Back to Journals » International Medical Case Reports Journal » Volume 18
Autosomal Recessive Limb-Girdle Muscular Dystrophy Type 10 (LGMD,10), Caused by a Novel Homozygous Variant in the TTN Gene
Authors Alghamdi OA ![]() , Obaid O
, Obaid O ![]() , Sayed AG, Farhan H
, Sayed AG, Farhan H ![]() , Sayed J
, Sayed J ![]()
Received 22 July 2024
Accepted for publication 31 May 2025
Published 4 June 2025 Volume 2025:18 Pages 683—689
DOI https://doi.org/10.2147/IMCRJ.S483508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
Omar Ahmed Alghamdi,1 Osama Obaid,2 Ahmed Gamal Sayed,3 Hania Farhan,3 Jamal Sayed1
1Department of Pediatrics, Security Forces Hospital Makkah, (SFHM), Makkah, Saudi Arabia; 2Department of Pediatrics, Maternity and Children Hospital, Makkah, Saudi Arabia; 3College of Medicine, Alfaisal University, Riyadh, 11533, Saudi Arabia
Correspondence: Omar Ahmed Alghamdi, Email [email protected]
Abstract: Limb-girdle muscular dystrophy was first introduced in the 1950s as a distinct family of unusual genetic diseases. The prevalence of the disease is about 4– 7/1000, with a spectrum of onset at different ages. LGMD has a cluster of symptoms varying in severity and presentation in patients. Limb-girdle muscular dystrophy`s inheritance is unique as it can be autosomal dominant or recessive. We report a young boy with autosomal recessive limb-girdle muscular dystrophy (LGMD), presented with distal muscle weakness in all four limbs for three years and thinning of legs, arms, and thighs. Our gene of focus is TTN, which is associated with muscle elasticity and myogenesis. Our subtype reported is currently associated with a new homozygous TTN variant; thus, we are writing a novel variant mutation causing limb-girdle muscular dystrophy type 10.
Keywords: LGMD, muscular dystrophies, TTN gene, titinopathy
Introduction
Limb-girdle muscular dystrophy (LGMD) was introduced in the early 1950s as a term for a heterogeneous group of rare progressive genetic disorders comprising more than 25 genetic diseases and 30 plus different subtypes.1–3 LGMDs have a broad spectrum of clinical manifestations, from gradually progressive muscle deterioration to severe subtypes in infants, diminishing their ability to ambulate without assistance.4 The prevalence of LMGDs is about 4–7/1000, with childhood, teenage, or adult-onset.5 The various subtypes of LMGD are either autosomal dominant or recessive inheritance patterns, out of which the most significant subtype in our reported case is LGMD type 10 (LMGD2J).1,4,6 LGMD type 10 is due to a titin gene (TTN) defect, which encodes large multifunctional polypeptides required to maintain muscle elasticity and myogenesis.7,8 A mutation in the TTN gene is responsible for many conditions, such as congenital centronuclear myopathy, early-onset myopathy with fatal cardiomyopathy, and typically adult-onset muscular dystrophy.8 Herein, we report the case of a 6-year-old male presenting with LGMD who was found to have a new missense homozygous mutation in the TTN gene.
Case Report
A 6-year-old male presented with foot drop in both lower limbs since 18 months of life, at the same time he began to walk. He had a static course with no significant deterioration in gait or improvement. He had difficulty climbing stairs, running, and getting up from a squatting position without assistance, and he ran with a waddling gait. He has a thin body habitus, including his trunk’s upper and lower limbs. There was atrophy of the calves noted bilaterally as well (Figure 1) and winging of the scapula (Figure 2), There is no history of distal muscle weakness, twitching of the muscles, abnormal movements, periodic paralysis or muscle spasms, facial or cranial nerve involvement, and no bladder or bowel disturbances. He is the first child born of a consanguineous marriage, with a younger healthy female sibling of 3 years. Antenatal and perinatal history was insignificant for complications during pregnancy or delivery. His parents, in their fourth decade of life, reveal no symptoms and are completely normal on examination. Neurological examination revealed symmetrical, predominant limb-girdle weakness [score of 4/5 at knee flexion, 4/5 at knee extension, 2/5 at plantar dorsiflexion, 5/5 plantar flexion, 5/5 at hip flexion, 4/5 at hip extension, 5/5 hip adduction, 5/5 hip abduction, 5/5 upper limb hand flexion and extension, 5/5 extension at both elbows and 4/5 shoulder abduction and adduction], Ocular, facial, bulbar, and neck muscles were spared. All deep tendon reflexes were diminished; there were no fasciculations, upper motor signs, calf pseudo-hypertrophy, or joint contractures. Sensation (light touch, pinprick, position sense) was also normal. No intellectual disability was noted. The patient could tell a story, eat with a spoon, color match, play with peers, and understand 3 step commands. Respiratory, Cardiovascular, and Abdominal system was unremarkable. Investigations showed normal creatinine phosphokinase (CK) 160 IU/l. (Reference: 24–190 IU/l.), Whole exome sequencing was performed in a commercial lab after genetic consultation, and the results showed; a homozygous missense variant of unknown significance in the TTN gene, (NM_001267550.2): c.48430G>C (p. Asp16144His), Chr2(GRCh37): g.179480398C>G, 179,479,798–179,480,998, related to the group of neuromuscular diseases and myopathies. This variant with 0% allele frequency in the general population (geno MAD: no frequency) and a deleterious effect of more than 20 CAD scores is highly supported by biochemical evidence of very mild elevation in creatine kinase level. The variant in our patient has not been reported before in any case with a similar phenotype. Titinopathies are a genetically and clinically heterogeneous group of disorders with different modes of inheritance. It can follow autosomal dominant and autosomal recessive mode. The titin protein is crucial in muscle assembly, force transmission at the Z line, and maintaining resting tension in the I band region.9 Sanger sequencing and family segregation analysis were conducted, revealing that both parents were heterozygous for the same missense variant (Figure 3). At that time, both parents were asymptomatic. Unfortunately, no other siblings could be screened for further segregation analysis in this family. extensive neuromuscular evaluation suggests apparent and combatable phenotype with limb-girdle muscular dystrophy in the form of distal muscle weakness, muscle atrophy, and myopathic changes on EMG. We consider our variant as a novel variant for limb girdle muscular dystrophy AR type 10 (Omim #608807).10
 |
Figure 1 Winging of the scapula. |
 |
Figure 2 Thinning of the lower limb. |
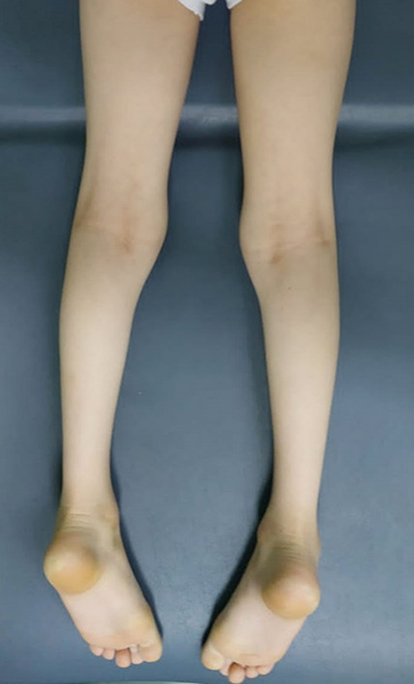 |
Figure 3 Atrophy of the calves’ muscles. |
We started physical therapy in conjunction with the use of an ankle-foot orthosis (AFO). Which significantly improved his gait and helped prevent high foot rise (steppage gait). However, once the physical therapy sessions concluded, the foot drop and frequency of falls increased again. During long-term follow-up, we noted some limitations in ankle dorsiflexion, with a range of motion reduced by about 10 degrees. Nonetheless, his overall upper and lower limb strength remained stable compared to findings from the initial visit, and he is able to walk independently two years after his initial presentation.
Discussion
LGMDs are divided into autosomal dominant (LGMD1) and autosomal recessive (LGMD2). Every time a new locus is discovered, it is added chronologically as a letter under the two distinct types. There are 26 subtypes of LGMD type two and eight subtypes of LGMD type 1 as of 2018.11 The clinical presentation of LGMD is progressive, with the usual initial presentation being atrophy and weakness of the shoulder, thigh, and hip. Due to this, patients complain of frequent falls, toe walking, and the development of a waddling gait, which can then later lead to hyperlordosis.12 As the disease progresses, mobility becomes increasingly difficult, such as rising from the floor and climbing stairs. However, facial muscles seem to be spared.13,14 The calves may be found hypertrophied as a result of compensation.3,15 Eventually, most patients are indicated for wheelchair use in the later years and may experience distal muscle weakness, such as the arms.6 The patients are at further risk of joint contractures, cardiac issues, and respiratory insufficiency.14 Making a diagnosis based on particular clinical criteria in individuals with a distinct LGMD presentation may be possible. On the other hand, it is crucial to note that for practically any of the several types of LGMD, there will be individuals with atypical symptoms, and a definitive diagnosis cannot be anticipated or ruled out for these patients. Regarding age of onset and degree of advancement, there are significant variances across the various forms of LGMD and within the same form. One should also be careful about rushing to a diagnosis because several LGMDs have only recently been characterized, and the phenotypic range for many still needs to be well defined. When assessing a patient with an unknown type of LGMD, it is critical to retain an open mind.14 There is very little research or cases reported regarding LGMD10. However, from what was found in the literature, we know the usual presentation begins with proximal muscular weakening in the first decade of life, followed by all the cases being wheelchair reliant before the age of 25. The blood CK activity of all LGMD10 patients was elevated, as was the dystrophic pattern of their muscle biopsies.14 Our patient did not see significant disease progression, as the course was pretty static from the onset at 18 months. However, the usual symptoms of difficulty in climbing stairs, running, getting up from squatting position without assistance, and a waddling gait were seen. The blood CK levels were not particularly elevated either (160 IU/l). Thus, molecular genetic testing is indicated, and Whole exome sequences are requested (WES). Our genetic testing methodology was that nuclear DNA was isolated then exonic regions were enriched using the Agilent SureSelect V6 kit (Agilent Technologies, USA). Then the sequencing was performed on Illumina NovaSeq to an average coverage depthing of 100–130X. The alignment of sequencing raw data to the hg 19 genome assembly, variant calling, and annotation were performed by bioinformatics tool. Assessment and classification of filtered variant were carried out using ACMG guidelines. The titin protein is a ligand-binding site for several other muscle proteins, such as myosin, telethonin, calpain-3, -actinin, and myomesin. Western blot investigation cannot reveal a decrease in titin expression, although all LGMD10 patients have an almost complete loss of calpain-3. Such finding can be explained by the calpain binding location mutation in the titin gene.14 Titin interacts with about 25 additional proteins. This connectivity serves several functions, including the optimization of myocyte contractility, the formation and maintenance of a stable sarcomeric structure, and viscoelasticity, the quality regulation of myofilamentary proteins, and transcriptional repression or activation in the context of mechanosensing.15 Titin, a large sarcomeric protein, spans a half-sarcomere from the sarcomeric Z-disc to the M-band and creates a continuous filament system in striated muscle myofibrils. Most alternative splicing events occur in the I-band area of titin, although they may also influence the Z-disc and M-band regions. The Z-disc area has longer isoforms in cardiac muscle, whereas,
I-band titin has longer isoforms in skeletal muscles. The participation of distinct titin isoforms in anatomically confined myopathies may be linked. The M-band of titin is a hotspot mutation area near its extreme C-terminus.16 Muscular dystrophies induced by pathogenic TTN variants are diverse and counts as a broad group. LGMD10 and tibial muscular dystrophy are two of them. Furthermore, the TTN gene, is one of the primary culprits involved in dilated cardiomyopathies, accounting for 17% of all the cases. Given that the physiopathology of these illnesses is unclear, it becomes critical to record such disorders to enhance diagnosis and gain a better genotype-phenotype correlation for variations.17 In the human gene mutation database (HGMD), 346 TTN disease-causing mutations have been associated with at least ten different conditions, including isolated cardiomyopathies, purely skeletal muscle phenotypes, and infantile diseases affecting both types of striated muscles.7 Despite being one of the least-contributing genes, many unique pathogenic variants (more significant allelic heterogeneity). suggesting that if whole genome sequencing is used instead of panel testing, an even higher number of distinct pathogenic variants, such as deep intronic/deletion/duplications, might be found in the TTN gene with confirmed diagnosis of LGMD10. As a result, LGMD may continue to be clinically underdiagnosed entity overall.18
WES showed a mutation in TTN (NM_001267550.2):c.48430G>C (p.Asp16144His), Chr2(GRCh37):g.179480398C>G. Based on in silico prediction tool of polyphen 2 algorithm and mutation assessor, this variant predicted to be pathogenic. This variant not present in general population and not present in geno MAD and this indicate very low (0%) allele frequency in general population.
Conservation Scores of this variant is positive based on phyloP which also suggest deleterious effect. Based on ACMG criteria this variant classified as variant of uncertain significant.
Titin contains 6 M band-encoding exons at the C terminus, exons 358 to 363, referred to as Mex1 to Mex6. These exons are constitutively expressed in both skeletal and cardiac muscleIn.18 Another study, a homozygous missense mutation (c.98807G > A; p.Arg32936His), in exon 353 of the gene was discovered, which has been linked to symptoms of LGMD.7 One common variation has been identified as causing the LGMD (the recessive 11-bp insertion/deletion seen in LGMD10). Such variants are frequently regarded as accidental and/or variants of unknown clinical significance (VUS).19 Previous reported cases of homozygous TTN by Dabby et al reported a 29-year-old man of Romanian and Hungarian descent who presented with slowly progressive lower limbs proximal muscle weakness resulting in difficulty in climbing stairs and getting up from a seated position. He also had mild weakness of the shoulder girdle muscles.20 Another report from Zheng et al reported 3 adult sibs from a consanguineous Han Chinese family with LGMD10. The patients presented between 13 and 16 years of age with difficulty running and climbing, frequent falls, and Gowers sign. They developed proximal muscle weakness and atrophy affecting the lower and upper limbs. The disorder was progressive, and they developed elbow and ankle joint contractures. It is difficult to compare our patient with these reported cases due to lack of long term follow up and early onset disease.16
Conclusion
LGMDs are a distinct family of muscular dystrophies that require more clinical care by the practitioners in the medical field. The disease can be a challenging entity to diagnose, given that can mimic multiple hereditary muscular dystrophies, requiring the necessary clinical suspicion to reach an appropriate diagnosis. As illustrated, a detailed physical examination is definitive for reaching an accurate diagnosis. It can prevent extensive investigations that burden families with high economic cost, thus allowing for an appropriate treatment plan. Our experience in regards to the TTN genetic variants-related disorders do further recommend to be enhanced by more studies; for that, our case, both clinically and genetically, aims to have documentation for a presentation of one of the LGMD subtypes, particularly the LGMD10. Our novel variant can aid physicians in detecting such a rare entity to reach a legitimate clinical diagnosis with proper genetic counseling for the disease control.
Ethics Approval
We confirm that this study adheres to the journal’s ethical standards, and institutional approval from Security Forces Hospital Makkah is not required for publishing the case details.
Informed Consent Statement
Written informed consent for publication was obtained from the patient’s parent for publication of the case details, including the history, physical findings, laboratory reports, and images.
Acknowledgment
We hereby thank Dr. Heba Mohammed El masry the genetic director of Jeddah mega lab for her contribution and support in the analysis of our genetic study in our case.
Funding
This research received no external funding.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Itoh-Satoh M, Hayashi T, Nishi H, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291(2):385–393. doi:10.1006/bbrc.2002.6448
2. OMIM® and Online Mendelian Inheritance in Man® Johns Hopkins University. Available from: https://www.omim.org/clinicalSynopsis/608807.
3. Mitsuhashi S, Kang PB. Update on the genetics of limb girdle muscular dystrophy. Semin Pediatr Neurol. 2012;19(4):211–218. doi:10.1016/j.spen.2012.09.008
4. Liu J, Harper SQ. RNAi-based gene therapy for dominant limb girdle muscular dystrophies. Curr Gene Ther. 2012;12(4):307–314. doi:10.2174/156652312802083585
5. Lee SJ, Choi E, Shin S, Park J. Genetically confirmed limb-girdle muscular dystrophy type 2B with DYSF mutation using gene panel sequencing: a case report. Medicine. 2020;99(28):e20810. doi:10.1097/MD.0000000000020810
6. Wang G, Lv X, Xu L, Zhang R, Yan C, Lin P. Novel compound heterozygous mutations in the TTN gene: elongation and truncation variants causing limb-girdle muscular dystrophy type 2J in a Han Chinese family. Neurol Sci. 2022;43(5):3427–3433. doi:10.1007/s10072-022-05979-z
7. Yiş U, Diniz G, Hazan F, et al. Childhood onset limb-girdle muscular dystrophies in the Aegean part of Turkey. Acta Myol. 2018;37(3):210–220.
8. Audhya IF, Cheung A, Szabo SM, Flint E, Weihl CC, Gooch KL. Progression to loss of ambulation among patients with autosomal recessive limb-girdle muscular dystrophy: a systematic review. J Neuromuscul Dis. 2022;9(4):477–492. doi:10.3233/JND-210771
9. Khan A, Wang R, Han S, et al. Homozygous missense variant in the TTN gene causing autosomal recessive limb-girdle muscular dystrophy type 10. BMC Med Genet. 2019;20(1):166. doi:10.1186/s12881-019-0895-7
10. Peddareddygari LR, Oberoi K, Grewal RP. Novel titin gene mutation causing autosomal dominant limb-girdle muscular dystrophy. Cureus. 2022;14(10):e30550. doi:10.7759/cureus.30550
11. Chu ML, Moran E. The limb-girdle muscular dystrophies: is treatment on the horizon? Neurotherapeutics. 2018;15(4):849–862. doi:10.1007/s13311-018-0648-x
12. Ramos E, Pardo S, Mas Rodríguez MF, Vélez J. Limb-girdle muscular dystrophy type 2A resulting from c.C479G and c.G1818A mutations in the calpain-3 gene. J Clin Neuromuscul Dis. 2015;17(2):59–62. doi:10.1097/CND.0000000000000097
13. Saylam E, Moore SA, Aravindhan A, et al. A novel noncoding FKRP mutation in early onset limb-girdle muscular dystrophy. Neurology. 2019;6(1):e388. doi:10.1212/NXG.0000000000000388
14. Straub V, Bushby K. The childhood limb-girdle muscular dystrophies. Semin Pediatr Neurol. 2006;13(2):104–114. doi:10.1016/j.spen.2006.06.006
15. Linke WA, Hamdani N. Gigantic business: titin properties and function through thick and thin. Circ Res. 2014;114(6):1052–1068. doi:10.1161/CIRCRESAHA.114.301286
16. Zheng W, Chen H, Deng X, et al. Identification of a novel mutation in the titin gene in a Chinese family with limb-girdle muscular dystrophy 2J. Mol Neurobiol. 2016;53(8):5097–5102. [PubMed: 26392295, related citations]. doi:10.1007/s12035-015-9439-0
17. Outtaleb FZ, Tazzite A, Gazzaz B, Dehbi H. Limb-girdle muscular dystrophy type 2J: case report. In: Endocrine Abstracts. Vol. 81. Bioscientifica; 2022.
18. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574–1587. doi:10.1002/acn3.649
19. Rich K, Morales A, Eck D, et al. Investigation of TTN variants in patients with limb-girdle muscular dystrophy identifies novel titinopathies (N2. 002). Neurology. 2019;92(15_supplement):N2–002.
20. Dabby R, Sadeh M, Hilton-Jones D, et al. Adult onset limb-girdle muscular dystrophy--a recessive titinopathy masquerading as myositis. J Neurol Sci. 2015;351:120–123. [PubMed: 25772186, related citations]. doi:10.1016/j.jns.2015.03.001
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.