Back to Journals » Hepatic Medicine: Evidence and Research » Volume 8
Autophagy in hepatocellular carcinomas: from pathophysiology to therapeutic response
Authors Dash S, Chava S, Chandra P, Aydin Y, Balart L, Wu T
Received 22 August 2015
Accepted for publication 30 November 2015
Published 22 February 2016 Volume 2016:8 Pages 9—20
DOI https://doi.org/10.2147/HMER.S63700
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Srikanta Dash,1,2 Srinivas Chava,1 Partha K Chandra,1 Yucel Aydin,2 Luis A Balart,2 Tong Wu1
1Department of Pathology and Laboratory Medicine, 2Department of Gastroenterology and Hepatology, Tulane University School of Medicine, New Orleans, LA, USA
Abstract: Autophagy is an intracellular lysosomal degradation process performed by the cells to maintain energy balance. The autophagy response plays an important role in the progression of liver disease due to hepatitis virus infection, alcoholic liver disease, nonalcoholic fatty liver disease, liver cirrhosis, and hepatocellular carcinoma (HCC). An increased autophagy response also contributes to the pathogenesis of liver disease through modulation of innate and adaptive immune responses; a defective cellular autophagy response leads to the development of HCC. Recent progress in the field indicates that autophagy modulation provides a novel targeted therapy for human liver cancer. The purpose of this review is to update our understanding of how the cellular autophagy response impacts the pathophysiology of liver disease and HCC treatment.
Keywords: hepatocellular carcinoma, macroautophagy, microautophagy, autophagy inhibitor, chloroquine, hydroxychloroquine, sorafenib
Introduction
Autophagy is a lysosomal degradation mechanism important for cell survival under conditions of starvation, stress, or infection. The mechanism evolved as a means of regenerating energy from intracellular materials (cytoplasm, organelles, protein aggregates, etc) to meet energy requirements in low-nutrient conditions.1 Autophagy, is one such mechanism and is induced by a variety of stimuli, including cytokine stimulation, stress, diverse pathogens, accumulation of misfolded proteins, and damaged organelles.2 The importance of autophagy in liver homeostasis and energy conservation has been verified in animal models. For example, inhibition of autophagy in mouse models has been observed to impair lysosomal degradation in hepatocytes, resulting in a fourfold increase in liver weight.3 Likewise, nutrient starvation experiments in mice have shown that autophagy is responsible for degradation of 35% of total proteins in the liver within 24 hours.4 These data illustrate the importance of autophagy in the maintenance of liver functions and liver weight. Autophagy also plays a major role in the modulation of innate and adaptive immune responses in the pathogenesis of chronic liver diseases, including diseases due to cancer, diabetes, neurodegeneration, and aging.
Initiation and termination of autophagy are linked to cellular nutrient-sensing mechanisms.5,6 For example, the molecule AMP-kinase (AMPK) senses cellular energy requirements through AMP to ATP ratios in the cell cytoplasm. High AMP levels reflect low energy states in the cell, and under these conditions, AMPK can initiate autophagy through inactivation of mTOR1 (a mechanistic target of rapamycin complex 1) or by phosphorylation of ULK1/2 protein. Another autophagy-inducing signal is related to inhibition of mTOR1 by depletion of amino acid levels in the cytoplasm. It is now believed that inhibition of mTOR1 due to low energy states in the cell activates autophagy, whereas activation of mTOR1 due to high energy states inhibits cellular autophagy.
Three different types of autophagy response have been described in the mammalian cells: macroautophagy, chaperon-mediated autophagy (CMA), and microautophagy.7 The differences among these three types of autophagy are illustrated in Figure 1. In macroautophagy, a portion of cytosol is engulfed by a double-membrane structure called an autophagosome, which fuses with a lysosome to become an autophagolysosome; the contents of the autophagolysosome are degraded by lysosomal enzymes (proteases, lipases, nucleases, and glycases) in a process coordinated by 37 ATG proteins. Several cellular compartments, including the endoplasmic reticulum (ER), Golgi/trans-Golgi apparatus, and plasma membrane, participate in autophagosome formation. CMA is responsible for the degradation of cytosolic proteins under conditions of stress. All CMA substrates contain a consensus pentapeptide motif (KFERQ) that is recognized by a cytosolic chaperone, for example, HSC70;8 HSC70 binds to Lamp2a, which results in the direct translocation of unfolded protein substrate across lysosomal membranes and subsequent degradation of the cytosolic proteins. In microautophagy, cytosolic material is directly engulfed by the lysosome via membrane rearrangement. Recently, microautophagy has been renamed on the basis of the cargo it degrades, as mitophagy, pexophagy, reticulophagy, and ribophagy.
 | Figure 1 Three different types of autophagy response allow degradation of cytosolic content and organelles in the lysosome. |
This review focuses mainly on macroautophagy (autophagy) and its role in the pathogenesis of liver diseases, cirrhosis, and hepatocellular carcinoma (HCC).
Molecular interactions in autophagy
In general, autophagy is coordinated by five different steps, known as initiation, nucleation, elongation, maturation, and degradation (Figure 2). Decreasing mTOR1 levels caused by low nutrient levels, such as low levels of amino acid, lipids, and sugars, activate autophagy signaling. During basal-level autophagy, Unc-51-like kinase (ULK1/2), ATG13, FIP200, and ATG101, exist in an inactive complex with mTOR1. During initiation, a decrease in mTOR activity leads to phosphorylation and translocation of the ULK complex (ULK1–ATG13–FIP200–ATG101) from the cytoplasm to the ER.6,9,10 The interaction of the ULK complex with ER-resident proteins leads to initiation of autophagy.6 The second step, nucleation, occurs as the ULK complex enlarges, due to interactions with class III phosphatidylinositol 3-kinase (PI3K) consisting of either Beclin 1–ATG14L–PI3KCIII–p150–Ambra1 or Beclin 1–UVRAG–PI3KCIII–p150–Bif1.11 Additional ER-resident proteins (DFCPI and WIPI) facilitate the nucleation and creation of a curved double-membrane structure. It has been shown that the autophagy process can be inhibited if Beclin 1 forms a complex with Rubicon or antiapoptotic protein (called Bcl-2).12 The third step, called elongation, is primarily mediated by ubiquitin-like protein conjugation systems. The ATG12–ATG5 complex associates with ATG16 to form ATG12–ATG5–ATG16 (ATG16L), which localizes at the autophagosomal membrane. The ATG16L complex then promotes LC3 lipidation by PE and membrane insertion. The LC3–PE is localized at the inner and outer membranes of the autophagosome. During elongation, the LC3 protein on the autophagosome can interact with misfolded and polyubiquitinated proteins through autophagy receptor proteins (p62, NBR1, or NIX).13,14 Proteins degraded through autophagy are recognized by autophagy receptors such as p62 and NBR1 that interact with ubiquitin-like protein LC3, which modifies the target proteins for delivery to the lysosome. The fourth step, maturation, is related to autophagosome completion. The autophagosome undergoes two maturation steps. First, the autophagosome fuses with multivesicular endosomes to form an amphisome, where proton pumps are acquired for acidification. Second, the amphisomes fuse with a lysosome to become an autolysosome. Finally, the cellular materials present inside the autolysosome are degraded by the action of different lysosomal enzymes into amino acids, lipids, and sugars. The degradation products, such as amino acids, lipids, and sugars, are released from the autolysosome via lysosome efflux transporters for reuse, for example, in the production of new proteins. The release of nutrients from the autophagolysosome reactivates mTOR, which triggers autophagy termination and the formation of nascent lysosomes. This process is called autophagic lysosome reformation. Under conditions of low nutrition, the cycle is repeated.
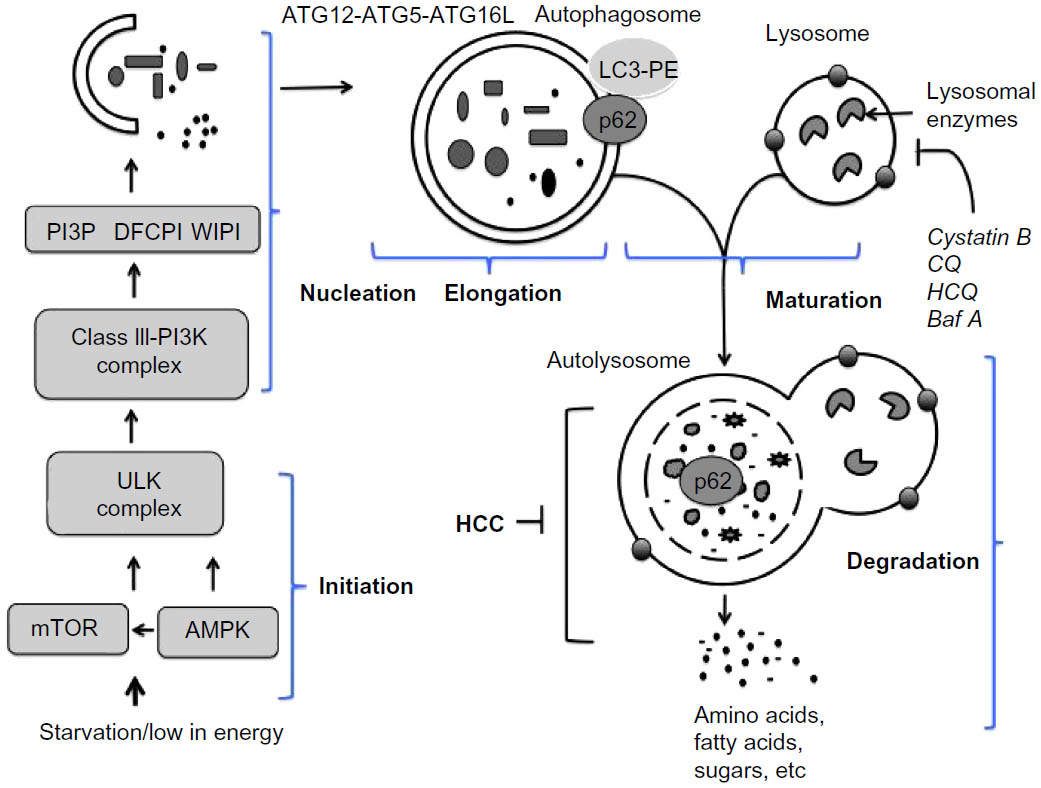 | Figure 2 Molecular signaling pathway involved in autophagy. |
The fusion of an amphisome with a lysosome requires Lamp2a and a small amount of GTPase Rab7.15,16 The retrieval of ATG proteins from the mature autophagosome is mediated by ATG2, ATG9, and ATG18.17 The ER is the most common site for the initiation of the autophagy membrane source, but autophagy initiation can involve Golgi apparati, endosomes, plasma membrane, and mitochondria. Different forms of autophagy can be induced by nutrient starvation, stress related to virus infection, and chemotherapy. Increased autophagic degradation can lead to cell death, which may be important in tissue homeostasis. A decreased autophagy response has also been linked to the development of cancer and neurological diseases. We here describe the important role of autophagy in chronic liver disease in humans.
Autophagy in chronic liver diseases
Mechanisms of acute and chronic liver injury in humans have been linked mainly to infection by hepatitis viruses, alcohol abuse, and fat deposition. These agents target mostly hepatocytes, as hepatocytes are the predominant cell type in the liver subject to acute and chronic injury. One of the host-related factors responsible for the evolution of chronic liver disease to liver cirrhosis and HCC is the degree of hepatocellular injury. An increase in serum aminotransferase has been used as a surrogate marker for assessing the extent of hepatic injury in patients with chronic liver diseases of both viral and nonviral etiology.
Liver injury is caused by three distinct types of cell death: apoptosis, necrosis, and autophagy. Apoptosis is a form of cell death mediated by an intracellular proteolytic cascade, in which cells die neatly. Apoptotic cells are usually phagocytosed either by neighboring cells or by a macrophage. Necrosis is a form of cell death mediated by acute injury in which the cell swells, bursts, and spills its contents into surrounding areas, causing an inflammatory response. Apoptosis and autophagy are interrelated biological processes important for maintaining tissue homeostasis and carcinogenesis. Hepatocellular apoptosis acts as a tumor suppressor or (prodeath) in the liver. It has been shown that the genes controlling apoptosis (Bcl-2) are involved in carcinogenesis. Oncogenic mutations in the Bcl-2 gene that inhibit apoptosis can lead to tumor initiation, progression, or metastasis. Alternatively, oncogenes that promote cellular apoptosis can initiate selective pressure to override apoptosis during multistage carcinogenesis. A number of excellent reviews have described the role of hepatocyte apoptosis in prodeath (tumor suppression) during chronic liver injury and carcinogenesis.18,19 Similarly, hepatocellular autophagy can also cause tumor suppression in chronic liver disease, and an impaired autophagy response can lead to malignant transformation and HCC. It is well known that HCC develops in the background of liver cirrhosis after many years of chronic liver disease due to hepatitis virus infection and alcoholic and nonalcoholic liver diseases. Available evidence suggests that the autophagy response is deregulated in chronic liver disease and liver cirrhosis, which can lead to HCC. To date, a consistent autophagic tumor suppressor mechanism causing the development of chronic liver disease, cirrhosis, and HCC has not been identified.
Infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) has been shown to induce an autophagy response both in vitro and in vivo in chronically infected liver.20 It has been observed that the autophagy response promotes HBV and HCV replication, whereas autophagy suppression inhibits replication. These results suggest that viral infection induces the autophagy response to degrade organelles and long-lived proteins needed to generate energy and sustain virus replication in hepatocytes, and, if uncontrolled, the induced autophagy response could lead to autophagic cell death and the elimination of infected hepatocytes in the liver. Actually, both viruses lead to chronic infection.
The prosurvival function of HBV and HCV infection has not been well established, with the exception of several studies that have used hepatoma cell lines to show that HBV or HCV infection inhibits autophagic degradation.21–23 The autophagy response is decreased in chronic liver disease on account of both alcoholic and nonalcoholic liver diseases. This conclusion is supported by the fact that suppression of autophagy by pharmacological agents or siRNA against ATG7 significantly exacerbates liver injury, whereas autophagy induction improves chronic liver disease caused by alcoholic and nonalcoholic fatty liver disease.24 The decreased autophagy response in alcoholic and nonalcoholic liver disease can lead to the accumulation of misfolded protein aggregates, which can increase oxidative stress, DNA damage, and genomic instability, all of which favor carcinogenesis. These results are consistent with human data showing that chronic liver disease and HCC develop in the presence of these factors. Since autophagy induction also improves alcoholic and nonalcoholic liver diseases, it is expected that autophagy induction should also improve liver cirrhosis related to alcoholic and nonalcoholic fatty liver disease. It appears that autophagy modulation (induction or inhibition) may be a potential therapeutic strategy for treatment of liver cirrhosis, but so far this strategy has produced mixed results, with the exception that autophagy induction using rapamycin has been shown to be beneficial for the treatment of hepatic fibrosis due to alpha-1 antitrypsin deficiency.25 Additional investigations to understand the role of autophagy in liver cirrhosis should guide whether autophagy inhibition or induction strategies will be beneficial for the treatment of liver fibrosis.
Taken together, the evidence indicates that the autophagy response increases in chronic liver disease, and persists in the stage of liver cirrhosis (Figure 3). An understanding of whether autophagy acts as a cell death pathway or a prosurvival pathway should allow the development of novel therapeutic strategies for treatment of liver cirrhosis and HCC.
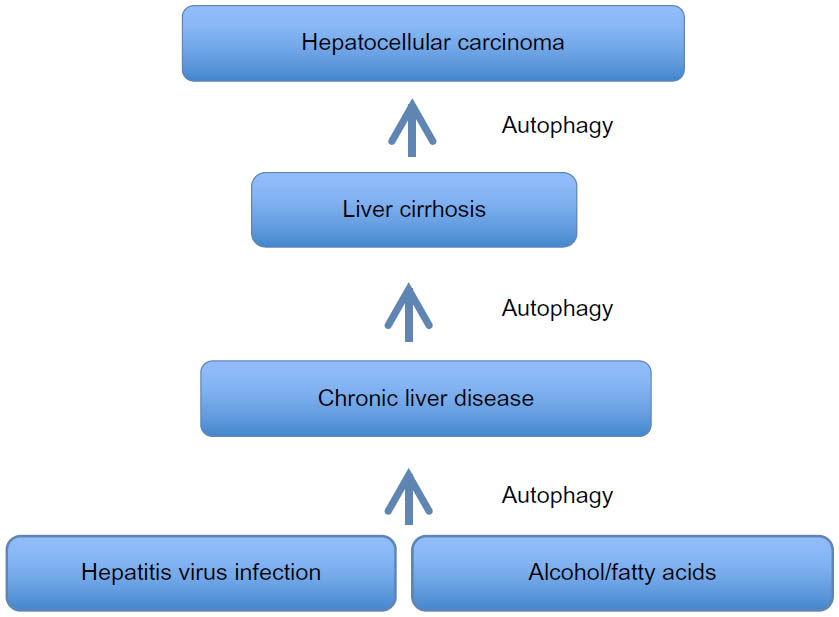 | Figure 3 Summary of autophagy response in chronic liver disease, liver cirrhosis, and hepatocellular carcinomas. |
Autophagy as a tumor suppressor mechanism in HCC
HCC accounts for more than 500,000 to 600,000 deaths per year worldwide.26 During the last 2 decades, significant progress has been made in understanding the role of autophagy in cancer development, including the development of HCC.27–29 Available evidence suggests that autophagy may serve as a tumor suppressor in cases of chronic liver disease and liver cirrhosis, and that autophagy deficiency may lead to HCC.
The following studies support the idea that HCC develops in the absence of autophagy. Abnormal expression of the autophagy gene Beclin 1 has been found associated with the development of a variety of cancers, including ovarian, breast, prostate, melanoma, colon, and brain.30–37 Heterozygous deletion of Beclin 1 increases susceptibility to spontaneous malignancies and accelerates HBV-related HCC.38 Mice lacking one copy of the gene encoding for the Beclin 1 regulator protein (called AMBRA1) also develop tumors.39 The role of other autophagy genes (UVRAG, Bif1, ATG4C, ATG5, and ATG7) in tumor suppression has been confirmed in mouse models.40–44
Studies by Takamura et al43 show that deletion of either the ATG5 or ATG7 gene in mice results in the development of hepatomegaly and hepatocellular adenoma at age 6–9 months. Takamura et al also found that accumulations of ubiquitinated proteins/aggregates are present in the hepatocytes of tumor-bearing mice but not in the hepatocytes of nontumor mice. They concluded that the tumor cells originated from autophagy-deficient hepatocytes, in association with mitochondrial swelling, p62 accumulation, oxidative stress, and an increased DNA damage response. Specific deletion of p62 expression in the hepatocytes of ATG7-deficient mice decreased tumor size, thus supporting the observation that hepatic p62 expression contributes to tumorigenesis.
Another study addressing p62 expression showed that p62 accumulation leads to hepatocellular adenoma through activation of Nrf2 target genes. The authors showed that p62 competed with the binding between Nrf2 and Keap1, resulting in enhanced transcriptional activation of Nrf2-specific genes in the autophagy-deficient hepatocytes.44 Liver-specific autophagy-deficient mice contained adenomas linked to the formation of p62- and Keap1-positive cellular aggregates, and activation of Nrf2 target genes.
To verify whether this mechanism operates in humans, we showed that high accumulations of autophagy flux protein (p62) are present in paraffin-embedded HCC tissues (Figure 4). Thus, p62 expression was found in the HCC samples but not in the surrounding nontumorous hepatocytes.45 The deposition of p62/ubiquitin/keratin-like protein aggregates observed in ATG7 knockout mouse is consistent with Mallory body detection in the hepatocytes of patients with alcoholic and nonalcoholic liver diseases.46 Decreased p62 expression, as determined by Western blot analysis, has been used as a reliable marker of autophagy flux. The p62 protein has been found to contribute to carcinogenesis through multiple signaling pathways, including NF-kB, Nrf2, Wnt/β-catenin, and mTOR.47–53 All of these reports provide evidence that autophagy plays a role in tumor suppression, and that autophagy deficiency could lead to HCC. Mechanistically, autophagy deficiency leads to the accumulation of misfolded proteins, dysfunctional mitochondria, the generation of reactive oxygen species, oxidative stress leading to increased DNA damage, the accumulation of double-stranded DNA breaks, increased DNA content, chromosomal instability leading to the cell transformation stage, and carcinogenesis (Figure 3). If this hypothesis is correct, then the mechanism by which autophagy deficiency is created in cirrhotic livers should be an interesting area of investigation.
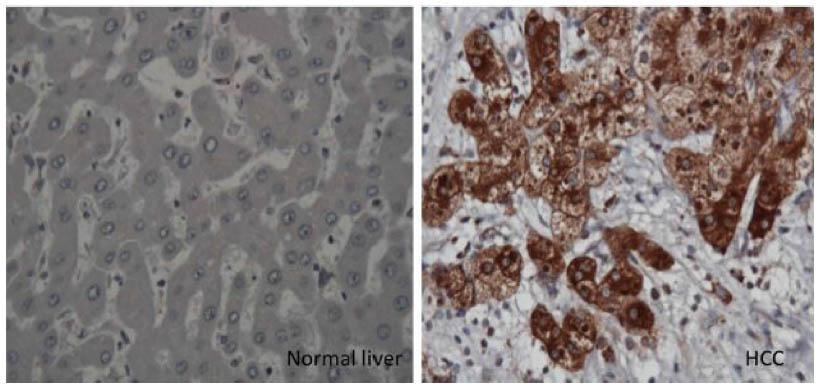 | Figure 4 Immunohistochemical staining of hepatocytes in HCC and cirrhotic liver. |
Autophagy as a prosurvival (oncogenic) mechanism in HCC
Cancer cells need autophagy to generate the energy required to sustain growth and survival under various metabolic as well as therapeutic stress conditions.54 Autophagy plays a prosurvival role during HCC development. Increased autophagy flux has been reported in advanced HCC,55,56 and an increased autophagy response has been found to correlate with malignant progression and poor prognosis of HCC.57 Autophagy was demonstrated to promote HCC invasion through activation of the epithelial–mesenchymal transition.58 In pancreatic cancer, autophagy promotes cell growth, survival, invasion, and metastasis.59–63 It has also been shown that in cells expressing oncogenic RAS, autophagy is required for promotion of cancer, as it maintains oxidative metabolism and facilitates glycolysis.59 Autophagy promotes tumor growth in a mouse model of RAS-driven pancreatic cancer by suppressing p53 activation.64 A similar observation has been made in HCC, indicating that the autophagy response is also needed to inactivate tumor suppressors to promote tumor development. Administration of dethylnitrosamine to wild-type mice inactivated p53-developed HCC, whereas liver-specific ATG5-knockout mice developed only benign hepatic adenoma due to induction of multiple tumor suppressors, including p53.65 Autophagy induction promotes growth of cancerous stem cell–derived mammary tumors.66 Available evidence suggests that 50%–60% of tumors grown under hypoxic conditions show an increased autophagy response.67 Tumor microenvironments, which are clearly different from those of normal tissue, have limited blood supply and are hypoxic, low in energy due to high mitotic activity, acidic, and inflammatory. These conditions induce autophagy by activating various pathways.68,69 All these lines of evidence support the hypothesis that autophagy induction is required for tumor progression, which could also explain why HCC develops more frequently in cirrhotic than in normal liver.
Altered autophagy signaling in HCC
The role of autophagy as a tumor suppressor or oncogenic inducer is unclear, as an increase or decrease in the autophagy response in cancer is often regulated by overactivation or inactivation of oncogenic signaling. This topic is highly complex, and this review therefore considers only selected pathways that are relevant to autophagic regulation in HCC. The interactions between antiapoptotic protein family Bcl-2 and autophagy protein Beclin 1 are of particular importance in the regulation of autophagy.70,71 The interaction of Bcl-2 with wild-type Beclin 1 inhibits the autophagy response, whereas mutant Beclin 1, which is defective in the Bcl-2 binding domain, can induce autophagy. Apoptosis and autophagic cell death are the two mechanisms of cell death that are controlled at the level of interactions between antiapoptotic protein Bcl-2 and autophagic protein Beclin 1. A minimal interaction between these two proteins (Bcl-2 and Beclin 1) favors an increased autophagy response, whereas a maximal interaction leads to autophagy inhibition. The dissociation of Bcl-2 from Beclin 1 is important for activation of autophagy, whereas their association inhibits autophagy. Therefore, the presence of mutant Beclin 1 protein or mutant multidomain protein members of the Bcl-2 family (Bcl-XL) could inhibit this interaction and induce the autophagy response. The interaction between Bcl-2 and Beclin 1 can also be affected by EGFR/mTOR signaling.
A significant number of cancers show high activations of receptor tyrosine kinases, such as epidermal growth factor (EGF). Epidermal growth factor receptor (EGFR) signaling is of key importance in liver injury, inflammation, fibrogenesis, and neoplastic transformation. The EGFR, a receptor tyrosine kinase in the ErbB family, consists of four members: EGFR (ErbB1, HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). The EGFR pathway becomes deregulated in HCC by a number of mechanisms, including overproduction of ligands, overproduction of receptors, and activation of receptors.72,73 During liver injury and regeneration, hepatocytes express high levels of ErbB1, and the expression of most ligands (eg, EGF and TGF alpha) is increased.74 The EGFR signaling is triggered when the ligand binds to the extracellular ligand-binding domain of the EGFR, which initiates receptor homo-/hetero-dimerization and autophosphorylation in specific tyrosine residues in the intracellular kinase domain of the receptor. Multiple phosphorylation at the kinase domain generates docking sites for a variety of signaling proteins, such as Shc, GRb2, Grb7, Crk, PLC, the kinase Src, PI3K, protein phosphatases SHP1 and SHP2, as well as the ubiquitin ligase Cbl E3.75 The EGFR signaling engages other signaling pathways generated from growth factors, cytokines, and inflammatory mediators to support growth and survival functions.
Studies supporting the role of EGFR-modulated autophagy have shown that overexpression of the EGFR represses autophagy, whereas silencing the EGFR in cancer cells leads to induction of autophagy.76 The EGFR signaling that is relevant to autophagy regulation includes the P13K–AKT–mTOR pathway and the EGFR–Beclin 1 axis. It has been well established that mTOR is the master regulator of autophagy, that mTOR is directly controlled by EGFR signaling,77 and that activation of EGFR inhibits autophagy by reducing Beclin 1 levels.78–80 Reports indicate that EGFR signaling also induces the autophagy response in some cancers, with the response being due to ligand-independent EGFR signaling via the truncated receptor. The EGFR mutant vIII, a naturally occurring EGFR mutant lacking 801 base pairs ligand-binding domain (exons 2–7), can stimulate ligand-independent activity in glycolytic tumors in the brain.81 Tumor-expressing EGFR vIII shows higher activation of the autophagy response due to upregulation of genes involved in cell metabolism, such as glucose transporters (GLUT1 and GLUT3), hexokinase 2 (HK2), and pyruvate dehydrogenase kinase (PDK1).78 The autophagy response is also induced in various models of RAS-induced lung and pancreatic tumors.72
Another explanation for the induced autophagy response in HCC is related to ERK signaling. Among the four classes of MAPK signaling in cancer cells, ERK signaling is activated in response to proliferation signals, while p38 and JNK are activated in response to various stresses.82 The EGFR signaling activates membrane-bound RAS, which interacts with RAF. The RAF phosphorylates two serine residues on the kinase mitogen protein kinase-1 and -2 (MEK1/2); MEK then activates ERK1/2 by phosphorylation of threonine and tyrosine residues, which are separated by one amino acid (threonine-183 and tyrosine-185 of ERK1/2). The activated ERKs phosphorylate numerous cytoplasmic and nuclear proteins to induce cell proliferation; ERK1/2 promotes cell proliferation, being overexpressed in human HCC. It has been reported that direct activation of ERK by MEK can promote autophagy without other signals.83 All of these lines of evidence indicate that altered EGF signaling could activate or inhibit the autophagy response in diverse cancers, including in HCC.
The p53 tumor suppressor, which is inactivated in more than 50% of human cancers, coordinates a wide varieties of responses, including DNA damage, transactivation of cell cycle arresting proteins, metabolism, proapoptotic function, and autophagy.84 Accumulating evidence indicates that p53 can modulate the autophagy response in cancer cells in a dual fashion, depending on its subcellular localization. Nuclear p53 induces the autophagy response by inducing transcription of two modulators: sestrin 1/2 and damage-regulated autophagy modulator (DRAM). Sestrin 1/2 activates autophagy by inhibiting mTOR signaling.85 Cytoplasmic p53 represses autophagy through inactivation of AMPK, which activates mTOR signaling.86 Cytoplasmic p53 can operate at the mitochondrial level to promote cell death and repress the autophagy response.
Autophagy induction in HCC therapy
Significant interest has emerged in the use of autophagy-inducing agents for the treatment and prevention of chronic liver diseases and HCC. As discussed earlier, autophagy plays a dual role in the pathophysiology of chronic liver disease, liver cirrhosis, and HCC, and it is therefore important to define the liver diseases appropriate for autophagy induction or inhibition therapy. The liver diseases that are expected to benefit from autophagy induction include alcoholic fatty liver disease and alpha-1 antitrypsin deficiency.87 Based on a “proof–principle approach”, autophagy induction using carbamazepine has been proven to benefit patients with alpha-1 antitrypsin deficiency.25 However, autophagy induction therapy may be problematic in the treatment of chronic viral hepatitis, as autophagy induction enhances the replication of HBV and HCV. Autophagy induction might also impair the host’s innate immunity and capacity to clear infection.88 Moreover, the autophagy induction therapy approach may not be applicable to those patients with alcoholic or fatty liver disease who are chronically infected with HBV or HCV.
Autophagy induction can be accomplished by the use of both pharmacological and nonpharmacological agents. The best nonpharmacological approach for autophagy induction may be calorie restriction and regular exercise. This approach provides protection against high fat diet–induced diabetes in mice.89 In addition to calorie restriction, other nutritional factors such as coffee and vitamin D intake may be used to improve health through autophagy induction.90–97 Consumption of caffeine induces autophagy, which reduces hepatic steatosis in mice with nonalcoholic fatty liver disease.93 Based on these reports, it is expected that calorie restriction, exercise, coffee consumption, and vitamin D can be adopted in the treatment of chronic liver disease due to alcoholic and nonalcoholic fatty liver disease in humans.
An alternative approach to inducing autophagy in a tissue-specific manner is by gene delivery of vectors that express autophagy genes. A study demonstrated that TFEB gene delivery improves the outcome of a variety of diseases, including obesity/diabetes and alpha-1 antitrypsin deficiency.98 The small-sized molecules currently approved by the US FDA to induce autophagy include carbamazepine, clonidine, lithium, metformin, rapamycin, rilmenidine, sodium valproate, verapamil, trifluoperazine, statin, and tyrosine kinase inhibitors.99,100 These agents can be used for the treatment of liver disease in the context of whether or not autophagy induction would be beneficial. Available evidence suggests that autophagy acts as a tumor suppressor, but is insufficient in HCCs. Therefore, induction of autophagy should help to reverse the malignant phenotype and improve chemotherapeutic treatment of HCC. Based on this reasoning, autophagy-inducing agents can be used along with the FDA-approved drug sorafenib for the treatment of liver cancer. Sorafenib is a multitargeted receptor tyrosine kinase inhibitor that has been approved as a standard therapy for advanced HCC.101 Sorafenib alone has only modest effects in prolonging the survival of HCC patients.102 Studies have shown that sorafenib itself induces the autophagy response and accumulates autophagosomes in HCC cells through inhibition of the mTOR pathway.103 Sorafenib also induces the expression of ER stress response genes (such as IRE-1 and CHOP), eIF2alpha phosphorylation, and the autophagy response in HCC cells.104 Whether chemotherapy drugs that induce autophagic cell death can be used in combination with sorafenib to improve the therapeutic response in HCC patients is currently under investigation, as the concept has been supported by several studies showing that small-molecule drugs inhibit HCC growth through autophagy induction.105–108 A number of chemotherapy drugs known to induce autophagic cell death (such as tamoxifen, etoposide, temozolomide, varinostat, arsenic trioxide, sodium selenite, and metformin) could be used in combination with sorafenib to inhibit HCC cells, especially in cases of defects in the apoptosis pathway. The success of combination therapies using sorafenib and other autophagy inducers needs further validation.
Autophagy inhibition in HCC therapy
Autophagy is required for tumor cell survival, and therefore autophagy inhibition could be explored as a potential therapeutic strategy for cancer treatment. A wide variety of pharmaceutical inhibitors that block different steps of the autophagy process are commercially available. Pharmaceutical inhibitors of HCC growth include 3MA, wortmannin, spautin-1, thapsigargin, vorinostat, chloroquine (CQ), hydroxychloroquine (HCQ), monensin, lucanthone, matrine, xanthohumol, azithromycin, bafilomycin A1, and concanamycin A.99 Among these, CQ and HCQ, which are used to treat malaria, are FDA-approved drugs commonly used as autophagy inhibitors in various experimental tumor models.109–111 Both CQ and HCQ are lysosomal lumen alkalizers that inhibit the activity of lysosomal hydrolases by neutralizing acidic pH in the lumen of lysosomal vesicles. Alkalization of lysosomal vesicles leads to the accumulation of autophagosomes by blocking lysosomal degradation.112 Based on this mechanism, CQ and HCQ have been used as anticancer drug candidates in humans.113
Another lysosomal inhibitor that has been developed, Lys05 (a dimeric CQ), accumulates in the lysosome and shows antitumor activity more potent than that of HCQ.114 A number of new potent autophagy inhibitors have been developed that inhibit autophagy by preventing fusion of autophagosomes with lysosomes, thus causing acidification of the lysosome and lysosomal degradation.115–117 At present, several ongoing cancer clinical trials include autophagy inhibitors along with other chemotherapy agents (http://www.clinicaltrials.gov). Autophagy inhibitors can enhance the effectiveness of oxaliplatin, cisplatin, 5-fluorouracil, and sorafenib in HCC models.103,118,119 Coadministration of sorafenib and CQ decreases tumor growth more significantly than administration of either agent alone. It has been demonstrated in experimental animal model that autophagy as inhibitors interact synergistically with either proteasome inhibitor or angiogenesis inhibitor to inhibit HCC growth.119,120
Cancer-initiating cells (ie, cancer stem cells) have been identified in a variety of cancers, but in only a small subpopulation of tumors.121 However, such cells in tumors can differentiate into multiple heterogeneous lineages of cancer cells. Available evidence indicates that current cancer treatments are ineffective in the elimination of the cancer stem cell population, thus resulting in tumor relapse and chemoresistance. Inhibition of autophagy by CQ was found to decrease the viability of liver cancer stem cells under conditions of hypoxia and nutritional starvation.122 It is anticipated that future research will clarify whether autophagy inhibition or induction will have a clinical benefit in the management of HCC chemotherapy.
Currently, there are more than 30 ongoing cancer treatment clinical trials using autophagy inhibitors (HCQ or CQ) in spite of the fact that many tumors, including HCC, show insufficiency in autophagy response (Figure 3).123 The mechanisms by which the autophagy inhibitors show strong antitumor response in the clinic are not well established. It has been reported that the anticancer mechanisms exhibited by HCQ or CQ are complex and involve more than one mechanism. Some studies reported that CQ sensitizes cancer cells to chemotherapy by inhibiting autophagy,124 inhibiting anticancer drug extrusion by blocking transporter P-glycoprotein,125 promoting apoptosis through lysosomal membrane permeabilization,126 and impairing DNA repair.127 The anticancer mechanism of CQ has been reported to be independent of autophagy inhibition.128 All these results indicate that further understanding of the anticancer mechanisms should establish the therapeutic potential of CQ in cancer.
Conclusion
Autophagy has been recognized as a tumor suppressor mechanism in the liver, and increased autophagy levels have been observed in cases of chronic liver disease, liver cirrhosis, and HCC. The hepatic autophagy response impairs the innate and adaptive immune response. Recent studies have shown that HCC may be associated with an insufficient autophagy response, and available evidence suggests that an insufficient response in HCC could be related to either impaired expression of autophagy genes or altered autophagy signaling. Future research will address whether an increased or decreased autophagy response is associated with the development of HCC related to liver cirrhosis. Autophagy inhibitors as chemotherapeutic agents have shown promising results in the treatment of HCC, by reducing cancer stem cell evolution and improving the immune response against HCC. In summary, autophagy modulation provides new prospects for anti-HCC therapies. We propose that more basic research is needed to further understand the detailed mechanisms of autophagy modulation, and to explore future applications of autophagy modulation to the treatment of liver disease.
Acknowledgments
We thank Samantha Hoekst for critically reviewing this manuscript and Troy Taliancich in the Pathology Department for assistance with image generation. This work was supported by NIH grants CA127481, CA089121, and AI103106.
Disclosure
The authors report no conflicts of interest in this work.
References
He C and Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Gsssenet. 2009;43:67–93. | |
Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. | |
Cuervo AM, Knecht E, Terlecky SR, Dice JF. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am J Physiol. 1995;269:1200–1208. | |
Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–434. | |
Efeyan A, Comb WC, Sabatini DM. Nutrient-sensing mechanisms and pathways. Nature. 2015;517:302–310. | |
Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. | |
Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451: 1069–1075. | |
Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011;23:184–189. | |
Jewell JL, Russell RC, Guan KL. Amino acid signaling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–139. | |
Mizushima N. The role of the ATG1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22:132–139. | |
Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol. 2011;8:528–539. | |
Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, ATG14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. | |
Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34:259–269. | |
Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res. 2012;2:397–413. | |
Jagar S, Bucci C, Tanida I, et al. Role of Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. | |
Tanaka Y, Guhde G, Suter A, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2 -deficient mice. Nature. 2000;406:902–906. | |
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. | |
Malhi H, Guicciardi ME, Gores GJ. Hepatocyte Death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. | |
Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. | |
Rautou PE, Cazals-Hatem D, Feldmann G, et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am J Pathol. 2011;178:2708–2715. | |
Taguwa S, kambara H, Fujita N, et al. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. J Virol. 2011;85:13185–13194. | |
Sir D, Chen WL, Choi J, Wakita T, yen TS, Ou J. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology. 2008;48:1054–1061. | |
Liu B, Fang M, Hu Y, Huang B, Li N, Chang C, et al. Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy. 2014;10:416–430. | |
Lin CW, Zhang H, Li M, et al. Pharmacological promotion of autophagy alleviates steatosis and injury in alcoholic and non-alcoholic fatty liver conditions in mice. J Hepatol. 2013;58:993–999. | |
Hidvegi T, Ewing M, Hale P, et al. An autophagy-enhancing drug promotes degradation of mutant alpha-1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. | |
El-Serag HB. Hepatocellular Carcinoma. N Eng J Med. 2011;365: 1118–1127. | |
Zhi X, Zhong Q. Autophagy in cancer. F1000 Prime Rep. 2015;7:18. | |
White E. The role for autophagy in cancer. J Clin Invest. 2015;125: 42–46. | |
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34:856–880. | |
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. | |
Saito H, Inazawa J, Saito S, et al. Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumors. Cancer Res. 1993;53: 3382–3385. | |
Gao X, Zacharek A, Salkowski A, et al. Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 1995;55:1002–1005. | |
Aita VM, Liang XH, Murty VV, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. | |
Shen Y, Li DD, wang LL, Deng R, Zhu XF. Decreased expression of autophagy related proteins in mammalian epithelial ovarian cancer. Autophagy. 2008;4:1067–1068. | |
Pirtoli L, Cevenini G, Tini P, et al. The prognostic role of beclin 1 protein expression in high grade glioma. Autophagy. 2009;5:930–936. | |
Li BX, Li CY, Peng RQ, et al. The expression of beclin 1 is associated with favorable prognosis in stage IIIB colon cancers. Autophagy. 2009;5:303–306. | |
Miracco C, Cosci E, Oliveri G, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumors. Int J Oncol. 2007;30:429–436. | |
Qu X, Yu J, Bhagat G, Furuya N, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. | |
Cianfanelli V, Fuoco C, Lorente M, et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat Cell Biol. 2015;17:20–30. | |
Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8: 688–699. | |
Takahashi Y, Coppola D, Matsushita N, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. | |
Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. | |
Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. | |
Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193: 275–284. | |
Bao L, Chandra PK, Moroz K, et al. Impaired autophagy response in human hepatocellular carcinoma. Exp Mol Path. 2014;96:149–154. | |
Zatloukal K, French SW, Stumptner C, et al. From Mallory to Mallory-Denk bodies: what, how and why. Exp Cell Res. 2007;313:2033–2049. | |
Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. | |
Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. | |
Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–1004. | |
Jin Z, Li Y, Pitti R, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–735. | |
Gao C, Cao W, Bao L, et al. Autophagy negatively regulates Wnt signaling by promoting Disheveled degradation. Nat Cell Biol. 2010;12: 781–790. | |
Duran A, Amanchy R, Linares JF, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. | |
Manley S, Williams JA, Ding W-X. Role of p62/SQSTM1 in liver physiology and pathogenesis. Exp Mol Med. 2013;238:525–538. | |
Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. | |
Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis and poor outcome. Clin Cancer Res. 2012;18:370–379. | |
Mikhaylova O, Stratton Y, Hall D, et al. VHL-regulated MiR-204 suppresses tumor growth inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell. 2012;21:532–546. | |
Wu DH, Jia CC, Chen J, et al. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumor Biol. 2014;35:12225–12233. | |
Li J, Yang B, Zhou Q, et al. Autophagy promotes HCC cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34:1343–1351. | |
Guo JY, Chen HY, Mathew R, Fan J, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. | |
Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22: 165–178. | |
Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. | |
Lock R, Kenific CM, Leidal AM, et al. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–479. | |
Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155:1216–1219. | |
Rosenfeldt MT, O’Pret J, Morton JP, et al. p53 status determined the role of autophagy in pancreatic tumor development. Nature. 2013;504:296–300. | |
Tian Y, Kuo CF, Wang L, Govindrajan S, Petrovic LM, Ou JH. Autophagy inhibits oxidative stress and tumor suppressors to exert its dual effect on hepatocarcinogenesis. Cell Death Differentiation. 2015;22:1025–1034. | |
Gong C, Bauvy C, Tonelli G, et al. Beclin 1 and Autophagy are required for the tumorigenicity of breast cancer stem-like progenitor cells. Oncogene. 2013;32:2261–2272. | |
Yang X, Yu DD, Yan F, et al. The role of autophagy induced by tumor microenvironment in different cells and stages of cancer. Cell Biosci. 2015;5:14. | |
Liu EY, Ryan KM. Autophagy and cancer-issues we need to digest. J Cell Sci. 2012;125:2349–2358. | |
Amaravadi RK, Lippincott-Schwartz J, Yin XM, et al. Principle and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res. 2011;17:654–666. | |
Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. | |
Pattingre S, Tessa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1 dependent autophagy. Cell. 2005;122:927–939. | |
Kenific CM, Debnath J. Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol. 2015;25:37–45. | |
Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev Mol Cell Biol. 2001;2:127–137. | |
Choi KJ, Baik IH, Ye SK, Lee Yh. Molecular targeted therapy for hepatocellular carcinoma: present status and future directions. Biol Pharm Bull. 2015;38:986–991. | |
McDonell LM, Kernohan KD, Boycott KM, Sawyer SL. Receptor tyrosine kinase mutations in developmental syndrome and cancer: two sides of the same coin. Hum Mol Genet. 2015;24(R1):R60–R66. | |
Carver RS, Stevenson MC, Scheving LA, Russell WE. Diverse expression of ErbB receptors proteins during rat liver development and regeneration. Gastroenterology. 2002;123:2017–2027. | |
Berasain C, Avila MA. The EGFR signaling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol. 2014;49: 9–23. | |
Jutten B, Rouschop MA. EGFR signaling and autophagy dependence for growth, survival and therapy resistance. Cell Cycle. 2014;13: 42–51. | |
LiX, Lu Y, Pan T, Fan Z. Role of autophagy in cetuximab-mediated cancer therapy against EGFR. Autophagy. 2010;6:1066–1077. | |
Wei Y, Zou Z, Becker N, Anderson M, et al. EGFR-mediated Beclin1 phosphorylation in autophagy suppression, tumor promotion and chemoresistance. Cell. 2013;154:1269–1284. | |
Babic I, Anderson ES, Tanaka K, et al. EGFR mutation induced alternative splicing of Max contribute to growth of glycolytic tumors in brain cancer. Cell Metab. 2013;17:1000–1008. | |
Hazzalin CA, Mahadevan LC. MAPK-regulated transcription: a continuously variable gene switch? Nat Rev Mol Cell Biol. 2002;3:30–40. | |
Corcelle E, Nebout M, Bekri S, et al. Disruption of autophagy at maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res. 2006;66:6861–6870. | |
Soussi T. p53 alterations in human cancers: more questions than answers. Oncogene. 2007;26:2145–2156. | |
Budanov AV, Karin M. p53 target genes sestrin 1 and senstrin 2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. | |
Tasdemir E, Maiuri MC, Galluzi L, et al. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10:676–687. | |
Levine B, Packer M, Codogno P. Development of autophagy inducers in clinical medicine. J Clin Invest. 2015;125:14–24. | |
Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. | |
He C, Bassik MC, Moresi V, et al. Exercise-induced BCL2 regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. | |
Handschin C, Spiegelman BM. The role of exercise and PGC1α in inflammation and chronic disease. Nature. 2008;454:463–469. | |
Jia K, Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007;3:597–599. | |
Watanabe T, Takemura G, Kanamori H, et al. Restriction of food intake prevents postinfarction heart failure by enhancing autophagy in the surviving cardiomyocytes. Am J Pathol. 2014;184:1384–1394. | |
Sinha RA, Farah BL, Singh BK, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology. 2014;59:1366–1380. | |
Moon JH, Lee JH, Park JY, et al. Caffeine prevents human prion protein-mediated neurotoxicity through the induction of autophagy. Int J Mol Med. 2014;34:553–558. | |
Freedman ND, Park Y, Abnet CC, Hollenbeck AR, Sinha R. Association of coffee drinking with total and cause-specific mortality. N Engl J Med. 2012;366:1891–1904. | |
Hoyer-Hansen M, Nordbrandt SP, Jaattela M. Autophagy as a basis for the health-promoting effects of vitamin D. Trends Mol Med. 2010;16:295–302. | |
Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M. Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ. 2005;12: 1297–1309. | |
Pastore N, Blomenkamp K, Annunziata F, et al. Gene transfer of master regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha 1-anti-trypsin deficiency. EMBO Mol Med. 2013;5:397–412. | |
Vakifahmetoglu-Norberg H, Xia H, Yuan J. Pharmacologic agents targeting autophagy. J Clin Invest. 2015;125:5–13. | |
Zhang MZ, Wang Y, Paueksakon P, Harris RC. EGFR inhibition slows progression of diabetic nephropathy in association with a decrease in endoplasmic reticulum stress and an increase in autophagy. Diabetes. 2014;63:2063–2072. | |
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Eng J Med. 2008;359:378–390. | |
Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. | |
Shimizu S, Takehara T, Hikita H, et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int J Cancer. 2012;131:548–557. | |
Shi YH, Ding ZB, Zhou J, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011;7:1159–1172. | |
Tai WT, Shiau CW, Chen HL, et al. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013;4:e485. | |
Gao M, Yeh PY, Lu YS, et al. OSU-03012, a novel celecoxib derivative induces reactive oxygen species-related autophagy in hepatocellular carcinoma. Cancer Res. 2008;68:9348–9357. | |
Yu HC, Lin CS, Tai WT, et al. Nilotinib induces autophagy in hepatocellular carcinoma through AMPK activation. J Biol Chem. 2013;288:18249–18259. | |
Bareford MD, Park MA, Yacoub A, et al. Sorafenib enhances pemetrexed cytotoxicity through an autophagy-dependent mechanism in cancer cells. Cancer Res. 2011;71:4955–4967. | |
Avalos Y, Canales J, Bravo-Sagua R, et al. Tumor suppression and promoting by autophagy. Biomed Res Int. 2014;2014:603980. | |
Jiang P, Muzushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. | |
Mahalingam D, Mira M, Sarantopous J, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic and pharmacodynamics analysis of hydroxychloroquine in combination with the HDAC inhibitor varinostat in patients with advanced solid tumors. Autophagy. 2014;10:1403–1414. | |
Schneider P, Korolenko TA, Busch U. A review of drug-induced lysosomal disorders of the liver in man and laboratory animals. Microsc Res Tech. 1997;36:253–275. | |
Gunja N, Robert D, McCoubrie D, et al. Survival after massive hydroxychloroquine overdoses. Anaesth Intensive Care. 2009;37:130–133. | |
Amaravadi RK, Winkler JD. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 2012;8:1383–1384. | |
Juhász G. Interpretation of bafilomycin, pH neutralizing or protease inhibitor treatments in autophagic flux experiments: novel considerations. Autophagy. 2012;8:1875–1876. | |
Carew JS, Espitia CM, Esquivel JA 2nd, et al. Lucanthone is a novel inhibitor of autophagy that induces cathepsin D-mediated apoptosis. J Biol Chem. 2011;286:6602–6613. | |
Wang Z, Zhang J, Wang Y, et al. Matrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases. Carcinogenesis. 2013;34:128–138. | |
Guo XL, Li D, Hu F, et al. Targeting autophagy potentiates chemotherapy-induced apoptosis and proliferation inhibition in hepatocarcinoma cells. Cancer Lett. 2012;320:171–179. | |
Hui B, Shi YH, Ding ZB, et al. Proteasome inhibitor interacts synergistically with autophagy inhibitor to suppress proliferation and induce apoptosis in hepatocellular carcinoma. Cancer. 2012;118: 5560–5571. | |
Guo XL, Li D, Sun K, et al. Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J Mol Med (Berl). 2013;91:473–483. | |
Lin Y-H, Huang Y-C, Chen L-H, Chu P-M. Autophagy in cancer stem/progenitor cells. Cancer Chemther Pharmacol. 2015;57:879–886. | |
Song YJ, Zhang SS, Guo XL, et al. Autophagy contributes to the survival of Cd133+liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Cancer Lett. 2013;339:70–81. | |
Siu X, Chen R, Wang Z et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death and Disease. 2013;4:e838. | |
Vlahopoulos S, Critselis E, Voutsas IF et al. New use for old drugs? Prospective targets of chloroquines in cancer therapy. Curr Drug Targets. 2014;15(9):843–851. | |
Yamagishi T, Sahni S, Sharp DM, Arvind A, Janson PJ and Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem. 2013;288(44):31761–31771. | |
Enzemuller S, Gonzalez P, Debatin KM, Fulda S. Chloroquine overcomes resistance of lung carcinoma cells to the dual PI3K/mTOR inhibitor PI103 by lysosome mediated apoptosis. Anticancer Drugs. 2013; 24(1):14–19. | |
Liu EY, Xu N, O’Prey J et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc Natl Acad Sci. 2015;112: 773–778. | |
Maycotte P, Aryal S, Cummings CT, Thorbum J, Morgan MJ, Thorburn A. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy. 2012;8(2):200–212. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.