Back to Journals » Research and Reports in Urology » Volume 6
Bilateral spermatocytic seminoma: a case report
Authors Narins H, Chevli K, Gilbert R, Duff M, Toenniessen A, Hu Y
Received 12 February 2014
Accepted for publication 7 April 2014
Published 12 June 2014 Volume 2014:6 Pages 63—65
DOI https://doi.org/10.2147/RRU.S62302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Hadley Narins,1 Kent Chevli,1,2 Richard Gilbert,2 Michael Duff,1,2 Andrew Toenniessen,2 Yan Hu3
1Department of Urology, State University of New York at Buffalo Medical School, Buffalo, NY, USA; 2Western New York Urology Associates, Cheektowaga, NY, USA; 3Department of Pathology, State University of New York at Buffalo Medical School, Buffalo, NY, USA
Abstract: Spermatocytic seminoma (SS) is a rare entity, accounting for 2%–12% of all seminomas; amongst those, fewer than 10% are bilateral. These may occur synchronously or metachranously. We report here a case of bilateral SS in a 63-year-old patient, who initially presented with bilateral testicular masses. In our search of the literature, this represents the fifth documented case of synchronous, bilateral SS.
Keywords: spermatocytic seminoma, bilateral seminoma, testicular, urology, scrotal swelling
Case report
A 63-year-old male was referred to the urology department (Western New York Urology Associates) with an 8 month history of painless scrotal swelling. A scrotal ultrasound revealed large bilateral solid testicular masses. The differential diagnosis included testicular lymphoma, leukemia, germ cell tumor, and granulomatous inflammation. He was evaluated with serum β-HCG, AFP, and lactate dehydrogenase which were all within normal ranges. As non-Hodgkin lymphoma is the most frequent testicular neoplasm in men older than 40 years,1 he was assessed with a positron emission tomography (PET)/computed tomography (CT) scan that was negative for lymphadenopathy or lymphomatous mass. After multidisciplinary review with urology, radiology, and oncology, the consensus was, left radical inguinal orchiectomy for tissue diagnosis.
Histopathology diagnosed a 7×6×4.2 cm mass contained to the left testicle with lymphovascular invasion and histological features of SS (Figures 1 and 2) stage 1B (T2N0M0S0). The tumor displayed pleomorphism, with small, medium, and large cell types. After receiving this diagnosis the patient was counseled to undergo orchiectomy of the right side; he underwent contralateral orchiectomy 15 days after the initial surgery. A 6×5.2×5.8 cm mass was again confined to the testes with pathology consistent with SS, stage 1 (T1N0M0S0) (Figures 1 and 2). There was no lymphovascular invasion identified in the pathologic review of the right testicle. The patient suffered a non-infected scrotal hematoma following his second surgery that was managed expectantly and subsequently resolved.
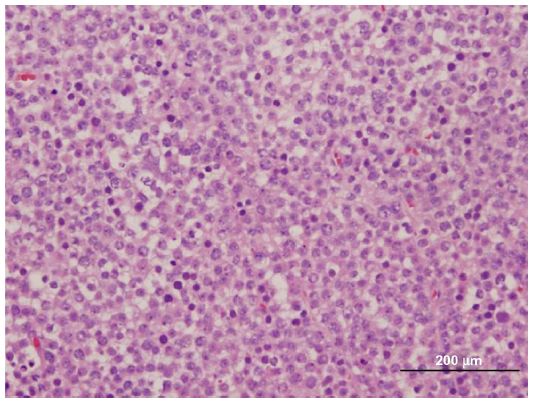 | Figure 1 Sheets of round cells with three cells types demonstrated (small, medium, and large). |
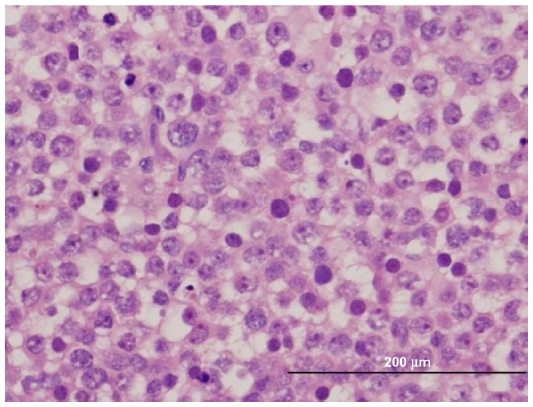 | Figure 2 Large cells with multiple nucleoli visible at this magnification; the intermediate cell type predominates. |
Postoperatively the patient was referred to radiation oncology for consideration of radiation therapy. After reviewing the options of adjuvant radiation therapy versus observation, the patient chose observation. At the time of writing this paper, the patient shows no signs of disease recurrence.
Comment
Although extremely rare, SS has been reported for approximately 70 years, and almost uniformly follows an indolent course following radical orchiectomy. The mean age of diagnosis is 54 years,2 and it has never been found in the pediatric population. Unlike other testicular neoplasms it has no ovarian homologue. Initially described in 1946, SS was thought to be a well differentiated form of seminoma, in which the tumor derives from spermatogonia rather than undifferentiated germ cells.3 More recent research by Rajpert-De Meyts et al, among others, suggests that the origin of SS may be a premeiotic germ cell.4 Similarly, Waheeb and Hoffman reviewed recent immunohistochemical data to refute the early claim of embryonic origin, arguing in favor of a postnatal germ cell as the cell of origin.5
Histopathological features of SS are characterized by three types of cells: small, medium, and large. The medium sized cell tends to be predominant. The small cell is 6–8 μm with a dark-stained nucleolus, similar to a lymphocyte. The medium cell is 15–20 μm with a round nucleus. The largest of the 3 cells is 80–100 μm, which may be mononucleated or multinucleated.6 As demonstrated in Figures 1 and 2, our patient’s tumor histology is consistent with these classic characteristics.
Traditionally, management of SS involved adjuvant radiation; however, over the past 2 decades this paradigm has changed. In the vast majority of cases this is an indolent tumor that may be managed with surgery alone and post-operative surveillance. Given the rarity of SS it is impossible to accrue sufficient cases for a randomized, prospective trial of postoperative radiation versus surveillance. However, in 2004 Chung et al retrospectively identified all 13 cases of SS identified over the course of 18 years, at their cancer center (1 patient excluded). Five received adjuvant radiation, while seven underwent surveillance only; none of these 12 patients experienced relapse.7 Similarly, in their study of ten patients, five of whom received adjuvant treatment and five surveillance only, Pendlebury et al showed no recurrence in either group at 8.5 years follow-up.8
The sarcomatous variant is the only SS subtype requiring further adjuvant treatment. This variant contains both a typical SS element, as well as a sarcomatous element, typically rhabdomyosarcomatous.2 In this case SS is transformed from an indolent to highly aggressive neoplasm, with most patients dying of their metastatic disease.9 Anaplastic SS is another variant that has a similar prognosis as classic SS. Histopathology demonstrates anaplastic appearing cells, interspersed with typical SS. The prognosis is equivalent to classic SS in multiple series; however, many of these patients with anaplastic SS received adjuvant chemotherapy-biasing the prognosis in favor of equivalence between these two histological variants.2
From the pool of greater than 200 known cases of SS, there were no confirmed cases of metastasis in the absence of sarcomatous elements until the last 30 years. Since 1988 there have been two case reports of patients with classic SS and metastasis. In the first documented case, Matoska et al, reported a 70-year-old patient with metastatic SS 19 months after radical inguinal orchiectomy; light microscopy, and immunohistochemistry of pathologically enlarged retroperitoneal nodes after node dissection was consistent with SS without any sarcomatous component. The patient subsequently died of complications of chemotherapy.10 In 2002 Steiner et al also identified a case of a pathologically enlarged lymph node in a 26-year-old patient 10 months after radical inguinal orchiectomy for SS. Like Matoska et al, they also identified SS in the metastatic site, confirmed both at their institution and at a second academic institution. The patient was treated with three rounds of cisplatin-based therapy and was disease free at the time of publication 3 years later.11 Although rare, sufficient cases of SS have accumulated to demonstrate that it is almost exclusively an indolent disease. In the two documented case of metastasis, it is unclear whether the patients were cured by retroperitoneal lymph node dissection alone, or whether radiation and chemotherapy has efficacy in these cases. Given the paucity of evidence, with the exception of transformation to the sarcomatous variant, orchiectomy followed by surveillance seems an appropriate standard of care, with postoperative imaging to rule out rare cases of metastatic disease.
Disclosure
The authors report no conflicts of interest in this work.
References
McDougal WS, Wein AJ, Kavoussi LR, Novick AW, Partin AW, Peters CA, editors. Campbell-Walsh Urology. 10th ed. Philadelphia. Elsevier Science Health Science Division, 2011. | |
Lombardi M, Valli M, Brisigotti M, Rosai J. Spermatocytic seminoma: review of the literature and description of a new case of the anaplastic variant. Int J Surg Pathol. 2011;19(1):5–10. | |
Talerman A. Spermatocytic seminoma: clinicopathological study of 22 cases. Cancer. 1980;45(8):2169–2176. | |
Rajpert-De Meyts E, Jacobsen GK, Bartkova J, et al. The immunohistochemical expression pattern of Chk2, p53, p19INK4d, MAGE-A4 and other selected antigens provides new evidence for the premeiotic origin of spermatocytic seminoma. Histopathology. 2003;42(3):217–226. | |
Waheeb R, Hofmann MC. Human spermatogonial stem cells: a possible origin for spermatocytic seminoma. Int J Androl. 2011;34(4 Pt 2):e296–e305. | |
Aggarwal N, Parwani AV. Spermatocytic seminoma. Arch Pathol Lab Med. 2009;133(12):1985–1988. | |
Chung PW, Bayley AJ, Sweet J, et al. Spermatocytic seminoma: a review. Eur Urol. 2004;45(4):495–498. | |
Pendlebury S, Horwich A, Dearnaley DP, Nicholls J, Fisher C. Spermatocytic seminoma: A clinicopathological review of ten patients. Clin Oncol (R Coll Radiol). 1996;8(5):316–318. | |
Gupta A, Pasricha S, Trivedi P. Spermatocytic seminoma associated with undifferentiated sarcoma: A rare case report. Indian J Pathol Microbiol. 2011;54(1):138–140. | |
Matoska J, Ondrus D, Hornák M. Metastatic spermatocytic seminoma. A case report with light microscopic, ultrastructural, and immunohistochemical findings. Cancer. 1988;62(6):1197–1201. | |
Steiner H, Gozzi C, Verdorfer I, Mikuz G, Bartsch G, Hobisch A. Metastatic spermatocytic seminoma-an extremely rare disease. Eur Urol. 2006;49(1):183–186. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.