Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 8
Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris
Authors Leight H, Zinn Z, Jalali O
Received 4 June 2015
Accepted for publication 30 July 2015
Published 15 September 2015 Volume 2015:8 Pages 485—488
DOI https://doi.org/10.2147/CCID.S89871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Hayley Leight, Zachary Zinn, Omid Jalali
Department of Dermatology, West Virginia University, Morgantown, WV, USA
Abstract: Here, we report a case of a middle-aged woman presenting with severe, long-standing, hyperkeratotic plaques of the lower extremities unrelieved by over-the-counter medications. Initial history and clinical findings were suggestive of an inherited ichthyosis. Ichthyoses are genetic disorders characterized by dry scaly skin and altered skin-barrier function. A diagnosis of ichthyosis vulgaris was confirmed by histopathology. Etiology, prevalence, and treatment options are discussed.
Keywords: filaggrin gene, FLG, profilaggrin, keratohyalin granules, hyperkeratosis
Introduction
Inherited ichthyoses are a diverse group of genetic disorders characterized by dry, scaly skin; hyperkeratosis; and altered skin-barrier function. While these disorders of cutaneous keratinization are multifaceted and varying in etiology, disruption in the stratum corneum with generalized scaling is common to all.1–4 Although not entirely known how each diverse genetic process of the ichthyoses could lead to a similar phenotype, a weakened epidermal barrier allowing for inflammation and subsequent hyperproliferation of the skin is hypothesized.1–4
Various ichthyoses can be differentiated from one another through history, clinical exam, histopathological analysis, electron microscopy, and genetics. Determining the inheritance pattern is an important part of the family history; this information is helpful in both formulating the differential diagnosis as well as in understanding the variable expressivity and severity of the disorder.1,3,4 It is also important to know: if the ichthyosis is congenital or was acquired later in adulthood; whether there is a presence or absence of erythroderma; and the features and specific appearance of the scale, as well as any other systemic manifestations of the disease.1,3,4 Although histopathological analysis can be useful to differentiate some of the ichthyoses, such as epidermolytic hyperkeratosis, other ichthyoses may not be able to be distinguished from one another via this manner alone.1,3,4
Ichthyosis vulgaris (IV), the most frequently occurring ichthyosis, is caused by mutations in the filaggrin gene (FLG).1,3–13 Typical hallmarks of IV, which encompass a wide clinical spectrum, include visible scaling and dryness that spares flexural surfaces, hyperlinearity of palms and soles, and strong predisposition for allergic disease comorbidities.1,3–11 Prevalence estimates for IV range from 4.0% to 7.7% in Europeans and 2.29% to 3.00% in Asians; observations of FLG mutations in darkly pigmented populations are low.4,5,12,13
Case report
A 57-year-old female with new-onset dyspnea was evaluated for severely hyperkeratotic skin of bilateral lower extremities (Figures 1 and 2). The lesions were reported as beginning during childhood, initially on the feet and progressing proximally to the level of the knee. The lesions, which were occasionally pruritic, had worsened over the prior 14 years and were not relieved by over-the-counter medications. Family history was significant for several generations of males and females with similar appearing skin as well as many unaffected family members. Our patient indicated improvement in symptoms when living in warmer, more humid environments with worsening of symptoms while living at more northern latitudes.
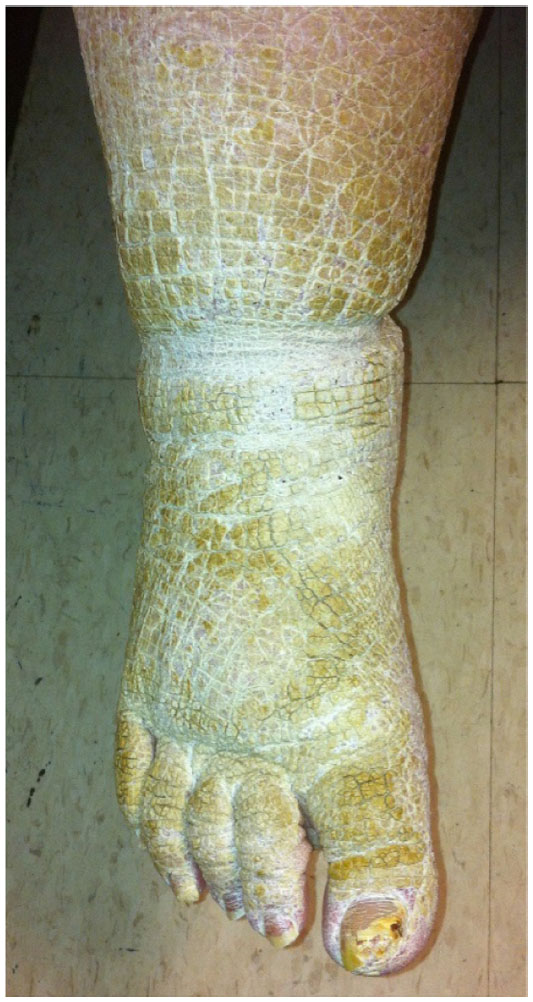 | Figure 1 Hyperkeratotic skin of the lower extremity. |
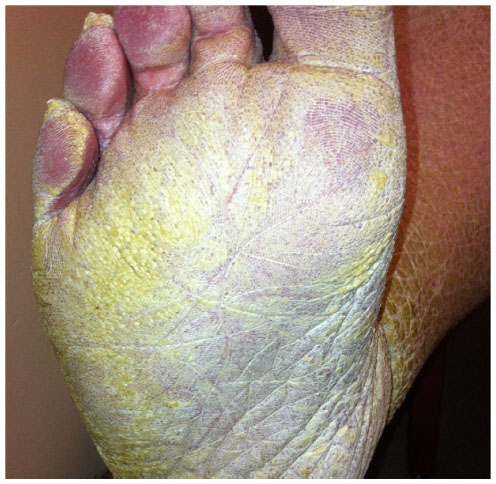 | Figure 2 Hyperkeratotic skin/hyperlinearity of the plantar surface. |
Discussion
IV, a genetically transmitted, autosomal semi-dominant disorder, exhibits varying penetrance of FLG mutations.4,6,8,13–19 Individuals heterozygous for the FLG mutation often have a less severe form of the disease than those homozygous for the mutation.4,7,8,10,14–19 FLG encodes for a protein called profilaggrin, which functions as a precursor to filaggrin (or filament-aggregating protein).7 Profilaggrin is stored in the granular layer of the epidermis as the major component of keratohyalin granules.3,4,7,15 Profilaggrin is cleaved into multiple filaggrin peptides that aggregate the keratin filaments. These keratin filament complexes are then cross-linked to the cell envelope and are important in maintaining the compact layer of skin and serving as a natural moisturizer.3–5,6,8,16–19
It has long been established that IV is associated with a decreased and even absent number of keratohyalin granules due to the deficiency of filaggrin proteins.4,7,15 A compensatory epidermal hyperplasia with inflammation, abnormal desquamation, and hyperkeratosis results in an impaired epidermal barrier, water loss, and dehydration.1,9,16,20 Chemicals and allergens are more prone to cross the impaired barrier, leading to sensitization and an increased propensity for inflammatory skin conditions.1,4,21–23
IV is usually not clinically present at birth and may have a variety of presentations later in life. With inherited IV, symptoms usually begin before age 1 year and alternate between exacerbation and remission. With milder presentations, patients present with flaky white scaling patches, often on extremities and extensor surfaces, typically sparing the flexural and more hydrated areas of the body.3,4,15 More severe cases of IV are rare and may show an increased hyperkeratosis of the palmoplantar skin with fissures forming on the heels of the feet.1,4,15 The flaking scales may be pronounced over the head, trunk, face, and extremities. Intertriginous areas of the body (armpits, groin, etc), are usually spared.1,4,15
Increased humidity explains the seasonal variation seen in IV. Symptoms often improve in the summer months and worsen in the cold and dry winter months, when the decreasing humidity further breaks down the residual filaggrin left in the skin.1,4,15 IV is frequently associated with the atopic triad of atopic dermatitis, hay fever, and asthma as well as with keratosis pilaris.4–7,11,16,18,19 It is hypothesized that the epidermal barrier abnormality continually exposes the body to pathogens and allergens, ultimately resulting in a T-helper 2 immunophenotype consistent with atopy.4
Histopathologically, IV demonstrates orthohyperkeratosis, diminished to absent stratum granulosum, and decreased or absent keratohyalin granules on electron microscopy.1,4 In patients heterozygous for the gene mutation, the keratohyalin granules may be present but appear small.3 Decreased or absent filaggrin on immunohistochemistry is characteristic as well.1,4
A histopathological analysis was done from a 4 mm punch biopsy of the right lower leg of our patient. Findings revealed orthohyperkeratosis with a markedly reduced stratum granulosum, consistent with IV. Figure 3 shows the histopathology of the punch biopsy specimen.
 | Figure 3 Histopathology slide of the punch biopsy specimen. |
In this case, the severity of the IV was hypothesized to be related to her obesity and chronic lymphedema. Obesity is associated with mild chronic inflammation as well as impaired lymphatic drainage due to tissue compression.24–27 Lymphedema, the result of an inability of the lymphatic vasculature to remove fluid and lipids, leads to their accumulation in interstitial spaces.24–32 Recent reports also note fibrotic changes in the extracellular matrix of adipose tissue decrease lymphatic clearance of inflammatory macromolecules from interstitial spaces.26,27
While lymphedema begins as spongy swelling, the chronic recurring inflammation induces an infiltrate of fibroblasts and keratinocytes thus transforming the epidermal layer leading to its subsequent thickening.28–32 Over time, the limb with untreated lymphedema gains a brawny and indurated texture and histopathologically reveals hyperkeratosis and papillomatosis.28–32 Lymphedema-induced hyperkeratosis superimposed on the epidermal thickening associated with IV likely played a role in the exaggerated presentation in this case.
Treatment of IV is similar to that of many of the other ichthyoses and is symptomatic, complex, and dynamic.9,20–23,33 Primary treatments are topical moisturizers and medications designed to reduce scaling, support skin-barrier function, reduce water loss, and decrease symptoms. Lessening the scale, providing hydration, and moisturization can be accomplished with emollients, humectants, bathing immediately followed with moisturizers, keratolytic agents, and topical retinoids.20,21 Management of bacterial skin infections, most commonly staphylococcal or streptococcal in origin, is done with topical mupirocin or bacitracin.21,23 On the horizon, researchers are attempting to use genetic pharmacology to increase the body’s own production of normal filaggrin.4,15
For this patient, first-line treatment for IV was begun using topical 0.1% tretinoin and 40% urea cream. At follow-up, warm bath soaks prior to medication application was added to the regimen to enhance absorption. At the present time, marked objective clinical improvement, as well as subjective patient satisfaction with treatment, is noted. She continues to be followed in the dermatology clinic.
Conclusion
Our patient presented with an extreme presentation of a common disease. Based upon the history, IV was considered. However, the severity of the clinical presentation of hyperkeratosis strongly suggested an alternative diagnosis. This case is a helpful reminder that IV is the most common inherited ichthyosis, and therefore should be a diagnostic consideration in even extreme and atypical presentations.
Acknowledgments
Special thanks to the Department of Dermatology at West Virginia University School of Medicine, Morgantown, WV, for providing medical student opportunity for case study research. Manuscript editing assistance was provided by Susan B Leight, EdD, FNP-BC, FAANP, Director, School of Nursing, West Virginia Wesleyan College. The authors advise that the West Virginia University Ethics Committee/Institutional Review Board does not require patient approval for case reports.
Disclosure
The authors report no conflicts of interest in this work.
References
Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012. | |
Styperek AR, Rice ZP, Kamalpour L, et al. Annual direct and indirect health costs of the congenital ichthyoses. Pediatr Dermatol. 2010; 27(4):325–335. | |
Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: McGraw Hill; 2008. | |
Thyssen JP, Godoy-Gijon E, Elias PM. Ichthyosis vulgaris: the filaggrin mutation disease. Br J Dermatol. 2013;168(6):1155–1166. | |
Li M, Cheng R, Shi M, et al. Analyses of FLG mutation frequency and filaggrin expression in isolated ichthyosis vulgaris (IV) and atopic dermatitis-associated IV. Br J Dermatol. 2013;168(6):1335–1338. | |
Wells RS, Kerr CB. Clinical features of autosomal dominant and sex-linked ichthyosis in an English population. BMJ. 1966;1(5493):947–950. | |
Liu P, Yang Q, Wang X, et al. Identification of a genetic locus for ichthyosis vulgaris on chromosome 10q22.3-q24.2. J Invest Dermatol. 2008;128(6):1418–1422. | |
Sandilands A, Terron-Kwiatkowski A, Hull PR, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39(5):650–654. | |
Bellew S, Del Rosso JQ. Overcoming the Barrier Treatment of Ichthyosis: A Combination-therapy Approach. J Clin Aesthetic Dermatol. 2010;3(7):49–53. | |
Perusquía-Ortiz AM, Oji V, Sauerland MC, et al. Complete filaggrin deficiency in ichthyosis vulgaris is associated with only moderate changes in epidermal permeability barrier function profile. J Eur Acad Dermatol Venereol. 2013;27(12):1552–1558. | |
Dreyfus I, Bourrat E, Maruani A, et al. Factors associated with impaired quality of life in adult patients suffering from ichthyosis. Acta Derm Venereol. 2014;94(3):344–346. | |
Dreyfus I, Pauwels C, Bourrat E, et al. Burden of inherited ichthyosis: a French national survey. Acta Derm Venereol. 2015;95(3):326–328. | |
Esparza-Gordillo J, Matanovic A, Marenholz I, et al. Maternal filaggrin mutations increase the risk of atopic dermatitis in children: an effect independent of mutation inheritance. PLoS Genet. 2015;11(3):1–16. | |
Mendes MS, Aquino TA, de Padua Lima A, Kouzak SS, Takano GH. Mosaic epidermolytic ichthyosis – case report. An Bras Dermatol. 2013;88(6 Suppl 1):116–119 | |
Brown SJ, McLean WH. One remarkable molecule: filaggrin. J Invest Dermatol. 2012;132(3 Pt 2):751–762. | |
Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol. 2010; 162(3):472–477. | |
Zhang X, Liu S, Chen X, et al. Novel and recurrent mutations in the filaggrin gene in Chinese patients with ichthyosis vulgaris. Br J Dermatol. 2010;163(1):63–69. | |
Nomura T, Akiyama M, Sandilands A, et al. Prevalent and rare mutations in the gene encoding filaggrin in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Invest Dermatol. 2009;129(5):1302–1305. | |
Winge MCG, Hoppe T, Berne B, et al. Filaggrin genotype determines functional and molecular alterations in skin of patients with atopic dermatitis and ichthyosis vulgaris. PLoS One. 2011;6(12):e28254. | |
Blanchet-Bardon C, Tadini G, Machado Matos M, Delarue A. Association of glycerol and paraffin in the treatment of ichthyosis in children: an international, multicentric, randomized, controlled, double-blind study. J Eur Acad Dermatol Venereol. 2012;26(8):1014–1019. | |
Fleckman P, Newell BD, van Steensel MA, Yans, AC. Topical treatment of ichthyoses. Dermatol Ther. 2013;26(1):16–25. | |
Bodemer C, Bourrat, E, Mazereeuw-Hautier J, et al. Short- and medium-term efficacy of specific hydrotherapy in inherited ichthyosis. Br J Dermatol. 2011;165(5):1087–1094. | |
Chan A, Godoy-Gijon E, Nuno-Gonzalez A. et al. Cellular basis of secondary infections and impaired desquamation in certain inherited ichthyoses. JAMA Dermatol. 2015;151(3):285–292. | |
Divyashree RA, Naveen KN, Pai VV, Athanikar SB, Gupta G. Cutaneous manifestations of obesity among dermatology patients in a tertiary care center. Indian J Dermatol Venereol Leprol. 2014;80(3):278. | |
Greene AK, Grant FD, Slavin SA. Lower-extremity lymphedema and elevated body-mass index. N Engl J Med. 2012;366(22):2136–2137. | |
Weitman ES, Aschen SZ, Farias-Eisner, et al. Obesity impairs lymphatic fluid transport and dendritic cell migration to lymph nodes. PLoS One. 2013;8(8):e70703. | |
Arngrim N, Simonsen L, Holst JJ, Bülow J. Reduced adipose tissue lymphatic drainage of macromolecules in obese subjects: a possible link between obesity and local tissue inflammation? Int J Obes (Lond). 2013;37(5):748–750. | |
Scheinfeld, NS. Obesity and dermatology. Clin Dermatol. 2004; 22(4):303–309. | |
Boza JC, Trindade EN, Peruzzo J, Sachett L, Rech L, Cestari TF. Skin manifestations of obesity: a comparative study. J Eur Acad Dermatol Venereol. 2012;26(10):1220–1223. | |
Lu S, Tran TA, Jones DM, et al. Localized lymphedema (elephantiasis):a case series and review of the literature. J Cutan Pathol. 2009;36(4):1–20. | |
Kaba H, Bakar Y, Ozdemir Oç, Sertel S. Complex Decongestive Physiotherapy Treats Skin Changes like Hyperkeratosis Caused by Lymphedema. Case Rep Dermatol Med. 2012;2012:416421. | |
Linnitt N. Complex skin changes in chronic oedemas. Br J Community Nurs. 2007;12(4):S10–S15. | |
Nayak S, Acharjya B, Mohanty P. Ichthyosis hystrix. Indian Dermatol Online J. 2013;4(1):47–49. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.