Back to Journals » Clinical and Experimental Gastroenterology » Volume 10
Autoimmune hepatitis: current challenges and future prospects
Received 31 August 2016
Accepted for publication 11 December 2016
Published 19 January 2017 Volume 2017:10 Pages 9—18
DOI https://doi.org/10.2147/CEG.S101440
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Andreas M. Kaiser
Yoshio Aizawa, Atsushi Hokari
Division of Gastroenterology and Hepatology, Department of Internal Medicine, Jikei University Katsushika Medical Center, Katsushika-ku, Tokyo, Japan
Abstract: Autoimmune hepatitis (AIH) is a chronic progressive liver disease characterized by high levels of aminotransferases and autoantibodies, hypergammaglobulinemia, and interface hepatitis. AIH affects all races and all ages worldwide, regardless of sex, although a preponderance of females is a constant finding. The etiology of AIH has not been completely elucidated, but immunogenetic background and environmental parameters may contribute to its development. The most important genetic factor is human leukocyte antigens (HLAs), especially HLA-DR, whereas the role of environmental factors is not completely understood. Immunologically, disruption of the immune tolerance to autologous liver antigens may be a trigger of AIH. The diagnosis of classical AIH is fairly easy, though not without pitfalls. In contrast, the diagnosis of atypical AIH poses great challenges. There is confusion as to the definition of the disease entity and its boundaries in the diagnosis of overlap syndrome, drug-induced autoimmune hepatitis, and AIH with concomitant nonalcoholic fatty liver disease (NAFLD) or chronic hepatitis C. Centrilobular zonal necrosis is now included in the histological spectrum of AIH. However, the definition and the significance of AIH presenting with centrilobular zonal necrosis have not been examined extensively. In ~20% of AIH patients who are treated for the first time with standard therapy, remission is not achieved. The development of more effective and better tolerated novel therapies is an urgent need. In this review, we discuss the current challenges and the future prospects in relation to the diagnosis and treatment of AIH, which have been attracting considerable recent attention.
Keywords: autoimmune hepatitis, immunogenetic background, overlap syndrome, centrilobular zonal necrosis, antinuclear antibody, immunosuppressive therapy
Introduction
Autoimmune hepatitis (AIH) was described for the first time early in the 1950s, under the name of lupoid hepatitis, as a disease prone to afflicting young women.1 Since then, the boundaries of AIH have expanded to all races and to all ages.2,3 The clinical presentations of AIH at the time of diagnosis vary considerably, from acute liver failure (ALF) or acute hepatitis, which are relatively rare, to chronic hepatitis, cirrhosis, or hepatocellular carcinoma, which represent the primary diagnosis in ~60% of patients.4 This may indicate that the majority of AIH cases tend to progress insidiously. AIH is classified as type 1 or type 2, according to differences in their autoantibodies. Type 1 is the major type of AIH and presents with antinuclear antibodies (ANAs) and/or antismooth muscle antibodies (ASMAs), whereas the less common type 2 AIH is characterized by antiliver kidney microsome 1 (LKM 1). The titer of LKM is associated with disease activity of type 2 AIH.5 Adult type 1 and type 2 AIHs share similar profiles with respect to clinical, biochemical, and histological features, and genetic predisposition.6
The variety of clinicopathological features of AIH may be partly due to differences in immunogenetic background.7 The immunological mechanism of AIH is considered to be an abnormal T lymphocyte reaction to autologous hepatocytes,8 caused by a failure of immunological tolerance. The diagnosis of classical AIH is fairly easy after exclusion of other known liver diseases. Nowadays, the definition and the diagnostic procedure for significant liver diseases other than AIH are firmly established. Though revised international diagnostic criteria proposed by the International Autoimmune Hepatitis Group (IAIHG) have been widely used for the diagnosis of AIH, the gold standard for diagnosis is an empiric judgment by an experienced hepatologist. The IAIHG criteria are based on the clinical, biochemical, serological, and histological characteristics of AIH and the response to immunosuppressive therapy.9 It may be beneficial to form an objective judgment as to the probability of AIH. However, the diagnostic criteria are not necessarily reliable for the diagnosis of atypical AIH, including acute onset AIH, overlap syndrome,10 drug-induced autoimmune hepatitis (DIAIH), and AIH coincident with other liver diseases, including nonalcoholic fatty liver disease (NAFLD). Histologically, centrilobular zonal necrosis (CZN) is recognized as part of the spectrum of AIH.11
In the treatment of AIH, corticosteroids alone or combined with azathioprine are standard.12 Remission can be achieved in ~80% of AIH patients who are treated by standard therapy. In the case of poor response or intolerance, alternative immunosuppressive therapy should be introduced. Mycophenolate mofetil may become a first-line drug as a replacement for azathioprine.13 In the future, novel drugs that can act specifically on the immune mechanisms of AIH will be developed.
Recent rapid progress in antiviral drugs against hepatitis C virus (HCV) and hepatitis B may eradicate viral hepatitis in the near future. AIH will then attract increasing attention as one of the major issues in the area of hepatology. In this review, the current challenges and future prospects of AIH are discussed from the viewpoint of immunopathophysiology (immunogenetic background and immune mechanisms), diagnosis, and treatment.
Immunogenetic background of AIH
The close association between AIH and human leukocyte antigens (HLAs) has been recognized in Caucasian14 (HLA-DR3 and DR4) and Japanese15 (HLA-DR4) patients. As an immunogenetic factor other than HLA, the cytotoxic T-lymphocyte antigen-4 (CTLA-4) +49 A/G polymorphism may be associated with a susceptibility to AIH.16 However, a recent genome-wide association study of genetic susceptibility to AIH showed a strong association only between the HLA locus and AIH, while dozens of weak susceptibility loci were determined.17 Therefore, the impact of the CTLA-4 polymorphism may be considerably limited.
In Latin Americans, DRB1*1301 is correlated with susceptibility to type 1 AIH.18 DRB1*1301 is also correlated with child type 1 AIH in Germany.19 In Pakistan, HLA-DR6, DRB1*14, and DRB1*13 were more prevalent in AIH.20
HLA may affect not only disease susceptibility but also the clinical manifestations and outcome of AIH. In Japan, HLA-DR4 is related to higher levels of serum immunoglobulin G (IgG), the appearance of ASMA,21 and age at the onset of AIH. HLA-DR4 is less frequent in childhood or advanced age (>70 years).22 In Caucasians, HLA-DR4 is associated with less severity and older age at onset compared to HLA-DR3. In addition, patients with HLA-DR3 may have more progressive disease than those with DR4. Similarly, HLA-DRB1*0301 and DRB1*0401 alleles are both independently associated with the aggregate diagnostic IAIHG score in type 1 AIH patients, and HLA-DRB1*0301 strongly influences the severity of AIH.23
The association between the HLA-DR or HLA-DRB1 allele and AIH is widely accepted. However, another locus of HLA (A, B, C, or DQ) may be closely associated with AIH. As there is a strong linkage disequilibrium in the HLA locus, it has been claimed that the haplotype of HLA is more closely involved in susceptibility to AIH and more strongly modifies the clinical manifestations of type 1 AIH.7 Meanwhile, the immunogenetic background of type 2 AIH has not been extensively studied, because of the relatively small number of patients. A possible relation between HLA-DQB1*0201, HLA-DRB1*07, or HLA-DRB1*03 and type 2 AIH has been reported.24 A future, large-scale study will be needed to distinguish the differences in immunogenetic background between type 2 and type 1 AIHs.
The significance of HLA in the onset and progression of AIH should be examined worldwide, because the distribution of HLA in the background population is different among different races. This may indicate that the type of HLA involved in the susceptibility to AIH and its progression is different in different races. The clinical characteristics of type 1 AIH in Asia and in European or American countries show some differences, in the age at onset and the prevalence of advanced liver disease. Differences in immunogenetic backgrounds may be related to the differences in clinical presentation.25 In Asian countries, the clinical features of AIH in India and Pakistan are reported to differ considerably from those in East Asia. A high prevalence of advanced liver disease at first diagnosis and a poor outcome of AIH were the features of AIH in India and Pakistan. The differences may be attributed to differences in HLA.
Immunological mechanisms participating in the onset and persistence of AIH
Although the factors triggering the onset of AIH have not been clearly identified, AIH is considered to be initiated by an immunological reaction against autologous liver antigens.26–28 Presumably, an epitope of liver autoantigen binds with the paratope of an HLA class II antigen and is exhibited on the surface of antigen-presenting cells. Nevertheless, the immune reaction to autoantigens is not evoked under normal conditions, because of immune tolerance; autoantigens can be recognized by naive CD4-positive T cells. Once a liver autoantigen is recognized via costimulatory signals, naive T cells are activated and the immune reaction is initiated. Naive T cells can be activated and differentiated into Th1, Th2, or Th17 cells, depending on the immunological microenvironment and the nature of the antigen epitope. These T cells then begin to trigger immune cascades.
Cytokines secreted from differentiated T cells are the key molecules for triggering immune reactions. Interferon-gamma, which is mainly produced by Th1 cells, may play a central role in liver cell damage by stimulating CD8-positive cells and enhancing the expression of HLA class I and class II molecules. Tumor necrosis factor alpha, secreted from activated macrophages, may be a key cytokine in the occurrence of inflammatory disease. Interleukin (IL)-4, which is secreted by Th2 cells, is a major cytokine for the maturation of B cells into plasma cells, which leads to the production of autoantibodies. Th17 is differentiated under the stimulation of both transforming growth factor beta and IL-6. Excessive Th17 cells are considered to be a major cause of autoimmune diseases. IL-23 is involved in the maintenance of Th17 cells and thus may play a role in AIH. Th17 cells secrete IL-17 and suppress regulatory T cells (Tregs). Tregs are essential for maintaining the homeostasis of the immune system and suppress excessive immune reactions in autoimmune diseases. Although the issue is still controversial,29 defective Tregs have been implicated in the pathogenesis of AIH,30 triggering its onset and persistence.
Pitfalls in the diagnosis of classical AIH
Although the diagnosis of classical AIH is not difficult, several potential pitfalls need to be taken into account. First of all, the presence of ANA should not be overemphasized. ANA is a nondisease-specific autoantibody and is frequently detected in the sera of chronic liver diseases other than AIH. A low or middle titer of ANA is not decisive in the differential diagnosis. In some particular cases, a high titration of ANA may be found in non-AIH patients. Similarly, prominent hypergammaglobulinemia and/or high serum IgG may be found in non-AIH chronic hepatitis or cirrhosis.
Moreover, the level of transaminases may not indicate the severity of interface hepatitis. Some AIH patients with slight elevation of transaminases may have severe interface hepatitis in liver histology. Contrarily, in a few patients who exhibited hypergammaglobulinemia, a high titer of ANA, and significant elevation of transaminases, necroinflammatory change was not observed in liver histology. Therefore, liver biopsy is essential for establishing the diagnosis of AIH. It should be kept in mind that CZN is part of the histological spectrum of AIH, even if interface hepatitis is not observed.11 Thus, a liver biopsy may provide essential information regarding the diagnosis of AIH.
Another important issue in the diagnosis of AIH is exclusion of known etiologies of liver diseases other than AIH, because not only the laboratory findings but also the histological features of AIH may largely resemble those of other chronic liver diseases, including drug-induced liver injury (DILI), chronic viral hepatitis, and other known chronic liver diseases. Among these, chronic active Epstein–Barr virus infection (CAEBV) must be carefully distinguished from AIH. Although there is a report of AIH being induced in a patient with preexisting CAEBV and successfully treated with a corticosteroid,31 CAEBV is generally a life-threatening progressive disease if adequate management is neglected. In some cases of CAEBV, the clinical, laboratory, and histological features are quite similar to those of AIH, but the prognosis is extremely poor.32,33 CAEBV should be included in the differential diagnosis of patients who do not respond sufficiently to immunosuppressive therapy. All AIH patients who exhibit an unusual clinical course should be thoroughly reexamined for the possibility of other liver disease.
Diagnosis and management of atypical AIH
Overlap syndrome
The concept of overlap syndrome applies to patients who present the features of primary biliary cholangitis (PBC) or primary sclerosing cholangitis (PSC) along with the features of AIH. PBC-AIH or PSC-AIH overlap syndrome denotes a condition in which the primary (dominant) disorder is PBC or PSC, and the features of AIH are defined concomitantly. In other words, a small number of PBC or PSC patients present the clinicopathological features of AIH, including high levels of IgG, positivity of ANA/ASMA, a prominent increase in transaminases, and interface hepatitis in liver histology, at the time of first diagnosis or during the treatment of PBC or PSC. However, there is no objective standard by which to judge whether PBC/PSC or AIH is the dominant disease in the same patient.
The overlap syndrome is conceptually divided into four categories (Table 1). The majority of overlap syndrome cases are considered to be PBC-AIH overlap. It is a debatable issue whether PBC-AIH overlap is a distinctive disease entity or only a variant form of PBC. ANA is frequently detectable in PBC patients.36 In the liver pathology of PBC, mild to moderate interface hepatitis is not an uncommon feature.37 Thus, the distinctive differentiation between a variant form of PBC (PBC exhibiting active hepatitis) and PBC-AIH overlap syndrome is rather uncertain. Nonetheless, prominent active hepatitis and the features of AIH can be seen in patients with PBC, though such a condition is rare.
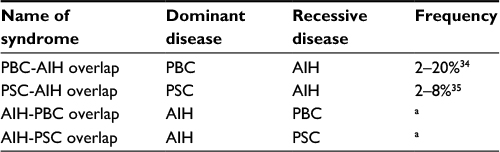 | Table 1 Classification of overlap syndrome Note: aThe frequency of this type of overlap syndrome is not clarified. Abbreviations: AIH, autoimmune hepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis. |
The Paris criteria are often applied to the definition of PBC-AIH overlap syndrome,38 though these are not universally established. In the Paris criteria, two of three features associated with AIH are required in addition to two of three features related to PBC: for AIH, a serum alanine aminotransferase (ALT) level ≥5 times the upper limit of normal (ULN), an IgG level ≥2 times the ULN, or the presence of ANA or ASMA and interface hepatitis on liver biopsy; for PBC, a serum alkaline phosphatase level ≥2 times the ULN or a gamma-glutamyl transpeptidase level ≥5 times the ULN, the presence of antimitochondrial antibodies, and florid duct lesions or destructive cholangitis on liver histology. In any case, distinctive clinical, laboratory, and pathological features of AIH on top of a definitive diagnosis of PBC are required for the diagnosis of PBC-AIH overlap.
PBC with marked elevation of transaminases has been treated empirically with ursodeoxycholic acid (UDCA) combined with a short-duration corticosteroid. From the viewpoint of the management of PBC-AIH overlap, the results of a meta-analysis suggested that the combination of corticosteroid and UDCA is a first-line therapy, because the combination therapy was more effective than UDCA alone.39 However, the efficacy of UDCA alone may depend on the histological severity of the interface hepatitis in PBC patients with features of AIH. The efficacy of UDCA alone and in combination therapy is similar in patients with low to moderate interface hepatitis, whereas the efficacy of UDCA alone is significantly lower than that of combined therapy in patients with severe interface hepatitis. Thus, UDCA alone is the treatment of choice in mild to moderate interface hepatitis, whereas UDCA combined with a corticosteroid should be preferred for the treatment of severe interface hepatitis.40 It would be interesting to know whether withdrawal of the corticosteroid exacerbates the liver inflammation in PBC-AIH overlap patients who are exhibiting prominent interface hepatitis. A high frequency of exacerbation after the withdrawal of a corticosteroid could denote the existence of PBC-AIH overlap in the true sense of overlapping disease. In our experience, corticosteroids may be successfully discontinued without any flare-up of transaminases in these patients.
There are no guidelines for the diagnosis of other types of overlap syndrome. A high prevalence of PSC-AIH overlap syndrome has been noted among PSC patients.41 Long-term follow-up of patients treated with UDCA, or UDCA plus an immunosuppressive drug, indicated that the prognosis of PSC-AIH overlap is not worse than that of classical PSC.42
The diagnosis of AIH-PBC overlap should be viewed carefully. Bile duct injury or bile duct reaction is a common histopathological feature of AIH.43 The interlobular bile duct may be frequently affected and damaged by an intensive portal inflammation of AIH. Nevertheless, a typical PBC bile duct lesion is very hard to find in the liver tissue of AIH. Therefore, an easy diagnosis of AIH-PBC overlap based on the histological finding of bile duct injury should be strictly resisted.
DIAIH
The diagnosis of DIAIH is relatively easy if the particular drug that is known to be the cause of DIAIH is administered until the onset of AIH. Minocycline, nitrofurantoin, methyldopa, hydralazine, and herbal drugs are the major drugs closely associated with DIAIH.44 In addition, statins45 and other newly developed drugs are reported to be associated with DIAIH. Statins tend to be taken by many patients for long periods, as opposed to minocycline and nitrofurantoin. Nevertheless, the reported number of DIAIH cases caused by statins is fairly small. Statins are often given to middle-aged or elderly people who are susceptible to AIH. Therefore, it should be borne in mind that the onset of AIH may incidentally coincide with the use of statins. AIH appearing during statin use must be carefully investigated to determine whether it was induced by the statin or not.
DILI can be distinguished from AIH without difficulty.46 However, the clinical and histopathological features of DIAIH are sometimes quite similar to those of AIH.47 Response to corticosteroids is similar for both DIAIH and AIH.48 There is disagreement as to whether recurrence after discontinuation of corticosteroids occurs in DIAIH. In DIAIH, successful discontinuation of immunosuppressive therapy has been reported, without relapses.44 Contrarily, the rate of relapse has been reported by others to be similar for both idiopathic AIH and DIAIH.48 It must be noted that, the latter report included a significant number of statin-induced DIAIH cases, while patients with nitrofurantoin-induced DIAIH are less likely to relapse than other DIAIH patients after discontinuation of immunosuppression. This finding may raise the suspicion that most statin-induced DIAIH is not drug-induced disease but idiopathic AIH, which appeared incidentally during statin use. In any case, long-term follow-up of patients who are diagnosed with DIAIH is recommended to monitor any relapse.
AIH-like laboratory findings are found in a majority of cases of nitrofurantoin- or minocycline-induced liver injury and in about half the cases of methyldopa- or hydralazine-induced liver injury. This abnormality spontaneously improves with recovery from liver damage and is not associated with HLA-DR3 or DR4.49 Therefore, the immunopathological settings of DIAIH are quite different from those of idiopathic AIH. Although the pathogenesis of DIAIH has not been completely elucidated, studies of dihydralazine and tienilic acid (these drugs are not currently used) suggested that bonding of reactive drug metabolites and cellular proteins forms neoantigens that can be recognized by immune systems, and that immune reactions against neoantigens may induce DIAIH.50
Under the present circumstances, the diagnosis of DIAIH is left to personal subjective judgment. The establishment of objective diagnostic criteria for DIAIH is urgently required.
AIH accompanied by NAFLD and chronic hepatitis C (CHC)
AIH and coincident NAFLD is not a rare condition. Patients with coincident AIH and nonalcoholic steatohepatitis (NASH) are more likely to present with cirrhosis and more likely to develop an adverse clinical outcome with poorer survival compared to AIH without coincidence of NAFLD/NASH.51 This finding suggests that the simultaneous presence of AIH and NASH may confer an increased risk of progressive liver disease and death. This hypothesis is supported by the finding from a mouse model that preexisting NAFLD potentiates the severity of AIH.52 In patients with NAFLD/NASH, a high prevalence of ANA and ASMA has been noted.53 Therefore, liver biopsy is essential for the definition of AIH and coincident NAFLD/NASH. Generally, the histological features of NASH are quite different from those of AIH and coincident NASH. However, AIH with coincident NASH is sometimes very hard to distinguish from NASH, even after inspection of liver histology. In addition, the response to standard therapy for AIH does not always become the index of the diagnosis of AIH coincident with NASH. Corticosteroids may exaggerate the deposition of fat in hepatocytes and worsen NASH, while reducing the inflammatory activity of AIH. Therefore, the efficacy of corticosteroids in AIH and coincident NASH may be attenuated. Close surveillance of these patients is warranted; if remission is not achieved using standard therapy, the corticosteroid must be replaced by other immunosuppressive drugs.
In CHC, autoimmune phenomena are often found. ANA is present in ~30% of patients with CHC.54 LKM antibodies are found in some patients with CHC.55 In general, molecular mimicry could explain the positivity of LKM antibodies in patients with HCV infection.56 The autoantibody status is not useful for differentiating between CHC alone and AIH concurrent with CHC. The diagnosis of concurrent AIH and HCV infection may depend on the empiric judgment of an experienced hepatologist, because there are no diagnostic criteria. Although the revised diagnostic criteria are not intended to be used for the diagnosis of AIH and coincident CHC, the criteria may be useful to some degree in the differential diagnosis.57
Liver histology may play a role in the definition of AIH and coincident CHC. The liver pathology of autoimmune-predominant CHC cases exhibits histological characteristics of AIH: severe interface hepatitis, plasma cell infiltration, and extensive lymphocyte accumulation in the portal region. However, these findings may also be seen in CHC without coincident AIH. Therefore, the diagnosis of AIH coincident with CHC is fairly ambiguous. As AIH with coincident CHC is not a familiar condition, an easy diagnosis should be strictly resisted.
Most CHC patients are successfully treated by a rapidly progressing interferon-free regimen against HCV infection. It would be very interesting to know whether liver inflammation completely subsides after the elimination of HCV and whether relapse occurs after the discontinuation of immunosuppressive therapy in patients with AIH and coincident CHC.
Histologically atypical AIH: CZN
The most significant pathological feature of classical AIH is interface hepatitis, in which hepatocytes in the periportal zone are affected. In contrast, confluent necroinflammatory change in the centrilobular zone (zone 3) may be seen in histologically atypical AIH. The necroinflammatory change in zone 3 is called CZN, centrilobular (central) necrosis, or zone 3 necrosis. The characteristics of histologically atypical AIH were first reported using the name CZN and included marked confluent necrosis in zone 3 with relative sparing of the portal area.58 Subsequently, pathological changes in zone 3 have been described by many authors. However, the nomenclature and the definition of necroinflammatory changes in zone 3 are confused; thus, the significance of CZN has not been correctly evaluated.
Though some authors described significant but mild to moderate necroinflammation in zone 3 as zone 3 necrosis,59 these findings are frequently encountered in histologically typical AIH, especially in patients with significant lobular hepatitis and those with slight fibrosis. In contrast, others have described CZN as marked confluent necrosis in zone 3 with sparing of the portal area.60 However, typical CZN and significant interface hepatitis may coexist in the same sample of a liver biopsy.61 Therefore, histologically atypical AIH may be classified into two types: CZN with sparing interface hepatitis and CZN with significant interface hepatitis (Figure 1).
 | Figure 1 Two types of centrilobular zonal necrosis (CZN). Notes: (A) Marked CZN without evidence of portal inflammation/interface hepatitis. (B) CZN accompanied by portal inflammation and interface hepatitis. C, central vein; p, portal vein. Masson’s Trichrome staining, ×200. |
The idea that CZN may represent a very early histologic phenotype of typical AIH is derived from a case report in which a marked predominance of centrizonal injury in an initial liver biopsy was later transformed to a typical histological appearance over the course of several months.62 However, CZN can be found in the liver biopsies of patients who developed AIH >6 months earlier. In addition, CZN may coexist with significant portal fibrosis in chronic AIH. Histologically atypical AIH in Japanese patients is immunogenetically distinct from typical AIH.63 HLA-DR4 is a resistant, whereas DR9 is a susceptible, HLA phenotype of histologically atypical AIH. These findings strongly suggest that histologically atypical AIH is not a transient histological feature of very early typical AIH but a feature of a distinctive subtype of AIH.
On the whole, the prognosis of histologically atypical AIH is good and the response to immunosuppression is excellent. However, this type of AIH may rapidly develop into ALF, which can be a cause of death.64 Thus, quick and accurate diagnosis is required. However, the AIH score of this type of AIH is lower than that of classical AIH, chiefly because of the lower prevalence of ANA and lower levels of IgG. As this type of AIH responds well to corticosteroid therapy if there is no delay in treatment, appropriate examinations to determine a definite diagnosis should be performed as soon as possible and sufficient immunosuppression therapy should be started without delay.
The most important differential diagnosis of histologically atypical AIH is DILI, in which zone 3 hepatocytes are mainly affected. There are many drugs and chemicals that induce zone 3 necrosis. The histological findings of DILI are sometimes quite similar to those of AIH accompanying CZN. Therefore, liver biopsy is not sufficient to distinguish histologically atypical AIH from DILI.
CZN is a relatively rare condition that accounts for 10–20% of AIH cases.63 In the future, CZN must be clarified using unified criteria, and the clinicopathological features of this subtype of AIH should be confirmed by a large-scale study.
Treatment
The aim of treatment for AIH is to achieve complete remission of disease, followed by maintenance of remission and prevention of disease progression. Asymptomatic AIH is claimed not to require immunosuppressive therapy. No difference is observed in the prognosis of mild to moderate cases, irrespective of whether these cases are treated with immunosuppressive drugs.65 In contrast, the 10-year survival rate for mild, untreated cases is reported to be significantly lower than for treated cases.66 In mild to moderate cases, the condition can spontaneously resolve, but this happens infrequently and there is a risk that the disease will flare up during the clinical course. There are also some patients whose liver disease progresses asymptomatically, and the diagnosis is not established till severe fibrosis or cirrhosis has developed. Therefore, if medical intervention is deferred, the patient must be carefully monitored. When these cases are treated, potential adverse reactions caused by the therapeutic agent must be monitored.
The guidelines of the European Association for the Study of the Liver (EASL) state that treatment is necessary for advanced fibrosis, or in cases that are histologically active (hepatic activity index [HAI] score of 4/18 or higher).67 When the ALT level is less than three times the ULN, the HAI score is <4/18, and there is no advanced fibrosis, treatment should be based on the patient’s age and pathology; if no treatment is selected, ALT and IgG levels should be measured every 3 months. If these levels are elevated, a follow-up liver biopsy is recommended; if the disease has become active, treatment should be initiated. The current strategy for the treatment of adult AIH is summarized in Figure 2.
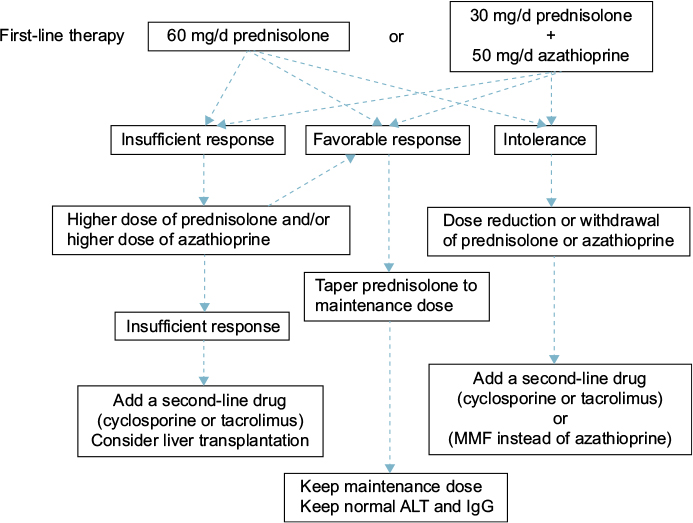 | Figure 2 Current treatment strategy for adult autoimmune hepatitis. Abbreviations: ALT, alanine aminotransferase; IgG, immunoglobulin G; MMF, mycophenolate mofetil. |
Prednisolone (PSL) monotherapy or PSL and azathioprine combination therapy is used for first-line AIH treatment. These two treatments have equivalent therapeutic effect.68 In combination treatment, the dose of PSL is lower than in PSL monotherapy, reducing the PSL-induced adverse reactions. Azathioprine combination therapy is preferred for postmenopausal women and for patients with osteoporosis, unstable diabetes, obesity, or hypertension. The recommended initial PSL dose is 60 mg (or 1 mg/kg of body weight) in monotherapy and 30–40 mg in combination with azathioprine. The azathioprine dose is generally 50 mg. Azathioprine is contraindicated in patients with severe cytopenia, thiopurine S-methyltransferase (TPMT) deficiency, pregnancy, or cancer.69 The TPMT genetic polymorphism is associated with reduced TPMT activity, so it is preferable to identify the genotype prior to the use of azathioprine to avoid myelosuppression.70
With PSL monotherapy, the dose should be decreased by 10 mg every week; the rate of dose reduction is eased from 30 mg downward to 5 mg every 1–2 weeks. Rapid reduction in the PSL dosage is an independent risk factor for recurrence.71 As the prognosis worsens in patients who have had two or more relapses, maintaining remission is very important.72 The PSL maintenance dose is ≤10 mg/d. Treatment should be continued until the transaminases, total bilirubin, gammaglobulin, IgG, and liver tissue have normalized. Therefore, it is essential to continue maintenance treatment over a long period.
Cases in which ALT level does not decrease to less than two times the ULN, relapsed cases, and cases that do not respond to high-dose PSL are considered PSL-resistant. In the case of PSL resistance, once drug compliance has been confirmed, increasing the PSL dose or adding or increasing the azathioprine dose should be considered. In patients who do not tolerate PSL or azathioprine, dose reduction, or withdrawal of the drug should be considered. As an alternative therapy, administration of cyclosporine,73 tacrolimus,74 or mycophenolate mofetil13 has been shown to be effective. There are a few reports on the efficacy of cyclophosphamide, methotrexate, rituximab, and infliximab, but no consensus has been reached on the usage of these drugs.
Budesonide may be considered for combination therapy with azathioprine instead of PSL, but there is little likelihood of efficacy in cases resistant to PSL therapy.75 Budesonide is metabolized by the liver and has a 90% first-pass effect; it has few systemic effects and is characterized by a lack of adverse drug reactions.76 However, as it is metabolized by the liver, it is contraindicated in patients with cirrhosis or patients with a liver shunt. In a prospective study of the additive effect of UDCA for PSL-treated patients, UDCA did not facilitate PSL dose reduction or reduce histological activity.77 In Japan, UDCA is administered to patients who have mild AIH with low disease activity and preserved liver function, and it has been found to have a positive effect in decreasing transaminase levels.78 However, the long-term effect of UDCA is unknown.
Mycophenolate mofetil (2 g daily orally) is administered to patients as an alternative to azathioprine when the patient has not responded to standard treatment. As mycophenolate mofetil and PSL combination therapy has a higher rate of inducing complete remission than azathioprine and PSL, and remission is maintained after PSL is discontinued, mycophenolate mofetil may come to replace azathioprine as the standard treatment.79 However, this treatment is problematic in terms of the large number of adverse drug reactions, including rash, alopecia, nausea, vomiting, and diarrhea.80
Elderly AIH patients respond well to PSL therapy, with less likelihood of treatment resistance, so PSL should be used. It is essential to be aware of the adverse effects of PSL in older patients. More than the prescribed dose should not be taken. With severe AIH, there is no difference in sepsis rate between PSL treatment and nontreatment groups. Even if PSL therapy fails, PSL does not adversely affect the prognosis.81 Therefore, adequate doses of steroids should be administered urgently in these patients.
The 5-year survival rate for AIH living-liver transplantation is good, at ~80%, with no evidence of recurrence in transplanted livers in Japan,82 although a significant recurrence rate has been reported in Europe and the US.83 The recurrence rate after transplantation may differ in different races; this question requires further investigation. Splenectomy may inhibit the recurrence of AIH after liver transplantation.84 In a mouse AIH model, by removing central Treg cells, splenectomy prolonged the effects of a corticosteroid.85 Therefore, splenectomy may be an additional option for the treatment of AIH.
As regards future therapies, correction of disturbed immunity in AIH, in particular the restoration of failed Treg cells30 has shown promise. In a type 2 AIH mouse model, remission was introduced by transferring ex vivo expanded autologous CXCR3-positive Tregs.86 Therefore, retrieval of peripheral tolerance by adoptive transfer of Tregs could be a promising therapy for refractory AIH.
Conclusion
The clinical spectrum of AIH is widely distributed and the diagnosis of atypical AIH is not necessarily easy. AIH may overlap with PBC or PSC, a condition that is known as PBC/PSC-AIH overlap syndrome. However, whether overlap syndrome is a distinctive disease entity or merely a variant of PBC/PSC is an issue to be solved in the future. Similarly, whether DIAIH is a distinctive disease entity or merely a subtype of DILI that exhibits autoimmune phenomena is controversial. An increase in the incidence of NAFLD/NASH may lead to an increase in the cases of AIH with coincident NAFLD/NASH. Coincidence of NAFLD/NASH may be problematic, because the outcome may be aggravated and the efficacy of corticosteroids may be attenuated. Histologically, the significance of CZN should be examined extensively. In the treatment of AIH, development of novel drugs based on the immunological mechanisms of AIH is awaited.
After viral hepatitis is eliminated, AIH will become one of the central themes of hepatology. In the future, AIH will attract more and more attention because many serious issues related to its management remain to be resolved.
Disclosure
The authors report no conflicts of interest in this work.
References
Cowling DC, Mackay IR, Taft LI. Lupoid hepatitis. Lancet. 1956;271(6957):1323–1326. | ||
Gregorio GV, Portmann B, Reid F, et al. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997;25(3):541–547. | ||
Newton JL, Burt AD, Park JB, Mathew J, Bassendine MF, James OF. Autoimmune hepatitis in older patients. Age Ageing. 1997;26(6):441–444. | ||
Omagari K, Kinoshita H, Kato Y, et al. Clinical features of 89 patients with autoimmune hepatitis in Nagasaki Prefecture, Japan. J Gastroenterol. 1999;34(2):221–226. | ||
Gregorio GV, McFarlane B, Bracken P, Vergani D, Mieli-Vergani G. Organ and non-organ specific autoantibody titres and IgG levels as markers of disease activity: a longitudinal study in childhood autoimmune liver disease. Autoimmunity. 2002;35(8):515–519. | ||
Muratori P, Lalanne C, Fabbri A, Cassani F, Lenzi M, Muratori L. Type 1 and type 2 autoimmune hepatitis in adults share the same clinical phenotype. Aliment Pharmacol Ther. 2015;41(12):1281–1287. | ||
Umemura T, Ota M. Genetic factors affect the etiology, clinical characteristics and outcome of autoimmune hepatitis. Clin J Gastroenterol. 2015;8(6):360–366. | ||
Heneghan MA, Yeoman AD, Verma S, Smith AD, Longhi MS. Autoimmune hepatitis. Lancet. 2013;382(9902):1433–1444. | ||
Alvarez F, Berg PA, Bianchi FB, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929–938. | ||
Chazouillères O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology. 1998;28(2):296–301. | ||
Tiniakos DG, Brain JG, Bury YA. Role of histopathology in autoimmune hepatitis. Dig Dis. 2015;33(suppl 2):53–64. | ||
Heneghan MA, McFarlane IG. Current and novel immunosuppressive therapy for autoimmune hepatitis. Hepatology. 2002;35(1):7–13. | ||
Zachou K, Gatselis NK, Arvaniti P, et al. A real-world study focused on the long-term efficacy of mycophenolate mofetil as first-line treatment of autoimmune hepatitis. Aliment Pharmacol Ther. 2016;43(10):1035–1047. | ||
Donaldson PT. Immunogenetics in liver disease. Baillieres Clin Gastroenterol. 1996;10(3):533–549. | ||
Seki T, Kiyosawa K, Inoko H, Ota M. Association of autoimmune hepatitis with HLA-Bw54 and DR4 in Japanese patients. Hepatology. 1990;12(6):1300–1304. | ||
Eskandari-Nasab E, Tahmasebi A, Hashemi M. Meta-analysis: the relationship between CTLA-4 +49 A/G polymorphism and primary biliary cirrhosis and type I autoimmune hepatitis. Immunol Invest. 2015;44(4):331–348. | ||
Webb GJ, Hirschfield GM. Using GWAS to identify genetic predisposition in hepatic autoimmunity. J Autoimmun. 2016;66:25–39. | ||
Bittencourt PL, Goldberg AC, Cançado EL, et al. Genetic heterogeneity in susceptibility to autoimmune hepatitis types 1 and 2. Am J Gastroenterol. 1999;94(7):1906–1913. | ||
Junge N, Tiedau M, Verboom M, et al. Human leucocyte antigens and pediatric autoimmune liver disease: diagnosis and prognosis. Eur J Pediatr. 2016;175(4):527–537. | ||
Hassan N, Siddiqui AR, Abbas Z, et al. Clinical profile and HLA typing of autoimmune hepatitis from Pakistan. Hepat Mon. 2013;13(12):e13598. | ||
Umemura T, Katsuyama Y, Yoshizawa K, et al. Human leukocyte antigen class II haplotypes affect clinical characteristics and progression of type 1 autoimmune hepatitis in Japan. PLoS One. 2014;9(6):e100565. | ||
Furumoto Y, Asano T, Sugita T, et al. Evaluation of the role of HLA-DR antigens in Japanese type 1 autoimmune hepatitis. BMC Gastroenterol. 2015;15:144. | ||
van Gerven NM, de Boer YS, Zwiers A, et al; Dutch Autoimmune Hepatitis Study Group. HLA-DRB1*03:01 and HLA-DRB1*04:01 modify the presentation and outcome in autoimmune hepatitis type-1. Genes Immun. 2015;16(4):247–252. | ||
Djilali-Saiah I, Fakhfakh A, Louafi H, Caillat-Zucman S, Debray D, Alvarez F. HLA class II influences humoral autoimmunity in patients with type 2 autoimmune hepatitis. J Hepatol. 2006;45(6):844–850. | ||
Yang F, Wang Q, Bian Z, Ren LL, Jia J, Ma X. Autoimmune hepatitis: east meets west. J Gastroenterol Hepatol. 2015;30(8):1230–1236. | ||
Buxbaum J, Qian P, Allen PM, Peters MG. Hepatitis resulting from liver-specific expression and recognition of self-antigen. J Autoimmun. 2008;31(3):208–215. | ||
Longhi MS, Ma Y, Mieli-Vergani G, Vergani D. Aetiopathogenesis of autoimmune hepatitis. J Autoimmun. 2010;34(1):7–14. | ||
Zierden M, Kühnen E, Odenthal M, Dienes HP. Effects and regulation of autoreactive CD8+ T cells in a transgenic mouse model of autoimmune hepatitis. Gastroenterology. 2010;139(3):975–986. | ||
Peiseler M, Sebode M, Franke B, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol. 2012;57(1):125–132. | ||
Grant CR, Liberal R, Holder BS, et al. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology. 2014;59(3):1007–1015. | ||
Wada Y, Sato C, Tomita K, et al. Possible autoimmune hepatitis induced after chronic active Epstein-Barr virus infection. Clin J Gastroenterol. 2014;7(1):58–61. | ||
Chiba T, Goto S, Yokosuka O, et al. Fatal chronic active Epstein-Barr virus infection mimicking autoimmune hepatitis. Eur J Gastroenterol Hepatol. 2004;16(2):225–228. | ||
Yamashita H, Shimizu A, Tsuchiya H, et al. Chronic active Epstein-Barr virus infection mimicking autoimmune hepatitis exacerbation in a patient with systemic lupus erythematosus. Lupus. 2014;23(8):833–836. | ||
Bonder A, Retana A, Winston DM, Leung J, Kaplan MM. Prevalence of primary biliary cirrhosis-autoimmune hepatitis overlap syndrome. Clin Gastroenterol Hepatol. 2011;9(7):609–612. | ||
Dienes HP, Erberich H, Dries V, Schirmacher P, Lohse A. Autoimmune hepatitis and overlap syndromes. Clin Liver Dis. 2002;6(2):349–362. | ||
Chou MJ, Lee SL, Chen TY, Tsay GJ. Specificity of antinuclear antibodies in primary biliary cirrhosis. Ann Rheum Dis. 1995;54(2):148–151. | ||
Kobayashi M, Kakuda Y, Harada K, et al. Clinicopathological study of primary biliary cirrhosis with interface hepatitis compared to autoimmune hepatitis. World J Gastroenterol. 2014;20(13):3597–3608. | ||
Kuiper EM, Zondervan PE, van Buuren HR. Paris criteria are effective in diagnosis of primary biliary cirrhosis and autoimmune hepatitis overlap syndrome. Clin Gastroenterol Hepatol. 2010;8(6):530–534. | ||
Zhang H, Li S, Yang J, et al. A meta-analysis of ursodeoxycholic acid therapy versus combination therapy with corticosteroids for PBC-AIH-overlap syndrome: evidence from 97 monotherapy and 117 combinations. Prz Gastroenterol. 2015;10(3):148–155. | ||
Ozaslan E, Efe C, Heurgué-Berlot A, et al. Factors associated with response to therapy and outcome of patients with primary biliary cirrhosis with features of autoimmune hepatitis. Clin Gastroenterol Hepatol. 2014;12(5):863–869. | ||
van Buuren HR, van Hoogstraten HJE, Terkivatan T, Schalm SW, Vleggaar FP. High prevalence of autoimmune hepatitis among patients with primary sclerosing cholangitis. J Hepatol. 2000;33(4):543–548. | ||
Tenca A, Färkkilä M, Arola J, Jaakkola T, Penagini R, Kolho KL. Clinical course and prognosis of pediatric-onset primary sclerosing cholangitis. United European Gastroenterol J. 2016;4(4):562–569. | ||
Verdonk RC, Lozano MF, van den Berg AP, Gouw AS. Bile ductal injury and ductular reaction are frequent phenomena with different significance in autoimmune hepatitis. Liver Int. 2016;36(9):1362–1369. | ||
Björnsson E, Talwalkar J, Treeprasertsuk S, et al. Drug-induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology. 2010;51(6):2040–2048. | ||
Alla V, Abraham J, Siddiqui J, et al. Autoimmune hepatitis triggered by statins. J Clin Gastroenterol. 2006;40(8):757–761. | ||
Suzuki A, Brunt EM, Kleiner DE, et al. The use of liver biopsy evaluation in discrimination of idiopathic autoimmune hepatitis versus drug-induced liver injury. Hepatology. 2011;54(3):931–939. | ||
Licata A, Maida M, Cabibi D, et al. Clinical features and outcomes of patients with drug-induced autoimmune hepatitis: a retrospective cohort study. Dig Liver Dis. 2014;46(12):1116–1120. | ||
Yeong TT, Lim KH, Goubet S, Parnell N, Verma S. Natural history and outcomes in drug-induced autoimmune hepatitis. Hepatol Res. 2016;46(3):E79–E88. | ||
de Boer YS, Kosinski AS, Urban TJ, et al; Drug-Induced Liver Injury Network. Features of autoimmune hepatitis in patients with drug-induced liver injury. Clin Gastroenterol Hepatol. Epub 2016 Jun 14. | ||
Beaune PH, Lecoeur S. Immunotoxicology of the liver: adverse reactions to drugs. J Hepatol. 1997;26(suppl 2):37–42. | ||
De Luca-Johnson J, Wangensteen KJ, Hanson J, Krawitt E, Wilcox R. Natural history of patients presenting with autoimmune hepatitis and coincident nonalcoholic fatty liver disease. Dig Dis Sci. 2016;61(9):2710–2720. | ||
Müller P, Messmer M, Bayer M, Pfeilschifter JM, Hintermann E, Christen U. Non-alcoholic fatty liver disease (NAFLD) potentiates autoimmune hepatitis in the CYP2D6 mouse model. J Autoimmun. 2016;69:51–58. | ||
Loria P, Lonardo A, Leonardi F, et al. Non-organ-specific autoantibodies in nonalcoholic fatty liver disease: prevalence and correlates. Dig Dis Sci. 2003;48(11):2173–2181. | ||
Hu KQ, Yang H, Lin YC, Lindsay KL, Redeker AG. Clinical profiles of chronic hepatitis C in a major county medical center outpatient setting in United States. Int J Med Sci. 2004;1(2):92–100. | ||
Ma Y, Peakman M, Lobo-Yeo A, et al. Differences in immune recognition of cytochrome P4502D6 by liver kidney microsomal (LKM) antibody in autoimmune hepatitis and chronic hepatitis C virus infection. Clin Exp Immunol. 1994;97(1):94–99. | ||
Kammer AR, van der Burg SH, Grabscheid B, et al. Molecular mimicry of human cytochrome P450 by hepatitis C virus at the level of cytotoxic T cell recognition. J Exp Med. 1999;190(2):169–176. | ||
Efe C, Wahlin S, Ozaslan E, et al. Diagnostic difficulties, therapeutic strategies, and performance of scoring systems in patients with autoimmune hepatitis and concurrent hepatitis B/C. Scand J Gastroenterol. 2013;48(4):504–508. | ||
Pratt DS, Fawaz KA, Rabson A, Dellelis R, Kaplan MM. A novel histological lesion in glucocorticoid-responsive chronic hepatitis. Gastroenterology. 1997;113:664–668. | ||
Miyake Y, Iwasaki Y, Terada R, et al. Clinical features of Japanese type 1 autoimmune hepatitis patients with zone III necrosis. Hepatol Res. 2007;37(10):801–805. | ||
Zen Y, Notsumata K, Tanaka N, Nakanuma Y. Hepatic centrilobular zonal necrosis with positive antinuclear antibody: a unique subtype or early disease of autoimmune hepatitis? Hum Pathol. 2007;38(11):1669–1675. | ||
Hofer H, Oesterreicher C, Wrba F, Ferenci P, Penner E. Centrilobular necrosis in autoimmune hepatitis: a histological feature associated with acute clinical presentation. J Clin Pathol. 2006;59(3):246–249. | ||
Singh R, Nair S, Farr G, Mason A, Perrillo R. Acute autoimmune hepatitis presenting with centrizonal liver disease: case report and review of the literature. Am J Gastroenterol. 2002;97(10):2670–2673. | ||
Aizawa Y, Abe H, Sugita T, et al. Centrilobular zonal necrosis as a hallmark of a distinctive subtype of autoimmune hepatitis. Eur J Gastroenterol Hepatol. 2016;28(4):391–397. | ||
Stravitz RT, Lefkowitch JH, Fontana RJ, et al; Acute Liver Failure Study Group. Autoimmune acute liver failure: proposed clinical and histological criteria. Hepatology. 2011;53(2):517–526. | ||
Feld JJ, Dinh H, Arenovich T, Marcus VA, Wanless IR, Heathcote EJ. Autoimmune hepatitis: effect of symptoms and cirrhosis on natural history and outcome. Hepatology. 2005;42(1):53–62. | ||
Czaja AJ. Features and consequences of untreated type 1 autoimmune hepatitis. Liver Int. 2009;29(6):816–823. | ||
European Association for the Study of the Liver. EASL clinical practice guidelines: autoimmune hepatitis. J Hepatol. 2015;63(4):971–1004. | ||
Lamers MM, van Oijen MG, Pronk M, Drenth JP. Treatment options for autoimmune hepatitis: a systematic review of randomized controlled trials. J Hepatol. 2010;53(1):191–198. | ||
Manns MP, Czaja AJ, Gorham JD, et al; American Association for the Study of Liver Diseases. Diagnosis and management of autoimmune hepatitis. Hepatology. 2010;51(6):2193–2213. | ||
Krynetski EY, Tai HL, Yates CR, et al. Genetic polymorphism of thiopurine S-methyltransferase: clinical importance and molecular mechanisms. Pharmacogenetics. 1996;6(4):279–290. | ||
Takahashi A, Ohira H, Abe K, et al. Intractable Liver and Biliary Diseases Study Group of Japan. Rapid corticosteroid tapering: important risk factor for type 1 autoimmune hepatitis relapse in Japan. Hepatol Res. 2015;45(6):638–644. | ||
Yoshizawa K, Matsumoto A, Ichijo T, et al. Long-term outcome of Japanese patients with type 1 autoimmune hepatitis. Hepatology. 2012;56(2):668–676. | ||
Sherman KE, Narkewicz M, Pinto PC. Cyclosporine in the management of corticosteroid-resistant type I autoimmune chronic active hepatitis. J Hepatol. 1994;21(6):1040–1047. | ||
Aqel BA, Machicao V, Rosser B, Satyanarayana R, Harnois DM, Dickson RC. Efficacy of tacrolimus in the treatment of steroid refractory autoimmune hepatitis. J Clin Gastroenterol. 2004;38(9):805–809. | ||
Czaja AJ, Lindor KD. Failure of budesonide in a pilot study of treatment-dependent autoimmune hepatitis. Gastroenterology. 2000;119(5):1312–1316. | ||
Danielsson A, Prytz H. Oral budesonide for treatment of autoimmune chronic active hepatitis. Aliment Pharmacol Ther. 1994;8(6):585–590. | ||
Czaja AJ, Carpenter HA, Lindor KD. Ursodeoxycholic acid as adjunctive therapy for problematic type 1 autoimmune hepatitis: a randomized placebo-controlled treatment trial. Hepatology. 1999;30(6):1381–1386. | ||
Miyake Y, Iwasaki Y, Kobashi H, et al. Efficacy of ursodeoxycholic acid for Japanese patients with autoimmune hepatitis. Hepatol Int. 2009;3(4):556–562. | ||
Zachou K, Gatselis N, Papadamou G, Rigopoulou EI, Dalekos GN. Mycophenolate for the treatment of autoimmune hepatitis: prospective assessment of its efficacy and safety for induction and maintenance of remission in a large cohort of treatment-naïve patients. J Hepatol. 2011;55(3):636–646. | ||
Hlivko JT, Shiffman ML, Stravitz RT, et al. A single center review of the use of mycophenolate mofetil in the treatment of autoimmune hepatitis. Clin Gastroenterol Hepatol. 2008;6(9):1036–1040. | ||
Yeoman AD, Westbrook RH, Zen Y, et al. Prognosis of acute severe autoimmune hepatitis (AS-AIH): the role of corticosteroids in modifying outcome. J Hepatol. 2014;61(4):876–882. | ||
Yamashiki N, Sugawara Y, Tamura S, et al. Living-donor liver transplantation for autoimmune hepatitis and autoimmune hepatitis-primary biliary cirrhosis overlap syndrome. Hepatol Res. 2012;42(10):1016–1023. | ||
Demetris AJ, Adeyi O, Bellamy CO, et al. Banff Working Group Liver biopsy interpretation for causes of late liver allograft dysfunction. Hepatology. 2006;44(2):489–501. | ||
Xu YT, Liu DJ, Meng FY, Li GB, Liu J. Possible benefit of splenectomy in liver transplantation for autoimmune hepatitis. Hepatobiliary Pancreat Dis Int. 2014;13(3):328–331. | ||
Maruoka R, Aoki N, Kido M, et al. Splenectomy prolongs the effects of corticosteroids in mouse models of autoimmune hepatitis. Gastroenterology. 2013;145(1):209.e–220.e. | ||
Lapierre P, Béland K, Yang R, Alvarez F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology. 2013;57(1):217–227. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.