Back to Journals » International Medical Case Reports Journal » Volume 9
Atypical scleromyxedema presenting with cutaneous and cardiovascular manifestations
Received 19 June 2016
Accepted for publication 5 August 2016
Published 19 September 2016 Volume 2016:9 Pages 295—299
DOI https://doi.org/10.2147/IMCRJ.S115315
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Sue-Ann Teh,1 David A Kandiah2
1Department of Health Western Australia, Bunbury Hospital, Bunbury, 2School of Psychiatry and Clinical Neurosciences, Faculty of Medicine, Dentistry and Health Sciences, University of Western Australia, Crawley, WA, Australia
Abstract: Scleromyxedema is part of a group of cutaneous mucinoses, characterized by a generalized papular eruption, dermal mucin deposition, and an increase in dermal collagen. This condition can be localized as discrete papular lichen myxedematous skin or as a systemic condition usually associated with paraproteinaemia. To date, there is no unifying treatment and is limited by rarity, small number of case reports, and the lack of randomized controlled trials. We describe the case of a 56-year-old gentleman with features of scleromyxedema who had cutaneous and cardiac involvement, and significant mediastinal lymphadenopathy without monoclonal gammopathy.
Keywords: scleromyxedema, heart failure, dermal mucin
Introduction
Scleromyxedema is a rare chronic cutaneous fibromucinous disorder of unknown etiology, commonly associated with monoclonal gammopathy, usually immunoglobulin G with lambda light chains.1 Dubreuilh first described this in 1906.2 This was further defined by Rongioletti and Rebora in 2001 as characterized by generalized papular and sclerodermoid eruption, mucin deposition, fibroblast proliferation and fibrosis, monoclonal gammopathy, and the absence of thyroid disorder.3
Case
The patient provided written informed consent for the publication of this paper and accompanying images. We report a case of a 56-year-old man who was referred by his general practitioner for assessment and investigations of dyspnea and bilateral lower limb swelling. This started after a tooth extraction for an abscess 2 months previously. Soon after, he developed lower limb swelling and painful lesions consistent but not diagnostic of erythema nodosum. He did not report any fever, night sweats, weight loss, or altered bowel habit. Initially, his condition was attributed to an immunological reaction to a tooth abscess with likely streptococcal bacteremia causing the systemic skin eruptions. He was prescribed a course of Augmentin Duo Forte 875/125 mg bid. Antistreptolysin titers were negative. The course of antibiotics settled the tooth infection but his dyspnea and lower limb edema worsened with associated skin tethering.
His past medical history was of benign heart murmurs diagnosed at the age of 2. He was not on any regular medications. He did not smoke but admitted to previous heavy alcohol intake, but currently limited to one beer per day.
Physical examination
His pulse was 86/min and regular and blood pressure was 150/90. There were a few 2–5 mm lesions with the appearance of lichenoid papules, which were tender over his abdomen and lower limbs. He had tight, tethered, and thickened skin up to his abdomen. Closer inspection of his skin showed that the skin was thickened in a linear distribution with the papules being skin-colored with scattered hyperpigmentation (Figure 1). The skin on his chest and arms was not affected. He had signs of biventricular heart failure. His heart sounds were dual with soft systolic murmurs. Ascites were present.
 | Figure 1 At presentation, there was linear thickening of skin of lower limbs with small discrete papules and areas of hyperpigmentation. |
Investigations
Full blood count and renal function were normal. Liver function test showed a mild cholestatic picture consistent with hepatic congestion. Serum and urine protein electrophoresis and immunoglobulin-free light chain assays were normal. Thyroid function tests and angiotensin converting enzyme levels were normal. Autoantibody screening was negative. His C-reactive protein was 38 mg/L (<5) with a normal erythrocyte sedimentation rate of 12 mm/h. Troponin I and creatinine kinase were both within normal limits. B type natriuretic peptide was 468 µg/L (<250). His chest X-ray showed marked globular cardiomegaly and interstitial pulmonary edema. Echocardiogram revealed a moderately dilated left ventricle with global impairment of systolic function with an estimated ejection fraction of 20%. There was a small to moderate pericardial effusion but no echo evidence of pericardial tamponade or constriction. Computed tomography of chest/abdomen/pelvis showed significant mediastinal lymphadenopathy with mildly enlarged abdominal and inguinal lymph nodes.
Magnetic resonance imaging of pelvis and legs showed widespread lower abdominal trunk, pelvic girdle, and bilateral lower limb subcutaneous edema with prominence of the dermal thickness (Figure 2). There was associated patchy tethering with increased T2 signal of the quadriceps, hamstring, and adductor muscle groups. Inguinal and external iliac lymph nodes were mildly enlarged. There was no hip or knee joint synovitis or effusions seen.
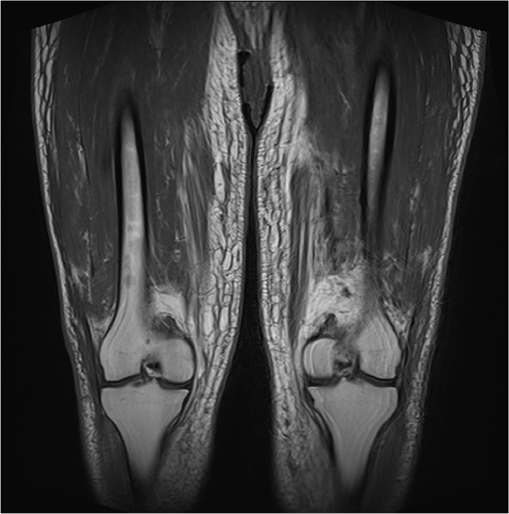 | Figure 2 Magnetic resonance imaging of pelvis and legs showed bilateral lower limb subcutaneous edema with prominence of the dermal thickness and patchy tethering with increased T2 signal of the quadriceps and adductor muscle groups. |
Clinical management
The patient was started on diuretics with frusemide 40 mg bid, and spironolactone 25 mg daily, and carvedilol 3.125 mg daily for heart failure and fluid restricted to 1.5 L/day. His heart failure improved with this treatment and he lost 9 kg of weight during the hospital admission in the first 5 days. Skin biopsy was taken from bilateral legs and abdomen. This showed compact orthokeratotic hyperkeratosis overlying mildly acanthotic epidermis. There was an increase in fibroblastic cells and interstitial connective tissue mucin in the dermis associated with perivascular lymphocytic infiltrate. There was thickening of the dermal collagen bundles with loss of adipose tissues around the eccrine glands (Figure 3A–D). The earlier findings were compatible with scleromyxedema.4 After the skin biopsy, he was encouraged to massage his skin regularly with sorbolene and vitamin E cream to improve vascular and lymphatic flow.
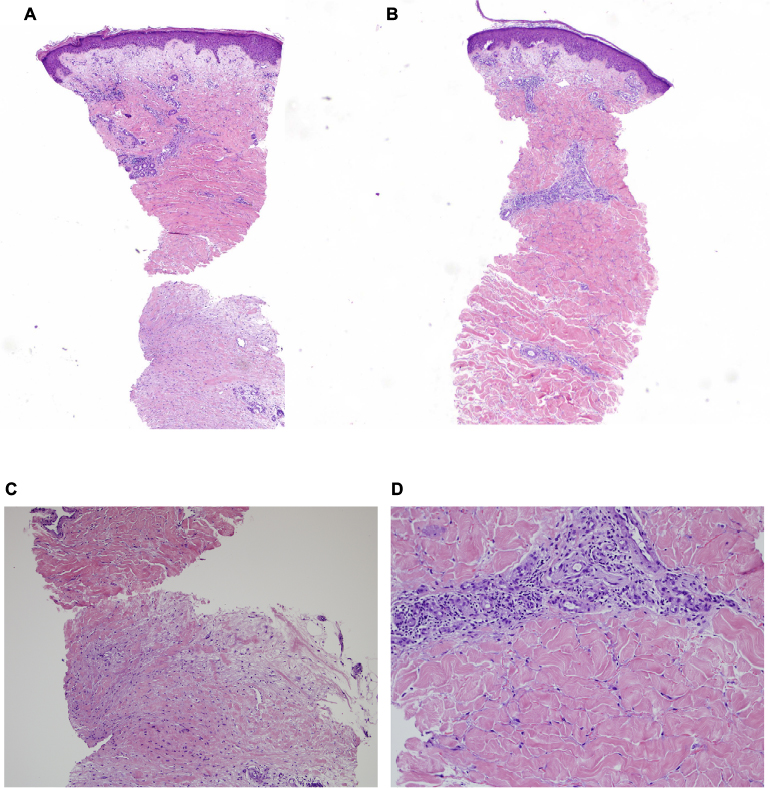 | Figure 3 (A) Skin biopsy – leg (×40 magnification), (B) skin biopsy – abdomen (×40 magnification), (C) deep skin biopsy – leg (×100 magnification), and (D) perivascular and thickened collagen – abdomen (×200 magnification). Note: The histology shows normal epidermis with proliferation of angular fibroblasts in the reticular dermis accompanied by disorganized collagen fibres (A–C). Increased interstitial connective tissue mucin in the dermis associated with perivascular lymphocytic infiltrate, there was thickening of the dermal collagen bundles with loss of adipose tissues around the eccrine glands (D). |
Progress over next 12 months
He subsequently lost another 8 kg in 8 weeks and went back to his normal average weight. His creatinine remained normal at 67 µmol/L and his estimated glomerular filtration rate (eGFR) was >90 mL/min/1.73m2. Frusemide was reduced to 40 mg/day but he remained on the same dose of spironolactone and carvedilol. Four weeks later, he became asymptomatic from congestive cardiac failure with no dyspnea, ascites, or peripheral edema. He developed two further episodes of a linear nontender papular mucinous rash in both his upper and lower limbs over the next 6 months with no flare of heart failure. His creatinine remained normal with an eGFR of 85 mL/min/1.73m2. His extractable nuclear antigen became equivocal from a previously negative result. His other autoantibodies were negative. The computed tomography scan of his chest and abdomen showed a significant improvement of his fluid retention. The ascites and mediastinal lymphadenopathy had resolved and the pericardial effusion had significantly reduced. Clinically, his lower limb skin was less tethered (Figure 4). Repeat echocardiogram revealed a mildly dilated left ventricle with an overall mild impairment of systolic function with an estimated ejection fraction of 45%.
 | Figure 4 Progress at 9 months: spontaneous regression of skin changes. |
Discussion
Scleromyxedema has been further subclassified by Rongioletti in 2006 into three categories: generalized form, localized form, including papular and nodular types, and atypical forms, which included scleromyxedema without monoclonal gammopathy.1 The skin eruption is commonly described as papular mucinosis or waxy papules with a predilection for sun exposed areas. This patient worked consistently outside in the sun. The firm papules can converge into indurated plaques, causing marked sclerosis, skin induration, and decreased joint mobility in the generalized form of scleromyxedema. The mucous membranes and scalp are usually spared as was the situation with this patient. Extra-cutaneous manifestations include neurological, gastrointestinal, respiratory, cardiovascular, rheumatological, renal, and hematological complications.3 In localized form, sclerotic features, systemic involvement, and monoclonal gammopathy are absent.
Our patient has an atypical form of scleromyxedema with cutaneous and cardiac involvement with prominent mediastinal lymphadenopathy. Cutaneous manifestations were the most common findings in scleromyxedema. Absence of paraproteinaemia has been described.3 A literature search of cutaneous manifestations of scleromyxedema showed that some patients exhibited red–brown discoloration of the involved skin.5 Scleromyxedema with cardiac involvement, for instance congestive cardiomyopathy, myocardial infarction, and inflammatory cardiomyopathy, has been previously reported.5 A cardiac biopsy could have confirmed myocardial involvement in this patient. This was considered too invasive but would have been performed if he had not responded adequately to cardiac failure treatment. As he is being monitored, this may be considered in future if his cardiac failure becomes refractory. In the absence of other causes of heart failure, normal troponins, and the relatively low B type natriuretic peptide, the cardiac failure was considered to be due to early myocardial fibrotic changes. Mucin deposition in the myocardium and heart vessels was noted in autopsy findings.6 Enlarged mediastinal lymphadenopathy was uncommon and was observed in a patient associated with monoclonal gammopathy.7
As our patient clinically improved with heart failure treatment, we opted to monitor him rather than commence the patient on immunological treatment for scleromyxedema. The current suggested treatments are based on anecdotal case studies and case series. Suggested treatments include systemic steroids,5 plasmapheresis,8 interferon alpha,9 stem cell transplantation,10 intravenous immunoglobulin,11 thalidomide,12 bortezomib,5,12 and chemotherapy.12
However, the side effects limit the use of many options. Studies show dramatic results with intravenous immunoglobulin as primary therapy11 but the effect may be short lasting as predicted from its mechanism of action. This requires regular maintenance and can be expensive. In our patient, the course of his disease has been favorable so far with spontaneous regression of skin changes.
Conclusion
In conclusion, scleromyxedema is rare and can be chronic and progressive with high morbidity and mortality. Hence, long-term follow-up of such patients is pivotal. The diversity of scleromyxedema clinical presentations means that clinicians should be aware of this possible diagnosis.
Disclosure
The authors report no conflicts of interest in this work.
References
Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perceptives for an old disease. Semin Cutan Med Surg. 2006;25(2):100–104. | ||
Dubreuilh W. Fibromes miliaires folliculaires: sclerodermie consecutive. Arch Dermatol Syphilol. 1906;7:569–570. | ||
Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44(2):273–281. | ||
Fernandez-Flores A, Gatica-Torres M, Ruelas-Villavicencio AL, Saeb-Lima M. Morphological clues in the diagnosis of sclerodermiform dermatitis. Am J Dermapathol. 2014;36(6):449–464. | ||
Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69(1):66–72. | ||
Loggini B, Pingitore R, Avvenente A, Giuliano G, Barachini P. Lichen myxedematosus with systemic involvement: clinical and autopsy findings. J Am Acad Dermatol. 2001;45(4):606–608. | ||
Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42(1):31–35. | ||
MacFarlane AW, Davenport A, Verbov JL, Goldsmith HJ. Scleromyxoedema – successful treatment with plasma exchange and immunosuppression. Br J Dermatol. 1987;117(5):653–657. | ||
Tschen JA, Chang JR. Scleromyxedema: treatment with interferon alfa. J Am Acad Dermatol. 1999;40(2 Pt 2):303–307. | ||
Feasel AM, Donato ML, Duvic M. Complete remission of scleromyxedema following autologous stem cell transplantation. Arch Dermatol. 2001;137(8):1071–1072. | ||
Blum M., Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine (Baltimore). 2008;87(1):10–20. | ||
Ataergin S, Arpaci F, Demiriz M, Ozet A. Transient efficacy of double high-dose chemotherapy and autologous peripheral stem cell transplantation, immunoglobulin, thalidomide, and bortezomib in the treatment of scleromyxedema. Am J Clin Dermatol. 2008;9(4):271–273. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.