauce to aid in swallowing,7,26 this method of administration may not be adequate for all patients. Consequently, we explored methods of administering dexlansoprazole capsule granules that would allow flexibility in dosing patients with difficulty swallowing the intact capsule. To that end, we evaluated the pharmacokinetics and safety of the 60 mg dexlansoprazole capsule when the capsule is opened, the granules are mixed with water, and the mixture is administered via an NG tube or orally via syringe.
Methods
Subject selection
To be eligible for the study, participants were healthy adults between ages 18 and 55, weight ≥50 kg, and body mass index ≥18 and ≤30 kg/m2. Participants were ineligible if they had a history of malignant disease or any clinically significant hematological, neurological, cardiovascular, pulmonary, hepatic, renal, metabolic, gastrointestinal, urological, immunological, endocrine, or psychiatric disorder. Potential participants who had recently received agents containing nicotine, caffeine, xanthine, or grapefruit products that could alter hepatic or renal clearance, or if they had evidence of either drug or alcohol consumption, were ineligible, similar to previous study.27 Further exclusion criteria included prior use of dexlansoprazole, lansoprazole, or any investigational compound for any indication within 30 days before check-in (day –1 of the first treatment period), or known hypersensitivity to any constituent of dexlansoprazole capsules or other PPIs. Male and female participants of childbearing potential had to agree to use contraception throughout the study and for the 30 days after the last dose. Participants failing to meet any entry criteria after randomization were removed at the investigator’s discretion and not replaced. Adverse events (AEs) could result in participant discontinuation in the study.
Study design
This Phase I, open-label study was conducted at Covance Madison Clinical Research Unit (Madison, WI, USA) between December 2011 and June 2012. The study required a screening period of up to 28 days. After the screening visit, eligible subjects returned to the study site on day –1 for baseline evaluations including an assessment of concomitant medications and AEs, a physical examination with vital signs, clinical laboratory tests, and a screen for illicit drugs of abuse and alcohol.
Participants in the study were randomized in a 1:1:1 ratio to 3 possible treatment sequences, each including a treatment period in which the participant received a single oral dose of Dexilant® (dexlansoprazole, Takeda Pharmaceutical Company Limited, Osaka, Japan) 60 mg capsule as follows: granules mixed with water and administered via NG tube (test regimen A), granules mixed with water and administered via oral syringe (test regimen B), or administered intact (reference regimen C). Participants were confined to the study site from the day before dose administration until all study procedures had been completed on day 2 similar to previous study.27 Predosing, participants were to have fasted for ≥10 hours, and participants continued fasting for 4 hours after dosing. Participants were able to drink water at any time outside of the 2-hour window ranging from 1-hour pre- and postdosing. On day 1 of each treatment period, participants were administered a single dose of dexlansoprazole 60 mg capsule according to the dosing regimen per the treatment sequence. Participants were discharged from the study site for a washout period of at least 5 days before subsequent treatment period. Any ongoing or emergent AEs were monitored by a follow-up phone call 5–10 days after the final dose of study drug.
Alternative routes of administration – test regimens
The bioavailability of dexlansoprazole capsule granules for test regimens A and B was examined when administered via a 16 French NG tube directly to the stomach or orally via syringe, respectively. In both regimens, the intact capsule was opened and the granules were added to a container with 20 mL of water. The granule/water mix was drawn up into a 60 mL catheter-tip syringe. In test regimen A, the syringe was attached to a 16 French NG tube, and the granule/water mix was administered directly into the participant’s stomach via the NG tube. In test regimen B, the granule/water mix was administered directly into the participant’s mouth for oral administration. Following initial administration, an additional 10 mL of water was used to rinse the dosing syringe and/or NG tube in each regimen. This rinse was performed a second time, and the water used for the rinse was administered to participants after each rinse similar to previous study.27
The bioavailability of administration of dexlansoprazole via NG tube and/or orally via syringe was compared to administration of the intact dexlansoprazole 60 mg capsule with 240 mL of water.
Pharmacokinetic evaluation
Blood samples (3 mL) were collected at ≤30 minutes before treatment and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 16, and 24 hours after dose administration per previously published protocol.27 Briefly, sample collection, processing, and evaluation with liquid chromatography-tandem mass spectrometry was done by PPD, Inc. (Middleton, WI, USA) as previously described.27 Plasma concentrations below the lower limit of quantitation (validated concentration range, 2.00–2,000 ng/mL) were set to 0 for pharmacokinetic analysis and summary statistics according to previous methods.27
Individual pharmacokinetic parameters were estimated using Phoenix WinNonlin Version 6.2.1 (Certara, Princeton, NJ, USA) as previously described.27 Briefly, primary pharmacokinetic endpoints included the maximum observed drug concentration in the plasma (Cmax) and the amount of systemic drug exposure, determined by the area under the plasma concentration–time curve (AUC) calculated using the linear trapezoidal rule.27 The AUC values presented were measured from time 0 extrapolated to infinity (AUC∞).27 Additional pharmacokinetic parameters included the rate of absorption (tmax), defined as the time to reach the first occurrence of Cmax, the apparent clearance after extravascular administration (CL/F), the terminal elimination half-life (t1/2), and the apparent volume of distribution after extravascular administration (Vz/F).
Since dexlansoprazole is metabolized by the polymorphic cytochrome P450 (CYP) 2C19 enzyme, increased plasma concentrations of dexlansoprazole may be observed in CYP2C19 poor metabolizers.28,29 As a result, a single blood sample was collected from all participants for the determination of the subject’s CYP2C19 metabolizer status, as has been previously described.27 Briefly, we screened for the 9 most common phenotypically relevant variations within the cytochrome CYP2C19 gene using the AutoGenomics INFINITI® CYP450-2C19+ Assay (AutoGenomics, Inc., Vista, CA, USA). Results were assessed using nomenclature outlined by the Human CYP/CYP450 Allele Nomenclature Committee.
Safety evaluations
Assessments of safety included AEs, clinical laboratory tests (serum chemistry, hematology, and urinalysis), vital signs, physical examinations, and electrocardiograms (ECGs). A treatment-emergent AE (TEAE) was defined as an AE that started or worsened any time after the first dose of study drug until 30 days after the last dose of study drug as previously described.27 AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) Version 15.0 (MedDRA, McLean, VA, USA).
Statistical analyses
A sample size of 51 participants (17 in each treatment sequence group) was planned for this study. This size provided at least 91% probability of concluding equivalence on dexlansoprazole Cmax between 2 regimens if the true difference between dexlansoprazole Cmax central values from 2 regimens was no more than 5%. The power for concluding equivalence on dexlansoprazole AUC between 2 regimens was expected to be >95%. This sample size was based on the intrasubject variance of 0.071 for log (Cmax) from previous clinical studies conducted with the dexlansoprazole capsule.
Descriptive statistics were calculated for the pharmacokinetic parameters. Because some participants did not complete all 3 regimens, separate descriptive statistics were calculated for: 1) participants who completed both the NG tube and the intact capsule regimens and 2) participants who completed both the syringe and the intact capsule regimens.
Individual pharmacokinetic parameters were analyzed with SAS Version 9.2 software (SAS Institute Inc., Cary, NC, USA) as previously described.27 Briefly, values for tmax and log-transformed Cmax and AUC∞ were analyzed using the analysis of variance model with sequence, period, and regimen as fixed effects and the study participant nested within sequence as a random effect. Pharmacokinetic parameters from participants who had data for the reference regimen and at least 1 of the test regimens were included in the statistical analyses.
Statistical comparisons evaluated the point estimate and 90% CIs for the Cmax and AUC∞ central value ratios of the test and reference regimens. Bioequivalence between regimens was declared if the CIs fell within the range of 0.80–1.25 for both Cmax and AUC∞.30
In the crossover study design, each participant received all regimens. While genetic variations between individuals, including CYP polymorphisms, can affect bioavailability estimates, the basic bioavailability/bioequivalence study design used here compared the formulations against each other within each individual. Since the bioavailability following administration of each regimen would be affected equally within an individual, a participant’s CYP2C19 genotype would not affect the statistical assessments of equivalence. Therefore, no formal statistical analyses were conducted based on CYP2C19 genotype.
Safety analyses
All subjects who received at least 1 dose of study drug were included in the safety analyses. Descriptive statistics were used to summarize safety assessments (TEAEs, clinical laboratory evaluations, vital signs, ECGs, and physical examination results) by regimen.
Ethics statement
The study was conducted according to established international and local guidance in accordance with the Declaration of Helsinki as reported elsewhere.27 The study was conducted at 1 study site, and the study protocol was approved by the Institutional Research Board affiliated to that site: Independent Investigational Review Board, Inc., 6,738 West Sunrise Boulevard, Suite 102, Plantation, FL 33313. Written informed consent was obtained from the participant before initiation of any study procedures. The study was designed according to the “Food and Drug Administration of the United States (FDA) Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products – General Considerations.”31 Since Phase I drug trials are excluded from US Food and Drug Administration Amendments Act 801 registration requirements, trial registration was not required for this study at the time of conduct.32
Results
Study population
Of the 62 participants enrolled into the study, 45 completed all 3 study drug regimens. Overall, enrolled participants were primarily male (68%), white (81%), and of non-Hispanic ethnicity (98%). Age ranged from 18 to 54 years, inclusive, with a median of 30.5 years. Mean body mass index values were comparable across all of the treatment sequence groups. None of the participants were current smokers, and the majority (73%) had never smoked. At the screening visit, 82% of the participants reported current alcohol consumption, and 77% reported caffeine consumption (Table 1).
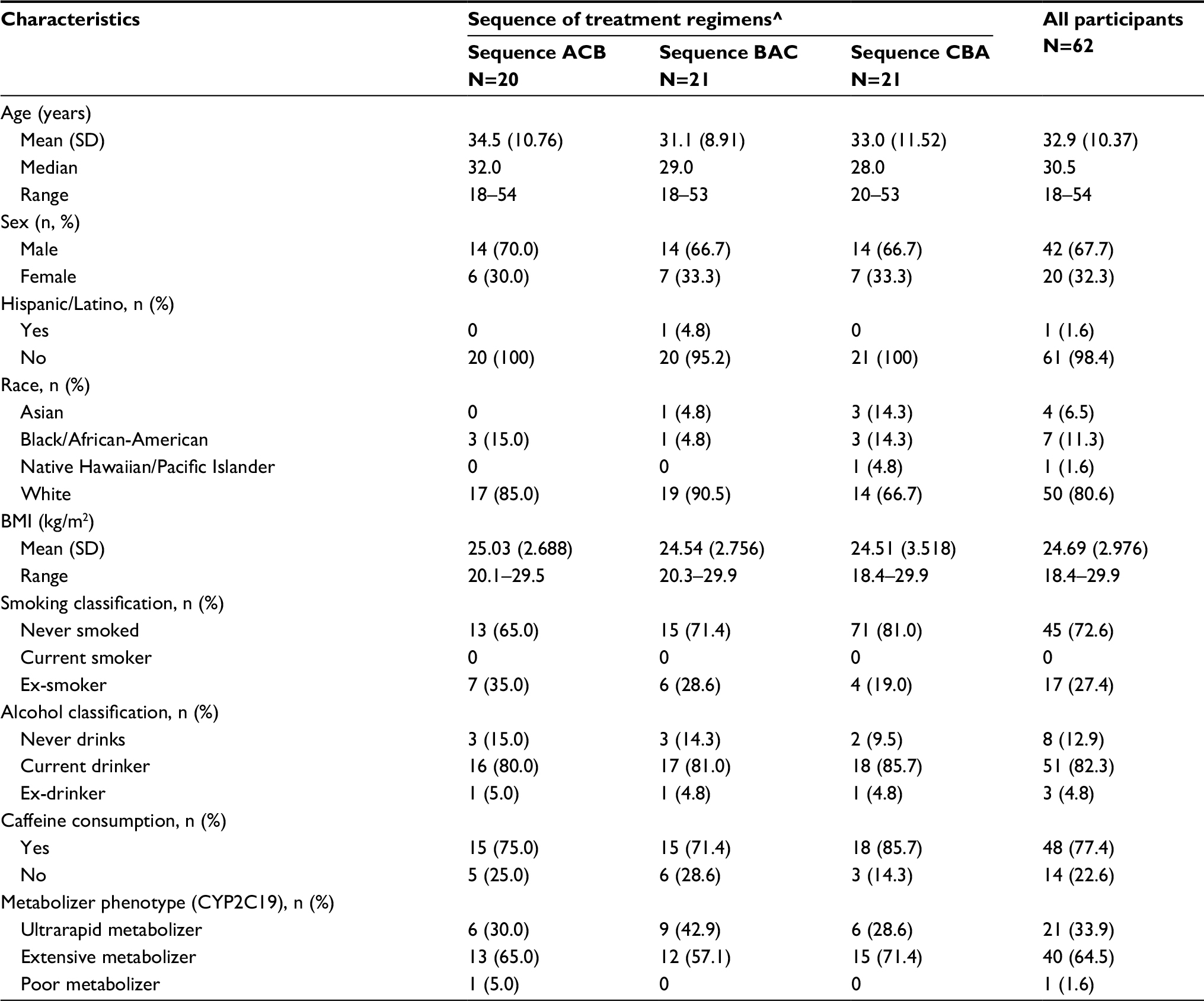 | Table 1 Subject demographics and other baseline characteristics Notes: ^Regimen A: dexlansoprazole 60 mg capsule granules mixed with water and administered via NG tube (Test). Regimen B: dexlansoprazole 60 mg capsule granules mixed with water and administered orally via syringe (Test). Regimen C: dexlansoprazole 60 mg capsule, administered intact (Reference). Abbreviations: BMI, body mass index; CYP2C19, cytochrome P450 2C19 enzyme; NG, nasogastric. |
The primary reason for discontinuing the study drug was inability to tolerate/place the NG tube in 8 of the 17 subjects who discontinued; the other discontinuations were due to loss to follow-up (1 subject), voluntary withdrawal (1 subject), AE (1 subject), and “other” (not related to study drug administration, in 6 subjects). Given the sequential crossover study design, once these discontinuations occurred, they affected the evaluable subject numbers for the subsequent protocol segments. Dosing errors led to the exclusion of 2 participants from the descriptive statistics for the plasma concentrations and pharmacokinetic parameters for regimen A for 1 participant and regimens A or B for the other.
Pharmacokinetic results
Following a single dose of dexlansoprazole 60 mg as an aqueous mixture of capsule granules administered via an NG tube or orally via syringe, or as an intact capsule administered with water, the mean plasma concentration–time profiles were similar regardless of regimen and reflect the dual delayed-release characteristics of the 2 granule types within the dexlansoprazole capsule (Figure 1).
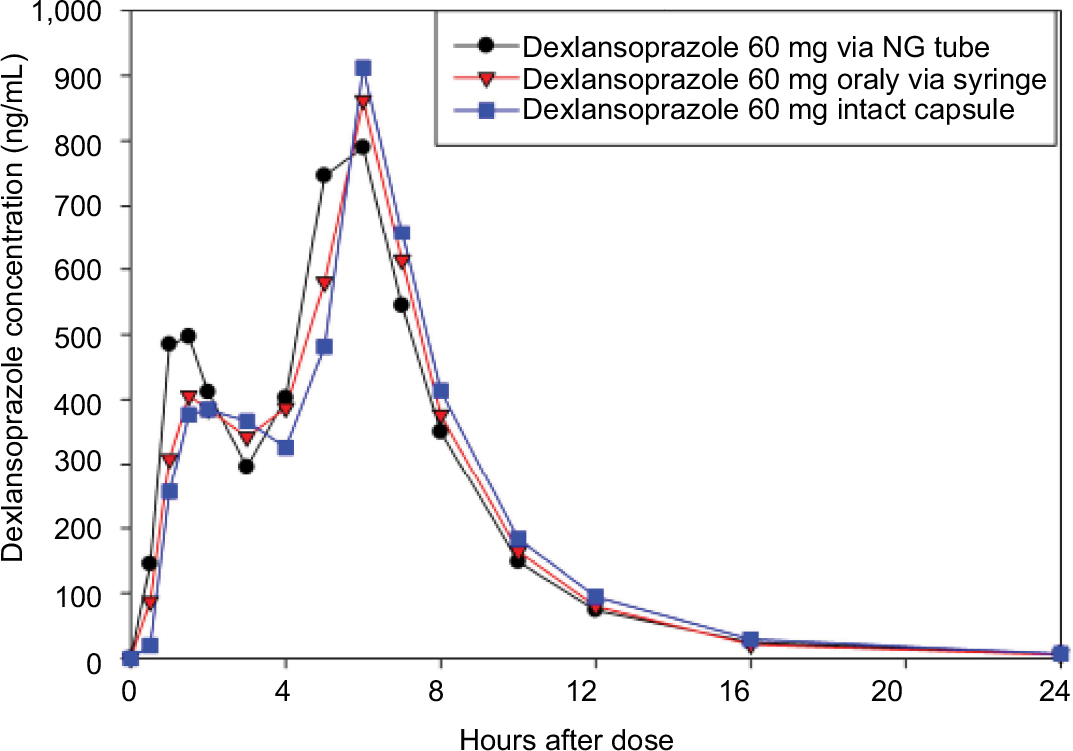 | Figure 1 Mean dexlansoprazole plasma concentration–time profiles (linear format) following a single dose of 60 mg dexlansoprazole capsule as the intact capsule or as granules mixed with water and administered via NG tube or orally via syringe. Abbreviation: NG, nasogastric. |
Following NG tube administration of dexlansoprazole as an aqueous mixture of capsule granules, the absorption of dexlansoprazole was slightly faster than from the intact capsule, with median tmax values of approximately 5 hours for the NG tube administration and 6 hours for the capsule. Dexlansoprazole mean Cmax following NG tube administration (997 ng/mL) was similar to the Cmax value for the capsule (1,083 ng/mL). As measured by dexlansoprazole AUC∞, the mean systemic exposure to dexlansoprazole following NG tube administration was also similar to the capsule. The mean AUC∞ value for dexlansoprazole following NG tube administration was 4,842 ng·h/mL, and was 5,006 ng·h/mL for the capsule. The mean dexlansoprazole t1/2, CL/F, and Vz/F values were similar for the 2 regimens. The descriptive statistics for the pharmacokinetic parameters from subjects who completed the NG tube and intact capsule regimens are presented in Table 2, and plasma concentration–time plots are shown in Figure S1.
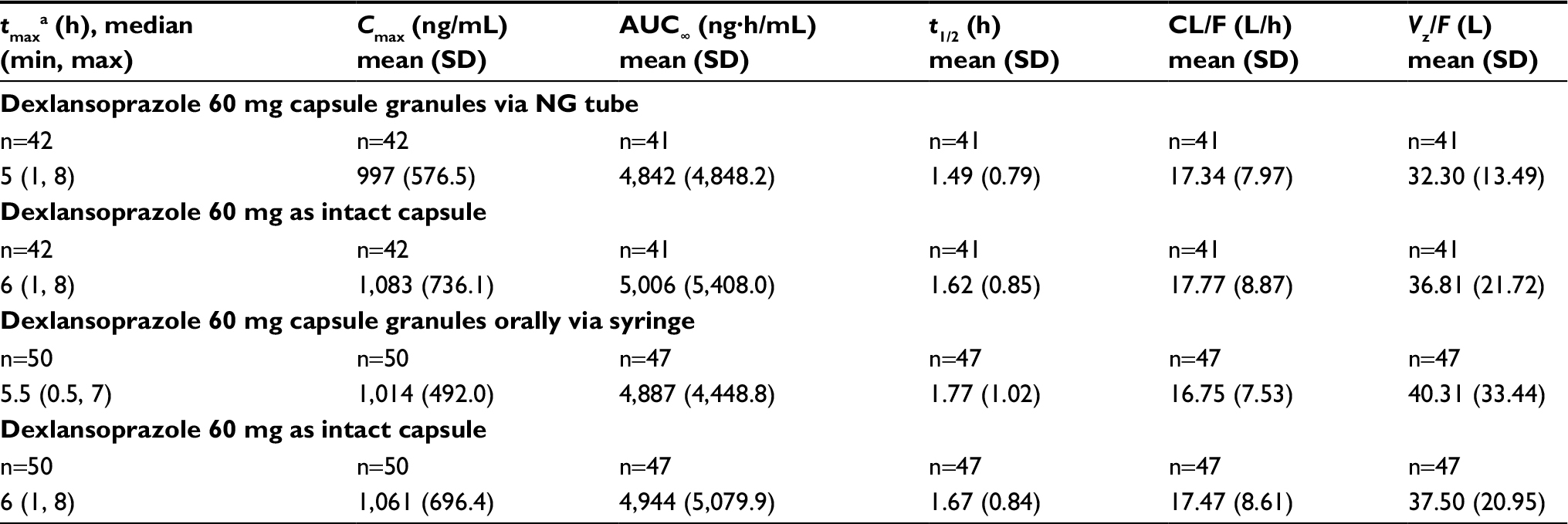 | Table 2 Dexlansoprazole pharmacokinetic parameters following administration of dexlansoprazole 60 mg as an aqueous mixture of capsule granules via an NG tube, orally by syringe, or as the intact capsule to healthy participants Notes: aMedian (minimum–maximum) reported for tmax. Because of variability in the terminal phase of the plasma concentration–time curve, the terminal elimination rate constant could not be determined for some subjects, and therefore, the pharmacokinetic parameters that use this constant in their calculations (ie, t1/2, AUC∞, CL/F, and Vz/F) could not be estimated. Abbreviations: AUC∞, area under the plasma concentration–time curve from time 0 to infinity; CL/F, apparent clearance after oral administration; Cmax, maximum observed concentration; NG, nasogastric; t1/2, terminal elimination half-life; tmax, time to first occurrence of Cmax; Vz/F, apparent volume of distribution after oral administration. |
Following administration of dexlansoprazole as an aqueous mixture of granules delivered orally via syringe, the absorption of dexlansoprazole was slightly faster than that for the capsule, with median tmax values of approximately 5.5 hours for the oral syringe administration and 6 hours for the capsule. Dexlansoprazole mean Cmax following syringe administration (1,014 ng/mL) was similar to the Cmax value for administration of the intact capsule (1,061 ng/mL). As measured by dexlansoprazole AUC∞, the mean systemic exposure to dexlansoprazole following oral syringe administration was also similar to the capsule. The mean AUC∞ value for dexlansoprazole following oral syringe administration was 4,887 ng·h/mL, and was 4,944 ng·h/mL for the capsule. AUC from time 0 to the time of the last quantifiable concentration closely matched the AUC∞ values for both treatment regimens. The mean dexlansoprazole t1/2, CL/F, and Vz/F values were similar for the 2 regimens. The descriptive statistics for the pharmacokinetic parameters from subjects who completed the syringe and intact-capsule regimens are presented in Table 2, and plasma concentration–time plots are shown in Figure S2.
The CYP2C19 genotype was determined for 62 participants. Of these, 32 participants were homozygous extensive metabolizers, 8 participants were heterozygous extensive metabolizers, 21 participants were CYP2C19 ultrarapid metabolizers, and 1 participant was a CYP2C19 poor metabolizer. As expected, the poor metabolizer participant had the highest plasma dexlansoprazole AUC∞ values in each regimen.
The 90% CIs for the relative bioavailability of central value ratios between dexlansoprazole 60 mg from an aqueous mixture of granules administered via an NG tube or orally via syringe relative to administration of the intact 60 mg capsule for dexlansoprazole Cmax and AUC∞ were contained within the bioequivalence range of 0.80–1.25. The bioavailability estimates indicated that dexlansoprazole Cmax and AUC∞ from the NG tube and oral syringe regimens were equivalent to the Cmax and AUC∞ from the intact capsule administration (Table 3).
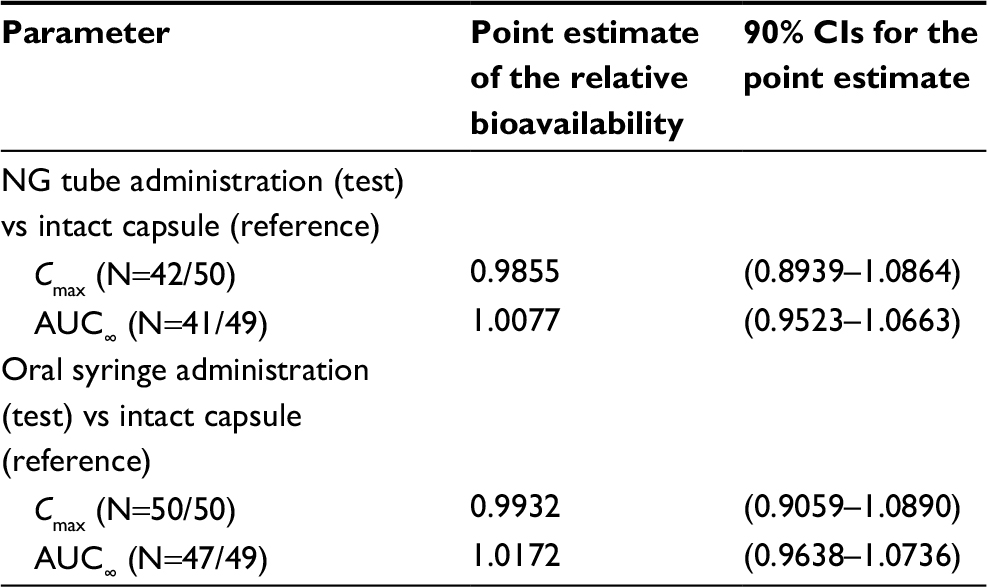 | Table 3 Statistical comparisons of pharmacokinetic parameter estimates for dexlansoprazole following administration of dexlansoprazole 60 mg as an aqueous mixture of capsule granules via NG tube or orally via syringe, or as the intact capsule to healthy participants Notes: N = (Test/Reference). The participants with valid estimates for intact capsule regimen and at least 1 other regimen were included in the analysis for that parameter. Abbreviations: AUC∞, area under the plasma concentration–time curve from time 0 to infinity; Cmax, maximum observed concentration; NG, nasogastric. |
Safety results
A total of 19 AEs were reported for 12 (24.5%) participants during NG tube administration, 22 events were reported for 10 (16.7%) participants during oral syringe administration, and 19 AEs were reported for 12 (23.1%) participants during intact capsule administration (Table 4). Most AEs were not related to study drug, and the majority were mild; 2 events of moderate severity were reported, and no AEs were classified as severe. One participant was discontinued from study drug following the oral syringe treatment regimen because of an AE (otitis externa); this was not considered to be related to study drug. No clinically important findings were noted in the physical examination, safety clinical laboratory, vital sign, or ECG data. No serious adverse effects or deaths were reported.
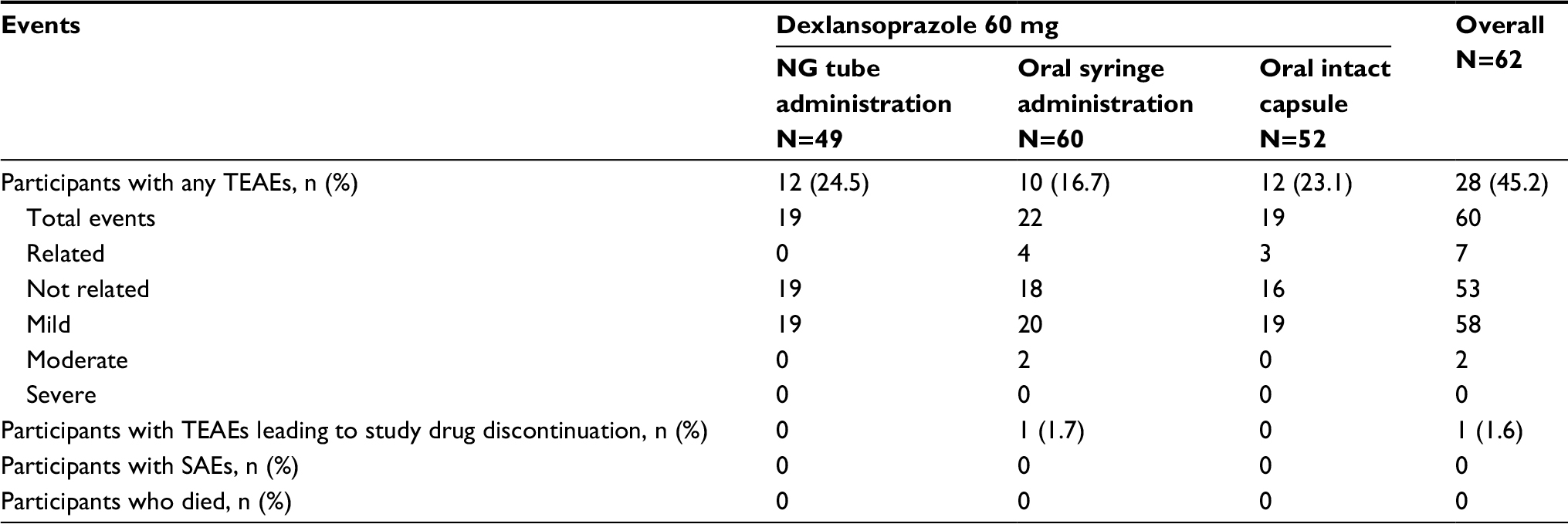 | Table 4 TEAEs, including SAEs Notes: A TEAE was defined as an AE whose date of onset occurred after the first dose of study drug and within 30 days after the last dose of study drug. Percentages were based on the number of participants in the safety set for each treatment group. Medical Dictionary for Regulatory Activities (MedDRA) Version 15.0 was used for coding AEs. Abbreviations: AE, adverse event; NG, nasogastric; SAE, serious adverse events; TEAE, treatment-emergent adverse event. |
Discussion
This study was designed to investigate the bioavailability of dexlansoprazole after administration via routes appropriate for patients for whom swallowing capsules is not feasible. Here, administration of dexlansoprazole by dispersing the granules in water and administering via 16 French NG tube or oral syringe was determined to be bioequivalent to administration of the intact capsule. The equivalent dexlansoprazole plasma exposures following the alternative routes of administration evaluated in the current study suggest similar levels of drug in the body, making dispersal of the granules in water a suitable alternative administration for patients unable to receive oral therapy. NG tube may be required for hospitalized patients with severe dysphagia as well as for traumatic medical events, severe malnutrition, or a range of neurological disorders.24 In inpatient settings, oral syringes are the current apparatus of choice for administering oral liquid medications.33 The granules contained in the dexlansoprazole capsule can be dispersed in water and administered via an NG tube (size 16 French or larger) or orally via syringe.
Conclusion
The administration of dexlansoprazole capsule granules via NG tube or via syringe is approved for the same indications as administration of the intact capsule: 60 mg for the healing of all grades of EE and 30 mg for maintaining healing of EE and relief of heartburn, and treating heartburn associated with symptomatic nonerosive GERD.7 The equivalence of systemic exposure achieved in this study between oral capsule ingestion and NG tube or oral syringe supports the use of these additional dosing pathways in patients for whom swallowing capsules is compromised.
Data sharing statement
All authors had access to the data and vouch for the veracity and completeness of the data and the data analysis.
Acknowledgments
Funding for this study was provided by Takeda Development Center Americas, Inc. The results of this study were presented at Digestive Disease Week, May 2013. The authors thank Dr Michael Cwik, an employee of Takeda, and the staff of PPD, for conducting the bioanalytical portion of the study. The authors also thank Ms Kelly Hanna for managing the conduct of the study.
Author contributions
MCP, SN, and MK contributed to the conception and design of the study. MK and SN contributed to the acquisition and interpretation of the data. SN performed the statistical analysis. MK, SN, and MCP contributed to the drafting and critical review of the article and approved the final version of the article before submission. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
All authors were employees of Takeda Development Center Americas, Inc., at the time of manuscript development; Michael Kukulka and Sai Nudurupati are no longer affiliated with Takeda Development Center Americas, Inc. The authors report no other conflicts of interest in this work.
References
Katz PO, Gerson LB, Vela MF. Guidelines for the diagnosis and management of gastroesophageal reflux disease. Am J Gastroenterol. 2013;108(3):308–328. | ||
Dent J, El-Serag HB, Wallander MA, Johansson S. Epidemiology of gastro-oesophageal reflux disease: a systematic review. Gut. 2005;54(5):710–717. | ||
Shin JM, Sachs G. Pharmacology of proton pump inhibitors. Curr Gastroenterol Rep. 2008;10(6):528–534. | ||
Howden CW. Review article: immediate-release proton-pump inhibitor therapy – potential advantages. Aliment Pharmacol Ther. 2005; 22(Suppl 3):25–30. | ||
Emerson CR, Marzella N. Dexlansoprazole: a proton pump inhibitor with a dual delayed-release system. Clin Ther. 2010;32(9):1578–1596. | ||
Metz DC, Vakily M, Dixit T, Mulford D. Review article: dual delayed release formulation of dexlansoprazole MR, a novel approach to overcome the limitations of conventional single release proton pump inhibitor therapy. Aliment Pharmacol Ther. 2009;29(9):928–937. | ||
Dexilant® (dexlansoprazole) delayed-release capsules [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2016. Available from: http://general.takedapharm.com/content/file.aspx?filetypecode=DEXILANTPI&cacheRandomizer=b1b7fc2b-e156-45b4-aa38-98b0bc2d4ec1. Accessed April 6, 2017. | ||
Vakily M, Zhang W, Wu J, Atkinson SN, Mulford D. Pharmacokinetics and pharmacodynamics of a known active PPI with a novel Dual Delayed Release technology, dexlansoprazole MR: a combined analysis of randomized controlled clinical trials. Curr Med Res Opin. 2009;25(3):627–638. | ||
Lee RD, Vakily M, Mulford D, Wu J, Atkinson SN. Clinical trial: the effect and timing of food on the pharmacokinetics and pharmacodynamics of dexlansoprazole MR, a novel dual delayed release formulation of a proton pump inhibitor – evidence for dosing flexibility. Aliment Pharmacol Ther. 2009;29(8):824–833. | ||
Kukulka M, Eisenberg C, Nudurupati S. Comparator pH study to evaluate the single-dose pharmacodynamics of dual delayed-release dexlansoprazole 60 mg and delayed-release esomeprazole 40 mg. Clin Exp Gastroenterol. 2011;4(4):213–220. | ||
Fass R, Johnson DA, Orr WC, et al. The effect of dexlansoprazole MR on nocturnal heartburn and GERD-related sleep disturbances in patients with symptomatic GERD. Am J Gastroenterol. 2011;106(3):421–431. | ||
Howden CW, Larsen LM, Perez MC, Palmer R, Atkinson SN. Clinical trial: efficacy and safety of dexlansoprazole MR 60 and 90 mg in healed erosive oesophagitis – maintenance of healing and symptom relief. Aliment Pharmacol Ther. 2009;30(9):895–907. | ||
Peura DA, Metz DC, Dabholkar AH, Paris MM, Yu P, Atkinson SN. Safety profile of dexlansoprazole MR, a proton pump inhibitor with a novel dual delayed release formulation: global clinical trial experience. Aliment Pharmacol Ther. 2009;30(10):1010–1021. | ||
Wittbrodt ET, Baum C, Peura DA. Delayed release dexlansoprazole in the treatment of GERD and erosive esophagitis. Clin Exp Gastroenterol. 2009;2:117–128. | ||
Liu F, Ranmal S, Batchelor HK, et al. Patient-centred pharmaceutical design to improve acceptability of medicines: similarities and differences in paediatric and geriatric populations. Drugs. 2014;74(16):1871–1889. | ||
Cho SY, Choung RS, Saito YA, et al. Prevalence and risk factors for dysphagia: a USA community study. Neurogastroenterol Motil. 2015;27(2):212–219. | ||
Locke GR 3rd, Talley NJ, Fett SL, Zinsmeister AR, Melton LJ 3rd. Prevalence and clinical spectrum of gastroesophageal reflux: a population-based study in Olmsted County, Minnesota. Gastroenterology. 1997;112(5):1448–1456. | ||
Vakil NB, Traxler B, Levine D. Dysphagia in patients with erosive esophagitis: prevalence, severity, and response to proton pump inhibitor treatment. Clin Gastroenterol Hepatol. 2004;2(8):665–668. | ||
Daniels SK. Neurological disorders affecting oral, pharyngeal swallowing. GI Motil Online. 2006. Available from: https://www.nature.com/gimo/contents/pt1/full/gimo34.html. Accessed August 29, 2018. | ||
Jaradeh S. Muscle disorders affecting oral and pharyngeal swallowing. GI Motil Online. 2006. Available from: https://www.nature.com/gimo/contents/pt1/full/gimo35.html. Accessed August 29, 2018. | ||
Sheehan NJ. Dysphagia and other manifestations of oesophageal involvement in the musculoskeletal diseases. Rheumatology (Oxford). 2008;47(6):746–752. | ||
Sonies BC, Dalakas MC. Dysphagia in patients with the post-polio syndrome. N Engl J Med. 1991;324(17):1162–1167. | ||
Yan BM, Shaffer EA. Eosinophilic esophagitis: a newly established cause of dysphagia. World J Gastroenterol. 2006;12(15):2328–2334. | ||
Ciocon JO. Indications for tube feedings in elderly patients. Dysphagia. 1990;5(1):1–5. | ||
Stroud M, Duncan H, Nightingale J; British Society of Gastroenterology. Guidelines for enteral feeding in adult hospital patients. Gut. 2003;52(Suppl 7):vii1–vii12. | ||
Czerniak R, Vakily M, Wu J. TAK-390MR, a Novel Dual Delayed Release Formulation of a PPI, is Bioequivalent When Administered as Granules Sprinkled Over Applesauce or as an Intact Capsule. Paper presented at: 73rd Annual Scientific Meeting of the American College of Gastroenterology; October 3–8; 2008; Orlando, FL. | ||
Kukulka M, Nudurupati S, Perez MC. Pharmacokinetics and safety of dexlansoprazole MR in pediatric patients with symptomatic gastroesophageal reflux disease. Clin Exp Gastroenterol. 2014;7:461–471. | ||
Aslam N, Wright R. Dexlansoprazole MR. Expert Opin Pharmacother. 2009;10(14):2329–2336. | ||
Grabowski B, Lee RD. Absorption, distribution, metabolism and excretion of [14C]dexlansoprazole in healthy male subjects. Clin Drug Investig. 2012;32(5):319–332. | ||
Food and Drug Administration. Guidance for industry: bioavailability and bioequivalence studies submitted in NDAs or INDs – general considerations. Bethesda, MD: Center for Drug Evaluation and Research (CDER), Food and Drug Administration; 2014. Available from: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm389370.pdf. Accessed August 29, 2018. | ||
Food and Drug Administration. Guidance for industry: bioavailability and bioequivalence studies for orally administered drug products – general considerations. Washington, DC: Food and Drug Administration; 2003. Available from: https://www.ipqpubs.com/wp-content/uploads/2014/04/BABEOld.pdf. Accessed August 29, 2018. | ||
FDAAA 801 Requirements. Clinicaltrials.gov Web site. Available from: https://clinicaltrials.gov/ct2/manage-recs/fdaaa. Accessed April 6, 2016. | ||
Grissinger M. Oral syringes: making better use of a crucial and economical risk-reduction strategy. P T. 2013;38(1):5–6. |
Supplementary materials
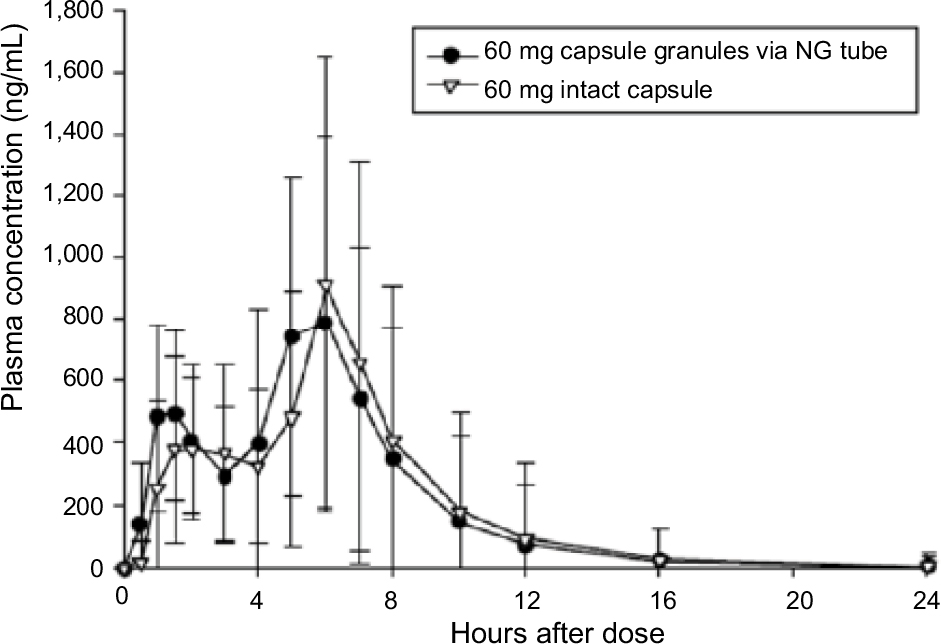 | Figure S1 Mean (±SD) plasma concentration–time plots (linear format) for dexlansoprazole following administration of dexlansoprazole 60 mg as an aqueous mixture of capsule granules via an NG tube or as an intact capsule to healthy subjects. Abbreviation: NG, nasogastric. |
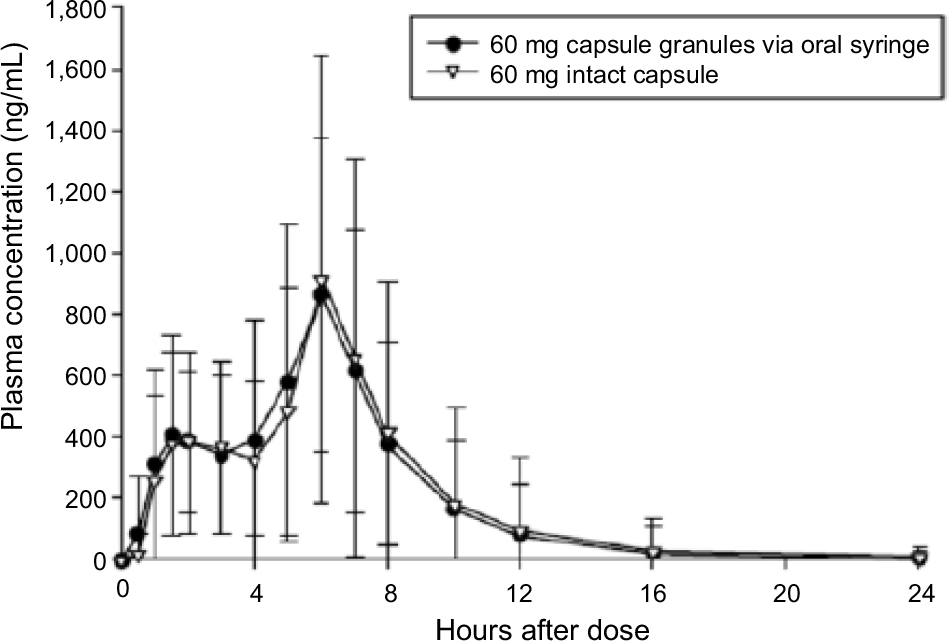 | Figure S2 Mean (±SD) plasma concentration–time plots (linear format) for dexlansoprazole following administration of dexlansoprazole 60 mg as an aqueous mixture of capsule granules via an oral syringe or as an intact capsule to healthy subjects. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.