Back to Journals » International Medical Case Reports Journal » Volume 19
Axenfeld-Rieger Syndrome with Negative Chromosomal Microarray and Whole-Exome Sequencing: A Case Report
Authors Moraes PDC, Montalli VAM, Sperandio M, Dos Santos AM, Rosa ACG ![]()
Received 2 January 2026
Accepted for publication 20 March 2026
Published 25 March 2026 Volume 2026:19 593292
DOI https://doi.org/10.2147/IMCRJ.S593292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Paulo de Camargo Moraes,1,* Victor Angelo Martins Montalli,1,* Marcelo Sperandio,1,* Ana Mondadori Dos Santos,2,* Ana Cláudia Garcia Rosa3,*
1Department of Oral Medicine and Pathology, Faculdade São Leopoldo Mandic Research Institute, Campinas, SP, Brazil; 2Department of Medical Genetics, Faculdade São Leopoldo Mandic Research Institute, Campinas, SP, Brazil; 3Department of General Pathology, Federal University of Tocantins, Palmas, TO, Brazil
*These authors contributed equally to this work
Correspondence: Ana Cláudia Garcia Rosa, Department of General Pathology, Federal University of Tocantins, Palmas, TO, Brazil, Tel +55 63 3229 4658, Email [email protected]
Abstract: Axenfeld-Rieger Syndrome (ARS) is a rare genetic disorder defined by ophthalmological abnormalities, frequently accompanied by craniofacial and dental anomalies. Although mutations in the PITX2 and FOXC1 genes are commonly identified, some patients do not receive molecular confirmation. This report describes a clinically compelling case of ARS with a typical phenotype but negative genetic testing. A 10-year-old male demonstrated physical abnormalities from early childhood and underwent multiple medical evaluations without a definitive diagnosis. He experienced difficulty and discomfort during mastication following the exfoliation of his deciduous teeth. Physical examination revealed short stature and ophthalmologic abnormalities, including strabismus and early-onset cataracts requiring bilateral phacoemulsification with intraocular lens implantation, as well as craniofacial and dental anomalies. Chromosomal microarray analysis identified inherited duplications in 4q26 and 7q31.2, but no pathogenic variants were detected. The diagnosis of ARS was established based on the characteristic phenotype. The patient remains under multidisciplinary care, receives orthodontic treatment, and continues regular ophthalmological monitoring. The recent onset of cataracts highlights the progressive nature of ocular involvement in ARS. Although molecular testing is essential for ARS, a subset of clinically diagnosed cases remains genetically unresolved. Advances in high-resolution and integrative genomic technologies are expected to improve diagnostic yield in these patients.
Keywords: Axenfeld-Rieger syndrome, PITX2, FOXC1, craniofacial anomalies, dental anomalies
Introduction
Axenfeld-Rieger syndrome (ARS) is a developmental disorder that primarily affects the anterior segment of the eye and results from abnormal migration of neural crest cells. This condition is characterized by anterior segment dysgenesis, including posterior embryotoxon, iris hypoplasia, and corectopia. Approximately 50% of affected individuals develop glaucoma during their lifetime. 1
The earliest description of these ocular abnormalities dates to 1920, when the German ophthalmologist Theodor Axenfeld identified an ocular anomaly. Specifically, he noted a white line on the posterior corneal surface near the limbus, with tissue strands extending from the peripheral iris. This alteration was called “posterior corneal embryotoxon” and later became known as Axenfeld anomaly.2
Subsequently, between 1934 and 1935, Austrian ophthalmologist Herwig Rieger reported cases with similar anterior segment abnormalities. These cases also showed additional iris alterations, such as hypoplasia, polycoria, and corectopia.3–6 Initially, these findings were described as dysgenesis mesodermalis iridis et corneae and later termed Rieger anomaly.6,7 In addition to ocular findings, some affected individuals had craniofacial and dental defects, including hypodontia, enamel defects, and maxillary hypoplasia, which followed an autosomal dominant inheritance pattern. The concurrence of ocular and craniofacial features subsequently led to the term Rieger syndrome.6–8
Currently, these findings are classified under Axenfeld–Rieger syndrome (ARS; OMIM #180500), reflecting shared features.9,10 ARS is rare, with an estimated prevalence of about 1:200,000. This prevalence may vary depending on the population studied and the diagnostic criteria used.10
Clinically, ARS may also include craniofacial and dental abnormalities, such as hypodontia, enamel defects, and maxillary hypoplasia.11,12 Genetically, the most common variants involve the paired-like homeodomain transcription factor 2 (PITX2) and forkhead box C1 (FOXC1) genes. Variants in PITX2 and FOXC1 account for a substantial proportion of cases; however, many patients remain without a definitive molecular diagnosis even after whole-exome sequencing (WES). Recent evidence suggests that alterations in non-coding regulatory regions, especially enhancer elements controlling PITX2 expression, may contribute to the phenotype but remain undetectable by standard genetic testing.13,14
Given the rarity and clinical and genetic heterogeneity of ARS, this report describes a clinically compelling case with characteristic craniofacial, dental, and ophthalmologic findings but without identifiable pathogenic variants on chromosomal microarray analysis (CMA) or whole-exome sequencing (WES). The diagnosis was initially suspected during dental evaluation based on distinctive craniofacial and dental features, highlighting the importance of phenotype-based recognition in genetically unresolved cases.
Case Report
All procedures followed the principles of the Declaration of Helsinki. According to institutional guidelines of Faculdade São Leopoldo Mandic (Campinas, Brazil), formal ethics committee approval was not required for the publication of a single case report. Written informed consent was obtained from the patient’s legal guardians, authorizing the publication of clinical data and images while ensuring confidentiality.
A 10-year-old male presented with difficulty and discomfort during mastication following exfoliation of the deciduous teeth. His mother reported noticing physical changes since early childhood. Previous assessments by multiple specialists did not yield a definitive diagnosis. The condition showed gradual progression, affecting facial, ocular, and dental development. Anthropometric assessment indicated short stature. General physical examination revealed an overweight status and a café-au-lait macule located near the left nipple. Craniofacial analysis identified multiple facial dysmorphisms, including a left-sided Darwin’s tubercle, prominent forehead, flattened and widened nasal base, bulbous nasal tip, bilateral epicanthal folds, telecanthus, double infraorbital folds, convergent strabismus, malar hypoplasia, and a predominance of the upper third of the face relative to the middle and lower thirds (Figure 1a and b). Intraoral examination revealed an anterior crossbite due to malocclusion (mandibular pseudo-prognathism), in association with oligodontia, microdontia, conical or malformed teeth, pyramidal dental roots, enamel hypoplasia, and hyperplasia of the upper labial frenulum (Figure 1c and d). Imaging studies, including frontal and lateral cephalometric radiographs, demonstrated hypoplasia of the nasal bone, maxillary hypoplasia, and underdevelopment of the maxillary sinuses. Panoramic and periapical radiographs showed teeth with pronounced root constriction and pseudotaurodontism (Figure 2a–c). The patient underwent comprehensive hormonal, metabolic, and hematological evaluation, including assessment of serum growth hormone, total and ionized calcium, phosphorus, magnesium, creatinine, alkaline phosphatase, insulin, fasting glucose, lipid profile, complete blood count, parathyroid hormone, 1,25-dihydroxyvitamin D, thyroid-stimulating hormone, thyroxine, luteinizing hormone, follicle-stimulating hormone, sex hormone-binding globulin, and urinary mineral metabolism. Laboratory results indicated elevated magnesium and thyroxine levels, while all other parameters were within normal limits. Thyroid and scrotal ultrasonography were unremarkable. Syndromic investigation using chromosomal microarray analysis (CMA) identified two duplications: a 2.4 Mb duplication in 4q26 and a 70 Kb duplication in 7q31.2, both initially classified as variants of uncertain significance (VUS). Parental segregation analysis revealed maternal inheritance of the first variant and paternal inheritance of the second. Subsequent whole-exome sequencing (WES) did not detect any clinically significant variants. Despite the absence of identifiable molecular alterations, the clinical presentation was consistent with ARS. The final diagnosis was established based on the constellation of clinical and radiographic findings and the characteristic phenotypic features. The patient continues to receive multidisciplinary care, including orthodontic treatment with a palatal expander and ongoing ophthalmological monitoring due to the risk of progressive ocular involvement. The recent development of cataracts further supports the pattern of progressive ophthalmological manifestations associated with ARS.
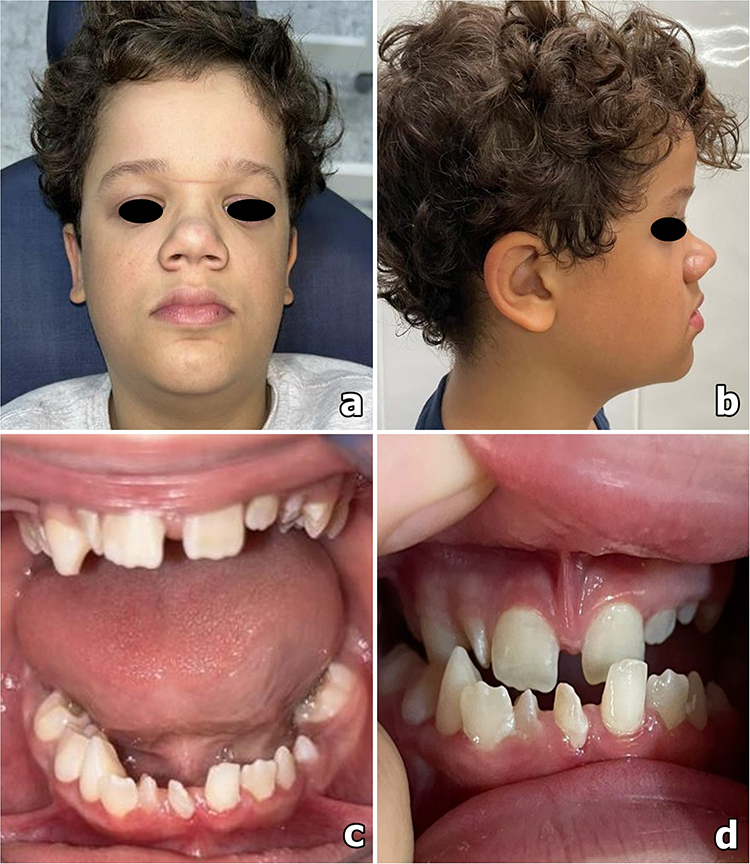 |
Figure 1 Craniofacial and intraoral clinical characteristics. (a and b) Frontal and lateral facial photographs demonstrate craniofacial dysmorphisms including telecanthus, prominent forehead, flattened nasal bridge with widened nasal base and bulbous nasal tip, and midface hypoplasia. (c and d) Intraoral examination shows oligodontia, microdontia, conical or malformed teeth, enamel hypoplasia, and anterior crossbite associated with mandibular pseudo-prognathism, as well as hyperplasia of the upper labial frenulum. |
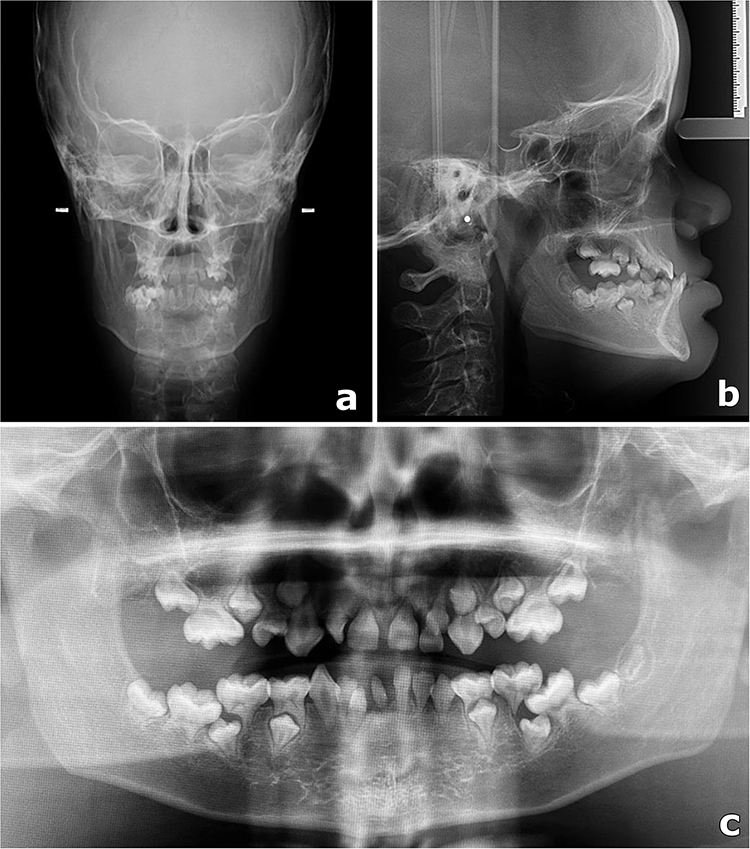 |
Figure 2 Radiographic findings. (a) Frontal cephalometric radiograph demonstrating craniofacial alterations, including nasal bone hypoplasia and maxillary hypoplasia. (b) Lateral cephalometric radiograph showing midfacial hypoplasia and altered maxillomandibular relationship. (c) Panoramic radiograph demonstrating oligodontia, microdontia, and teeth with short, constricted roots compatible with pseudotaurodontism. Small white dots and bars visible in the cephalometric images correspond to positioning markers generated by the radiographic equipment and do not represent anatomical structures. |
Discussion
ARS is a congenital autosomal dominant genetic condition characterized by high penetrance and variable expressivity, resulting from defects in the differentiation, development, or migration of neural crest cells.10,11,15 The clinical spectrum includes anterior segment ocular dysgenesis and systemic manifestations involving craniofacial, dental, cardiac, and neurological abnormalities.
Among the described ocular abnormalities are posterior embryotoxon, iris hypoplasia, polycoria, corectopia, anterior synechiae, and a high risk of glaucoma progression, observed in approximately 50% of patients throughout their lives.1,10 In addition to these structural abnormalities, other ophthalmologic findings such as strabismus and refractive errors have also been reported.1,16 In the present case, the patient exhibited strabismus and developed early-onset bilateral cataracts requiring phacoemulsification with intraocular lens implantation. Although glaucoma had not been diagnosed at the time of evaluation, the patient remains under regular ophthalmologic follow-up due to the known risk associated with ARS.
Beyond ocular involvement, individuals with ARS often exhibit facial dysmorphisms, including a wide and flat nasal bridge, maxillary hypoplasia, malar flattening, and midface alterations, as well as dental anomalies such as microdontia, hypodontia or oligodontia.12 Additional systemic manifestations may involve the musculoskeletal, neurological, auditory, and endocrine systems.10–14 The patient in this case showed extensive overlap with the clinical spectrum of ARS, including telecanthus, strabismus, short stature, bone alterations, and recently developed cataracts. Maxillofacial features such as oligodontia, microdontia, short dental roots with apical narrowing, maxillary hypoplasia, and pseudoprognathism were also present,10,11 supporting strong phenotypic compatibility with ARS.
Molecularly, ARS is classified into three subtypes: type 1 (associated with PITX2, 4q25), type 3 (associated with FOXC1, 6p25.3), and type 2 (linked to the 13q14 locus, which lacks a fully defined molecular basis).9,10,16,17 PITX2 variants are more frequently associated with dental defects and umbilical anomalies, while FOXC1 variants are related to auditory, cardiovascular, neurological, and skeletal alterations.11,16,17 The clinical features observed in this patient align with profiles described for both genes.
A subset of individuals exhibiting a phenotype consistent with ARS do not have detectable variants in PITX2 or FOXC1.13,14 For instance, Zhang et al (2021) identified 20 of 55 Chinese patients without variants in PITX2, FOXC1, or other anterior segment dysgenesis genes, while White et al (2023) reported that two of ten patients with ARS lacked a defined genetic diagnosis.13,14 Additional genes, including USP9X, CDK13, BCOR, and X-chromosome deletions involving HCCS and AMELX, have been implicated in overlapping phenotypes.17 Recent research suggests that structural alterations and non-coding regulatory mutations, particularly those affecting enhancers and chromatin interaction domains of PITX2, can reduce gene expression and result in typical ARS features, yet remain undetectable by CMA or WES.18,19 Farris et al (2024) described a balanced intrachromosomal rearrangement of chromosome 4 in a clinically compelling ARS1 patient, where even comprehensive testing, including whole genome sequencing (WGS), initially failed to identify pathogenic variants.20 The rearrangement disrupting PITX2 was detected only after advanced re-analysis.20 Collectively, these findings demonstrate that negative molecular results do not exclude ARS and that extended genomic strategies may be necessary in selected cases, supporting the rationale for negative testing in clinically compatible individuals.
In our case, the identified 4q26 duplication was observed only in the mother and is located outside the PITX2 (4q25) locus. Consequently, there is no evidence that this duplication is related to the patient's phenotype. In the absence of pathogenic variants in PITX2, FOXC1, or other associated genes, alternative mechanisms, including non-coding regulatory variants, alterations in chromatin organization, and epigenetic mechanisms, remain plausible.
The differential diagnosis of ARS encompasses conditions presenting with similar craniofacial and ocular anomalies, including 6p25 microdeletion syndrome, Alagille syndrome, De Hauwere syndrome, and oculodentodigital dysplasia.10,17,21 Souzeau et al (2021) identified gene-specific patterns of facial dysmorphism in patients with FOXC1 and PITX2 variants, indicating that distinct facial features can facilitate clinical diagnosis, particularly when genetic confirmation is not available.22 In the present case, alternative pathologies were excluded, as none fully accounted for the observed phenotype.
Beyond genetic testing, the evaluation of ARS should incorporate a thorough ophthalmological assessment and additional examinations tailored to the clinical context, such as neurodevelopmental evaluation, echocardiography, brain imaging, and hearing assessment.10 In this case, the patient underwent ophthalmological assessments, genetic testing, serological analyses, and imaging studies, and continues to be monitored clinically following the onset of cataracts.
Although CMA and WES did not identify clinically relevant pathogenic variants in this case, the absence of molecular confirmation does not preclude a diagnosis of ARS. The literature demonstrates that a subset of clinically well-characterized patients lack a definitive genetic diagnosis, underscoring that, in many cases, the diagnostic process relies on the integration of systemic, ocular, craniofacial, and dental findings.13,14,16 Recent studies indicate that structural alterations and non-coding regulatory disruptions, particularly those involving enhancers and chromatin-interaction domains of PITX2, as well as subtle complex rearrangements, may contribute to ARS but remain undetectable by CMA or WES.18–20 Therefore, the patient’s phenotype may result from variants not detectable by the tests performed.
The absence of definitive molecular confirmation, despite comprehensive evaluation with CMA and WES, limits this case study. This limitation illustrates the current boundaries of diagnostic technologies, as certain ARS cases may involve subtle structural variants, non-coding regulatory changes, or epigenetic mechanisms that conventional methods do not detect.16,18,19 Recent investigations into whole-genome sequencing (WGS) and advanced genomic reanalyses indicate that broader adoption of these techniques in clinical practice may, in the future, resolve cases that currently lack molecular confirmation.20,23
Conclusion
Although sequencing technologies have advanced considerably, some cases of ARS do not reveal identifiable pathogenic variants through conventional approaches. While PITX2 and FOXC1 are most commonly implicated, other genes and regulatory mechanisms have also been linked to overlapping phenotypes, highlighting the genetic heterogeneity of these cases. However, a subset of patients continues to lack a defined molecular diagnosis, either due to technological constraints or the presence of uncharacterized pathogenic mechanisms. As a result, the diagnosis of ARS continues to depend, at least partially, on thorough clinical evaluation, especially when the phenotype is strongly indicative.
AI Statement
Generative artificial intelligence assistance (ChatGPT, OpenAI; version GPT-5.2) was used to assist with language editing and improving clarity of the manuscript. The authors reviewed and approved all content and are responsible for its accuracy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Michels K, Bohnsack BL. Ophthalmological manifestations of Axenfeld-Rieger syndrome: current perspectives. Clin Ophthalmol. 2023;17:819–7. doi:10.2147/OPTH.S379853
2. Axenfeld TH. Embryotoxon corneae posterius. Ber Dtsch Ophthalmol Ges. 1920;42:301–302.
3. Rieger H. Demonstration von zwei Fällen von Verlagerung und Schlitzform der Pupille mit Hypoplasie des Irisvorderblattes. Z Augenheilk. 1934;84:98–99.
4. Rieger H. Beiträge zur Kenntnis seltener Missbildungen der Iris II. Über Hypoplasie des Irisvorderblattes mit Verlagerung und Entrundung der Pupille. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1935;133(4):602–635. doi:10.1007/BF01853793
5. Rieger H. Dysgenesis mesodermalis corneae et iridis. Z Augenheilk. 1935;86:333.
6. Schinzel A, Rieger H. 2 May 1898–1 February 1986. Am J Med Genet. 1987;27(4):749–752. doi:10.1002/ajmg.1320270402
7. Song W, Hu X. The rare Axenfeld-Rieger syndrome with systemic anomalies: a case report and brief review of literature. Medicine. 2017;96(33):e7791. doi:10.1097/MD.0000000000007791
8. Seifi M, Walter MA. Axenfeld-Rieger syndrome. Clin Genet. 2018;93(6):1123–1130. doi:10.1111/cge.13148
9. National Center for Biotechnology Information. Genetic Testing Registry: axenfeld–Rieger Syndrome. Available from: https://www.ncbi.nlm.nih.gov/gtr/conditions/C3714873/.
10. Tripathy K, Salini B. Axenfeld-Rieger syndrome. In: StatPearls. Treasure Island, FL: StatPearls Publishing; 2024. Available from https://www.ncbi.nlm.nih.gov/books/NBK538504/.
11. Fan Z, Sun S, Liu H, et al. Novel PITX2 mutations identified in Axenfeld-Rieger syndrome and the pattern of PITX2-related tooth agenesis. Oral Dis. 2019;25(8):2010–2019. doi:10.1111/odi.13196
12. Arte S, Pöyhönen M, Myllymäki E, et al. Craniofacial and dental features of Axenfeld-Rieger syndrome patients with PITX2 mutations. Orthod Craniofac Res. 2023;26(3):320–330. doi:10.1111/ocr.12631
13. Zhang Y, Chen X, Wang L, Sun X, Chen Y. Heterogeneity of Axenfeld-Rieger syndrome: molecular and clinical findings in Chinese patients. Front Genet. 2021;12:732170. doi:10.3389/fgene.2021.732170
14. White S, Taranath A, Hanagandi P, et al. Neuroimaging findings in Axenfeld-Rieger syndrome: a case series. AJNR Am J Neuroradiol. 2023;44(10):1231–1235. doi:10.3174/ajnr.A7995
15. Tümer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009;17(12):1527–1539. doi:10.1038/ejhg.2009.93
16. Reis LM, Maheshwari M, Capasso J, et al. Axenfeld-Rieger syndrome: more than meets the eye. J Med Genet. 2023;60(4):368–379. doi:10.1136/jmg-2022-108646
17. Reis LM, Amor DJ, Haddad RA, et al. Alternative genetic diagnoses in Axenfeld-Rieger syndrome spectrum. Genes. 2023;14(10):1948. doi:10.3390/genes14101948
18. Jiang Y, Peng Y, Tian Q, et al. Intergenic sequences harboring potential enhancer elements contribute to Axenfeld-Rieger syndrome by regulating PITX2. JCI Insight. 2024;9(9):e177032. doi:10.1172/jci.insight.177032
19. Jiang Z, Zhang Y, Wang L, et al. Complex genomic rearrangement with deletion of PITX2 in a Chinese family with Axenfeld-Rieger syndrome: a case report and literature review. Mol Vis. 2024;30:466–476.
20. Farris J, Khanna C, Smadbeck JB, et al. Complex balanced intrachromosomal rearrangement involving PITX2 identified as a cause of Axenfeld-Rieger syndrome. Am J Med Genet A. 2024;194(5):e63542. doi:10.1002/ajmg.a.63542
21. Maclean K, Smith J, Heaps LS, et al. Axenfeld-Rieger malformation and distinctive facial features: clues to a recognizable 6p25 microdeletion syndrome. Am J Med Genet A. 2005;132(4):381–385. doi:10.1002/ajmg.a.30274
22. Souzeau E, Siggs OM, Pasutto F, et al. Gene-specific facial dysmorphism in Axenfeld-Rieger syndrome caused by FOXC1 and PITX2 variants. Am J Med Genet A. 2021;185(2):434–439. doi:10.1002/ajmg.a.61982
23. Mitscherling J, Sczakiel HL, Kiskemper-Nestorjuk O, et al. Whole genome sequencing in families with oligodontia. Oral Dis. 2024;30(6):3935–3950. doi:10.1111/odi.14816
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.