Back to Journals » Drug Design, Development and Therapy » Volume 20
Beyond Oncogenes: Selectively Targeting Whole-Tumor Cell Growth Regulation
Authors Qin A ![]()
Received 17 October 2025
Accepted for publication 27 January 2026
Published 23 February 2026 Volume 2026:20 574224
DOI https://doi.org/10.2147/DDDT.S574224
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Albert Qin
Medical Research & Clinical Operations, PharmaEssentia Corporation, Taipei, Taiwan, Republic of China
Correspondence: Albert Qin, Medical Research & Clinical Operations, PharmaEssentia Corporation, Taipei, Taiwan, Republic of China, Email [email protected]
Abstract: Cell cycle-based surveillance system is an evolutionary adaptation aligned with the complex and heterogeneous nature of cancer in higher-order organisms and serves as a naturally existing model for potent anticancer therapy. It helps provide insights for the reasons underpinning the challenges facing oncogene-targeting in drug development, including cancer heterogeneity, difficulty in identifying “driver” oncogenes, and drug resistance due to additional mutations or other factors such as epigenetic changes, phenotypic adaptation, and microenvironmental influences. This perspective suggests the paradigm shift of targeting the whole cellular process instead of focusing on single-oncogene inhibition. Therefore, a potentially compelling strategy for cancer therapy is to selectively suppress tumor cells via targeting the whole-cell growth regulation while sparing normal cells. Future directions include precisely distinguishing and selectively interfering with tumor cell growth-regulatory networks from those of normally growing cells to achieve maximal clinical efficacy without compromising safety.
Keywords: targeted therapy, cell cycle-based anticancer surveillance, cellular growth-regulatory machinery, whole-cell growth regulation, selective anticancer therapy
Enormous progress has been made in developing targeted, anti-cancer drugs such as small-molecules, monoclonal antibodies and antibody-conjugated drugs.1,2 Newer immunotherapies and cell-based therapies via immunological responses against cancer cells have further revolutionized the treatment.3 However, cancer remains largely incurable. In anticancer drug development, targeting oncogenes is a logic and common approach but faces challenges including identification of “driver” oncogenes and drug resistance due to additional mutations or other factors including epigenetic changes, phenotypic adaptation, influences from the tumor microenvironment, and reconfiguration of intercellular signal networks.4–7 A natural anti-cancer system that selectively inhibits tumor cells by interfering with their growth-regulatory machinery may provide insightful clues for possible therapeutic approaches.
In humans, many de novo cancerous or transformed cells are eliminated before fully developing into cancer. One reason for this is a cell cycle-based anticancer surveillance system that monitors cancer cell development, alongside immunosurveillance.8,9 In this system, selective changes in the tumor cell growth-regulation, including activation of the cell cycle regulator p107 in response to interferon-beta and -alpha (IFNβ- and IFN-α) expression, impose a cell cycle checkpoint to suppress tumor cell growth and promote senescence, diminishing tumorigenicity.8,10 This system bypasses specific oncogenes and selectively inhibits tumor cells at the cellular level. Normally growing cells are largely unaffected because they are tightly controlled by the normal cell cycle-regulatory machinery, of which a key component is RB1 (or pRB).8,11 Additionally, IFN-β induces apoptosis of tumor cells when overexpressed,12 further enhancing the elimination of cancer cells. This network of actions reflects an effective natural anticancer mechanism at the cellular level.
The cell cycle-based surveillance system is an evolutionary adaptation aligned with the complex and heterogeneous nature of cancer in higher-order organisms. Figure 1 schematically illustrates the correlation between evolutionary development, cancer complexity/heterogeneity, and refinement of the surveillance system using four representative species, from Caenorhabditis (C). elegans to Homo sapiens. C. elegans does not develop cancer; it possesses only the RB1 ortholog LIN35 and lacks IFNs and p107.13,14 Drosophila can develop simple cancers and serves as a valuable model for studying oncogene-tumor suppressor gene interactions, but it possesses only the RB1 homologs Rbf1 and Rbf2, without p107.15 Although drosophila has the JAK/STAT pathway that can help restrict viral infection, it lacks IFNs.16 Mice develop more sophisticated cancers, yet their cell cycle-based anticancer surveillance is less robust than in humans. Murine IFN-β does not elicit as strong a cell cycle effect on mouse cancer cells as human IFN does on human cancer cells.17 In humans, cell cycle-based anticancer surveillance has evolved into a coordinated regulatory mechanism between tumor cells and normal cells. This system selectively suppresses cancer cells, regardless of their heterogeneity and driver oncogenes.8,11 It in turn suggests that a therapeutic approach, following this mechanism, can potentially override tumor cell heterogeneity and be applied to different types of cancer.
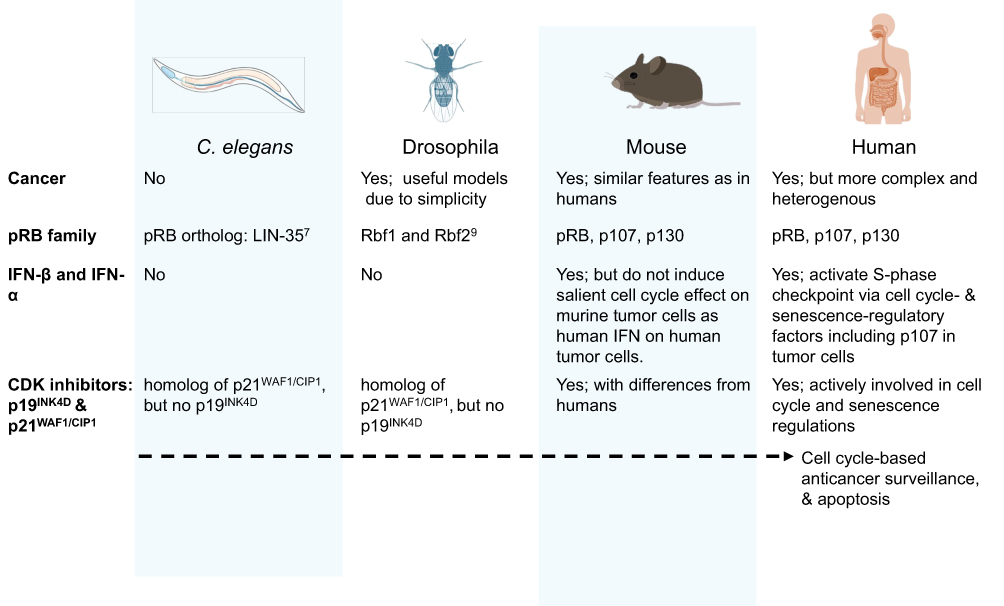 |
Figure 1 Schematic diagram illustrates the correlation between evolutionary development, cancer complexity/heterogeneity, and refinement of the surveillance system using four representative species, from Caenorhabditis (C.) elegans to Homo sapiens. |
The cancer cell surveillance and removal speak of the importance of targeting cancer at the level of whole-cell growth regulation rather than simply targeting oncogenes. Oncogenic activation or tumor suppressor loss-of-function initiates carcinogenesis, but once established, cancer cells acquire other irreversible alterations in their growth-regulatory machinery, including additional mutations and/or epigenetic changes (Figure 2). Consequently, targeting oncogenes that initiate tumorigenesis or reintroducing a tumor suppressor alone will not reverse oncogenesis. This limitation helps explain why many therapies aimed at individual oncogenes are clinically efficacious, leading to improved survival and quality of life, but still face gaps in durable benefits (ie, long-term cancer-free remission). Combination regimens that target known oncogenes yield notable responses, yet overall efficacy remains suboptimal. Consequently, targeted therapies that selectively address the entire cancer cell are essential, since cancerous cells harbor irreversible growth-regulatory defects that extend beyond the initiating oncogenic event. Therapies that selectively interfere with growth regulation of cancer cells would work differently from cell-based immune therapies, which target cancer cells via immune responses.
 |
Figure 2 Graph shows that once established, cancer cells have acquired irreversible alterations in their growth-regulatory machinery that extend beyond the initiating oncogenic event. |
Targeted therapy at the cellular level has advanced markedly in the management of myeloproliferative neoplasms. Polycythemia vera, a type of myeloproliferative neoplasm characterized by erythrocytosis, classically harbors the constitutively active driver mutation JAK2V617F. Here, ropeginterferon alfa-2b, a mono-PEGylated proline-interferon-α with an exposure-related response profile, represents a therapy targeting the neoplasm at the whole-cellular level.18,19 It exerts an anti-neoplastic effect by IFN receptor-mediated activation of JAK1/Tyk2 kinases, bypassing the mutated JAK2.20 Ropeginterferon alfa-2b treatment achieves high rates of hematologic and deep molecular responses, provides a survival advantage in patients with polycythemia vera, and has shown clinical benefits in other myeloproliferative neoplasms.21–24 The clinical results thus exemplify the promise of targeting whole-cell growth in oncology. Building on this backbone, combining such agents with complementary targeted therapies may yield even greater therapeutic gains. Consistently, combining epigenetic therapies with other treatments to overcome therapeutic resistance shows great potential in cancer therapy.25,26
In summary, targeting whole-cell growth regulation, thereby extending beyond focusing on single-oncogene inhibition, adds an important dimension to cancer treatment, either as monotherapy or potentially in combination with other mechanistic approaches. The clinical efficacy of ropeginterferon alfa-2b in myeloproliferative neoplasms provides a concrete illustration of this paradigm. A potential limitation of this strategy is that it remains a challenge to precisely distinguish and selectively interfere with tumor cell growth-regulatory networks in various cancer types without affecting normal cells.
Acknowledgments
The author would like to thank his colleagues for assistance during editing process of the manuscript. Albert Qin was formerly known as Xiao-Qiang Qin, with the name change when becoming a US citizen.
Disclosure
The author serves as chief medical officer of PharmaEssentia Corporation. The author reports no conflicts of interest in this work.
References
1. Zhong L, Li Y, Xiong L, et al. Small molecules in targeted cancer therapy: advances, challenges, and future perspectives. Sig Transduct Target Ther. 2021;6:201.
2. Diamantis N, Banerji U. Antibody-drug conjugates-an emerging class of cancer treatment. Br J Cancer. 2016;114:362–4. doi:10.1038/bjc.2015.435
3. Majzner RG, Heitzeneder S, Mackall CL. Harnessing the immunotherapy revolution for the treatment of childhood cancers. Cancer Cell. 2017;31(4):476–485. doi:10.1016/j.ccell.2017.03.002
4. Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16(3):280–296. doi:10.15252/embr.201439949
5. Emmons MF, Faião-Flores F, Smalley KSM. The role of phenotypic plasticity in the escape of cancer cells from targeted therapy. Biochem Pharmacol. 2016;122:1–9. doi:10.1016/j.bcp.2016.06.014
6. Keresztes D, Kerestély M, Szarka L, et al. Cancer drug resistance as learning of signaling networks. Biomedicine Pharmacother. 2025;183:117880. doi:10.1016/j.biopha.2025.117880
7. Panda M, Biswal BK. Cell signaling and cancer: a mechanistic insight into drug resistance. Mol Biol Rep. 2019;46:5645–5659. doi:10.1007/s11033-019-04958-6
8. Qin A. An anti-cancer surveillance by the interplay between interferon-beta and retinoblastoma protein RB1. Front Oncol. 2023;13:1173467. doi:10.3389/fonc.2023.1173467
9. Qin A. A plain language summary about a cell cycle-based, new surveillance mechanism against cancer. Future Oncol. 2024;20:3209–3212. doi:10.1080/14796694.2024.2402649
10. Kaynor C, Xin M, Wakefield J, Barsoum J, Qin XQ. Direct evidence that IFN-beta functions as a tumor-suppressor protein. J Interferon Cytokine Res. 2002;22:1089–1098. doi:10.1089/10799900260442511
11. Qin XQ, Runkel L, Deck C, DeDios C, Barsoum J. Interferon-beta induces S phase accumulation selectively in human transformed cells. J Interf Cytokine Res. 1997;17:355–367. doi:10.1089/jir.1997.17.355
12. Qin XQ, Tao N, Dergay A, et al. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci USA. 1998;95:14411–14416. doi:10.1073/pnas.95.24.14411
13. Lu X, Horvitz HR. lin-35 and lin-53, two genes that antagonize a C. elegans Ras pathway, encode proteins similar to Rb and its binding protein RbAp48. Cell. 1998;95:981–991. doi:10.1016/S0092-8674(00)81722-5
14. Batachari LE, Dai AY, Troemel ER. Caenorhabditis elegans RIG-I-like receptor DRH-1 signals via CARDs to activate antiviral immunity in intestinal cells. Proc Natl Acad Sci USA. 2024;121(29):e2402126121. doi:10.1073/pnas.2402126121
15. Du W, Vidal M, Xie JE, Dyson N. RBF, a novel RB-related gene that regulates E2F activity and interacts with cyclin E in Drosophila. Genes Dev. 1996;10(10):1206–1218. doi:10.1101/gad.10.10.1206
16. Dostert C, Jouanguy E, Irving P, et al. The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol. 2005;6:946–953. doi:10.1038/ni1237
17. Qin XQ, Beckham C, Brown JL, Lukashev M, Barsoum J. Human and mouse IFN-β gene therapy exhibits different anti-tumor mechanisms in mouse models. Mol Ther. 2001;4:356–364. doi:10.1006/mthe.2001.0464
18. Qin A, Wu D, Li Y, et al. Exposure-efficacy and exposure-safety analyses of ropeginterferon alfa-2b treatment in patients with polycythaemia vera. Br J Clin Pharmacol. 2024;90:1493–1502. doi:10.1111/bcp.16043
19. Qin A, Shimoda K, Suo S, et al. Population pharmacokinetics-pharmacodynamics and exposure-response of ropeginterferon alfa-2b in Chinese and Japanese patients with polycythemia vera. Pharmacol Res Perspect. 2025;13:e70109.
20. Qin A. Mechanism of action of ropeginterferon alfa-2b in polycythemia vera treatment. Clin Ther. 2024;46:439–440. doi:10.1016/j.clinthera.2024.03.005
21. Gisslinger H, Klade C, Georgiev P, et al. Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, Phase 3 trial and its extension study. Lancet Haematol. 2020;7(3):e196–e208. doi:10.1016/S2352-3026(19)30236-4
22. Suo SS, Fu RF, Qin A, et al. Molecular remission uncoupled with complete haematologic response in polycythaemia vera treatment with ropeginterferon Alfa-2b. Br J Haematol. 2024;205:2510–2514. doi:10.1111/bjh.19846
23. Mesa R, Gill H, Xiao X, et al. Ropeginterferon alfa-2b versus anagrelide for the treatment of essential thrombocythemia: topline results of the phase 3 SURPASS-ET trial. J Clin Oncol. 2025;53(16_suppl):6500. doi:10.1200/JCO.2025.43.16_suppl.6500
24. Gisslinger H, Klade C, Georgiev P, et al. Event-free survival in patients with polycythemia vera treated with ropeginterferon alfa-2b versus best available treatment. Leukemia. 2023;37:2129–2132. doi:10.1038/s41375-023-02008-6
25. Song J, Yang P, Chen C, et al. Targeting epigenetic regulators as a promising avenue to overcome cancer therapy resistance. Sig Transduct Target Ther. 2025;10:219.
26. Johnstone SE, Gladyshev VN, Aryee MJ, Bernstein BE. Epigenetic clocks, aging, and cancer. Science. 2022;378:1276–1277. doi:10.1126/science.abn4009
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.