Back to Journals » The Application of Clinical Genetics » Volume 7
Ataxia-telangiectasia: future prospects
Authors Chaudhary MW, Al-Baradie R
Received 6 March 2014
Accepted for publication 7 May 2014
Published 10 September 2014 Volume 2014:7 Pages 159—167
DOI https://doi.org/10.2147/TACG.S35759
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Mohammed Wajid Chaudhary, Raidah Saleem Al-Baradie
Pediatric Neurology, Neurosciences Centre, King Fahad Specialist Hospital, Dammam, Kingdom of Saudi Arabia
Abstract: Ataxia-telangiectasia (A-T) is an autosomal recessive multi-system disorder caused by mutation in the ataxia-telangiectasia mutated gene (ATM). ATM is a large serine/threonine protein kinase, a member of the phosphoinositide 3-kinase-related protein kinase (PIKK) family whose best-studied function is as master controller of signal transduction for the DNA damage response (DDR) in the event of double strand breaks (DSBs). The DDR rapidly recognizes DNA lesions and initiates the appropriate cellular programs to maintain genome integrity. This includes the coordination of cell-cycle checkpoints, transcription, translation, DNA repair, metabolism, and cell fate decisions, such as apoptosis or senescence. DSBs can be generated by exposure to ionizing radiation (IR) or various chemical compounds, such as topoisomerase inhibitors, or can be part of programmed generation and repair of DSBs via cellular enzymes needed for the generation of the antibody repertoire as well as the maturation of germ cells. AT patients have immunodeficiency, and are sterile with gonadal dysgenesis as a result of defect in meiotic recombination. In the cells of nervous system ATM has additional role in vesicle dynamics as well as in the maintenance of the epigenetic code of histone modifications. Moderate levels of ATM are associated with prolonged lifespan through resistance to oxidative stress. ATM inhibitors are being viewed as potential radiosensitizers as part of cancer radiotherapy. Though there is no cure for the disease at present, glucocorticoids have been shown to induce alternate splicing site in the gene for ATM partly restoring its activity, but their most effective timing in the disease natural history is not yet known. Gene therapy is promising but large size of the gene makes it technically difficult to be delivered across the blood–brain barrier at present. As of now, apart from glucocorticoids, use of histone deacetylase inhibitors/EZH2 to minimize effect of the absence of ATM, looks more promising.
Keywords: ATM, double-stranded DNA breaks, neurodegeneration
Introduction
The first description of patients with Ataxia-Telangiectasia (A-T) was published in 1926,1 but it was not until 1958 that the term ‘ataxia-telangiectasia’ was coined by Elena Boder and Robert P Sedgewick to distinguish it from other forms of early-onset truncal ataxia. They observed eight cases from five unrelated families, describing the heredofamilial nature of the disease and the susceptibility of these patients to sinopulmonary infections with characteristic dilated oculocutaneous blood vessels.2 The hallmark of the disease is progressive neurological dysfunction characterized by uncoordinated, ataxic movements as a result of cerebellar atrophy. Choreoathetosis and dystonias are variably present, and can be quite disabling in some cases. Other features include telangiectasia, immune and endocrine dysfunctions, genomic instability, premature aging, radiosensitivity, and predisposition to cancer. Since there is no definitive treatment available at present, supportive care is the mainstay of management. A-T is a life-limiting illness, and most patients die in the second or third decade of life, mainly with progressive neurodegeneration, pulmonary failure, or cancer.3
ATM gene
A-T is an autosomal recessive disorder, caused by mutations in the ataxia-telangiectasia mutated gene (ATM), a serine/threonine kinase that activates over a hundred proteins involved in DNA damage response, cell cycle regulation, and other pathways. The gene was first cloned by Savitsky et al and named ATM for A-T, mutated.4 Mutations in the ATM gene generally result in an absence of full-length, functional protein product. The human ATM gene is located at 11q22–23 and covers 160 kb of genomic DNA; the gene product, ATM protein, is produced from a 13 kb transcript that codes for a predicted 315 kDa protein. Patients with classical A-T manifest an early onset of the disease, severe progressively disabling cerebellar ataxia, dysarthria, oculomotor apraxia, chorea and dystonia, oculocutaneous telangiectasias, endocrine dysfunction, immunodeficiency, and cancer. Brain magnetic resonance imaging shows cerebellar atrophy. Laboratory tests reveal raised serum alpha-fetoprotein levels and chromosomal instability.3,5 Biochemical studies on cultured cells document a complete lack of functional ATM protein and genetic studies usually reveal homozygous or heterozygous “null” mutations in the ATM gene.3,5,6 On the other hand, variant A-T forms show a relatively mild neurological phenotype, often with normal brain magnetic resonance imaging and less frequently extraneurological features. These forms are usually caused by missense mutations leaving some detectable amount of functional ATM protein.5–7
The integrity of our genome is constantly under attack from both exogenous and endogenous sources. It has been estimated that a human cell is confronted with one million DNA lesions every day, one fifth of which may originate from the activity of reactive oxygen species alone,8,9 placing DNA damage response mechanisms in a position of paramount importance. The ability of a cell to correctly respond to and repair DNA damage to maintain genomic stability is critical for protection against development of cancer. Terminally differentiated neurons are highly active cells with very restricted, if any, regeneration potential.10 In addition, genome integrity and maintenance during neuronal development is crucial for the organism. Therefore, highly accurate and robust mechanisms for DNA repair are vital for neuronal cells.
The ATM protein, a serine/threonine protein kinase, is a member of the phosphoinositide 3-kinase-related protein kinase family. All members of this family are large serine/threonine protein kinases involved in signaling following cellular stress. Other members of the family are ATR (ATM and Rad3-related), DNA-PKcs (DNA-dependent protein kinase catalytic subunit), mTOR (mammalian target of rapamycin), and SMG1 (suppressor with morphological effect on genitalia family member), as shown in Figure 1. These atypical protein kinases regulate responses to DNA damage, nutrient-dependent signaling, and nonsense-mediated messenger RNA decay.11 These members all share common domain structures, including N-terminal HEAT Huntingtin, elongation factor 3, protein phosphatase 2A, and yeast kinase TOR 2 (HEAT) repeats, a FRAP, ATM, TRRAP (FAT) domain, a protein kinase domain, and a C-terminal FAT-C domain. A-T (ATM mutation), the related A-T-like disease (MRE11 mutation), Nijmegen breakage syndrome (NBS, NBS1/NBN mutation), and the more recently identified NBS-like disease (RAD50 mutation), all present with similar pathological outcomes in humans.12 ATM plays a central role in orchestrating molecular events involved in DNA double-strand break (DSB) signaling and repair.
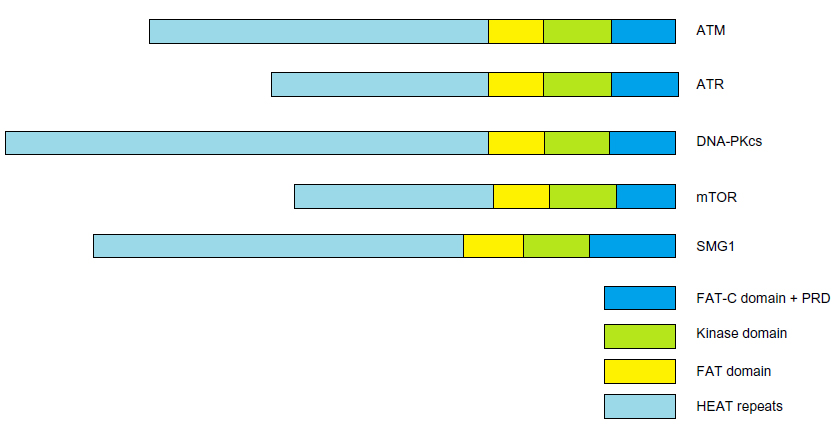 | Figure 1 PIKK family members, |
ATM is activated through one of the several mechanisms, the most significant of which is DNA DSBs, representing one of the most cytotoxic DNA lesions. Other activation mechanisms include reactive oxygen species, hypotonic stress, and chloroquine (Figure 2).
 | Figure 2 Activation of ATM. |
Role of ATM in DNA repair
Cells should be able to mount an appropriate response to millions of DNA lesions every day in each cell. This has to be a very precise and efficient system to prevent cells with damaged DNA from dividing further or being passed on to the next progeny through germ line mutation. The DNA DSB represents a particularly important threat to genome integrity. DSBs can be generated by exposure to ionizing radiation or various chemical compounds, such as topoisomerase inhibitors, that interfere with DNA replication and cell division. Generation of the antibody repertoire and maturation of germ cells both involve programmed generation and repair of DSBs via cellular enzymes.13,14 DSBs can also arise during DNA replication due to exposure to metabolites, such as reactive oxygen species, the activity of enzymes, such as topoisomerases, which break and rejoin DNA strands, and limitations in raw material needed for replication, such as nucleotides, that can promote fragile site expression and chromosomal breakage.15,16 Two major pathways of DSB are utilized in the cell, ie, nonhomologous end-joining, which is operative throughout the cell cycle, and homology directed repair, which is restricted to S/G2 when a sister chromatid is present as a template. The killing of cancer cells via generation of DSBs is a major strategy in cancer treatment and the cellular responses, and it is important to understand the mechanisms of repair and acquired resistance to these agents in order to improve the efficacy of current treatment regimens.17
ATM is the primary transducer of DSB-induced signaling.18 DSBs are recognized by the Mre11-Rad50-Nbs1 (MRN) also known as Mre11 complex, which is a sensor of DSBs. Capture of DNA ends by the Mre11 complex leads to rapid activation of the ATM kinase12 through autophosphorylation that promotes its monomerization and kinase activity,19–21 leading to a cascade of signals that determine the fate of the DNA and the cell involved through rapid post-translational modifications on many proteins and remodeling of chromatin structure around the break sites. Effector proteins, such as the Chk1 and Chk2 kinases, amplify the signal and cells can activate cell cycle checkpoints, regulate transcription, translation, and metabolism, and activate the appropriate DNA repair processes. In some circumstances, or in the face of irreparable lesions, cells can activate apoptosis and senescence. The collective result is prevention of genomic instability and the accompanying pathological outcomes.
Role of ATM in cell cycle regulation and fate
The cell cycle is divided into four sequential phases, ie, G1, S, G2, and mitosis (M). It is important that cellular regulation of progression through each of these phases be functioning correctly in order to maintain genomic integrity. Cell cycle checkpoints are pauses in cell cycle progression that allow the cell time to deal with physiological signals or challenges, such as DNA damage. One of the most prominent control points in the cell cycle is the entry into S-phase from G1, referred to here as the G1/S checkpoint. The tumor suppressor p53 was shown to play a critical role in the G1/S checkpoint following irradiation,22 and efficient induction of p53 after ionizing radiation requires ATM.23 The G1/S checkpoint is impaired in ATM-deficient cells. It is likely that defects in this checkpoint play a major role in predisposition to lymphoma24 as cells undergoing programmed rearrangements during V(D)J (variable [V], diversity [D], and joining [J]) recombination should not enter S-phase until repair takes place. Nbs1, the SMC1 component of cohesin, and activating transcription factor 2 (ATF2) have been identified as critical ATM targets in the intra-S checkpoint response,12,25 and cells in S-phase exposed to DNA damage activate the intra-S phase checkpoint, leading to a transient reduction in DNA synthesis. ATM plays a critical role in activation of the G2/M checkpoint that rapidly prevents G2 cells from entering mitosis after DNA damage. Deletion of the checkpoint kinase Chk2 with defects in the G2/M checkpoint results in tumor predisposition, indicating that the G2/M transition is important for tumor suppression.26,27 There is no general damage-induced response in mitosis phase.
There are three possible outcomes for a cell with DNA DSBs, ie, repair, apoptosis, or senescence. Apoptosis, or programmed cell death, is essential for development, particularly in the immune system, and represents an important mechanism for the clearance of cells with DNA damage. Apoptosis is triggered in response to a variety of DNA lesions, including DSBs, and defective apoptosis is considered a hallmark of cancer cells.28 Apoptosis in response to DSBs is regulated by p53 in many tissues, including lymphocytes. Stability of p53 is regulated through its phosphorylation by ATM and Chk2. ATM has also been implicated in regulation of senescence, ie, it inhibits senescence in response to oxidative stress. On the other hand, senescence induced by overexpression of some oncogenes is dependent upon ATM.29
Radiotherapy plays a central role in the treatment of cancer, and use of radiosensitizing agents can greatly enhance this modality. Given that ATM and the MRN complex play central roles in DNA repair and cell cycle checkpoints, these molecules are promising targets for radiosensitization. Following activation of ATM, downstream signaling is started, and p53 and Chk2 are undoubtedly the main targets of ATM. These in turn control the G1/S and G2/M checkpoints while interacting with each other. Inhibition of these checkpoints allows damaged cells to move to the mitotic phase without undergoing proper DNA repair, leading to mitotic catastrophe, which is currently considered a main cause of cell death induced by radiotherapy.30–32 Moreover, ATM is known to affect homology directed repair by directly or indirectly phosphorylating at least 12 targets, including breast cancer protein 1/2 and NBS1, and defects in ATM function lead to dysfunction in homology directed repair.33,34 These findings indicate that targeted ATM inhibition is an attractive approach to enhancing tumor radiosensitivity. Researchers have recently developed three ATM inhibitors and an MRN complex inhibitor (mirin) that have showed great potential as radiosensitizers of tumors in preclinical studies.35
Role of ATM in neuroprotection
One of the central pathologies associated with A-T, but unfortunately the most poorly understood aspect of it, is neurodegeneration and subsequent ataxia. This ataxia is already evident in the first years of life, and results in wheelchair dependency for most suffering children by the age of 10 years. Although the cerebellum in A-T patients is most profoundly affected with loss of Purkinje and granule cells, progressive neurodegeneration occurs also in other parts of the central nervous system.36–39 Notably, human diseases with the most similar neuropathology to A-T are caused by genes involved in the repair of diverse types of DNA lesions.40 The mice knockouts of ligase IV or XRCC4, proteins responsible for the nonhomogenous end-joining form of DNA repair, produce syndromes that lead to dysgenesis and death in the central nervous system followed by death of the embryo.41–43 Although the precise mechanism of neurodegeneration remains unknown, since pronounced neurodegeneration and ataxia are not observed in ATM null mice, it is thought that mitochondrial defects, accumulation of reactive oxygen species, transposon mobilization, innate immune responses, regulation of apoptosis, or specific repair pathway defects may contribute to triggering neuronal cell death.40,44,45 In rapidly dividing tissues, like bone marrow, cells with defective DNA repair undergo apoptosis and are replaced by nondefective ones, but in the central nervous system with terminally differentiated neurons that are highly active cells with, very restricted, if any, regeneration potential cannot be so replaced after apoptosis.
Abnormal neurogenesis is another contributor to the neurological manifestation of the disease. Although central nervous system pathology is a feature common to many diseases caused by mutations in genes that encode members of the ATM-dependent DNA damage response, including MRE11 (A-T-like disease), NBS1/NBN (NBS), RAD50 (NBS-like disease), and ATM (A-T), the relative contribution of neurodegeneration and abnormal neurogenesis depends on the underlying gene mutation. For example, mutations in MRE11 result in neurodegeneration, similar to what is observed in A-T patients, while other mutations in NBS1 or RAD50 cause microcephaly. Because of the complex interplay between these kinases, it is difficult to study their roles independently in animal models. Also, ATM-/- mice generated using gene targeting were characterized by motor learning deficiency, as well as ectopic and abnormal differentiation of Purkinje cells, although no obvious defects were found in their cerebella in one of the studies.46 Absence of typical neurodegeneration and ataxia seen in human A-T, among others, indicates that while many aberrations of human A-T can be observed and studied in mouse ATM-/- models, neurological findings in the human disease are only marginally observed.
ATM is involved in a much wider spectrum of cellular activities beyond DNA repair. Oxidative stress directly activates ATM by an independent mechanism47 and cells that are ATM-deficient are more sensitive to oxidative damage.47,48 ATM also has a role in cellular vesicle trafficking through its binding with β-adaptin and β-adaptin-related protein.49 This may be partly responsible for the neurological manifestation of A-T. Beyond vesicle release, the absence of ATM has profound effects on the systems-level circuitry of the brain.50
The role of ATM in development of the central nervous system has also been linked to transcriptional regulation in neurons. It is proposed that transcriptional defects caused by aberrant nuclear, rather than cytoplasmic, localization of histone deacetylase 4 (HDAC4) in ATM-deficient cells may contribute to neurodegeneration. HDAC4 accumulates in the nucleus in ATM-deficient neurons and causes global defects in histone acetylation and neuronal gene expression. Of particular interest, nuclear HDAC4 suppressed the activity of myocyte enhancer factor 2A and cyclic Adenosine Mono Phosphate (cAMP)-responsive element binding protein, which control prosurvival programs. Treatment with HDAC inhibitors reduced cell death and markers of cell cycle re-entry in the ATM-deficient cerebellum.51 Purkinje cell-specific deletion of the mouse males absent on the first (mMof) gene, which encodes a protein that specifically acetylates histone H4 at lysine 16 (H4K16ac) and influences ATM function, is critical for Purkinje cell longevity. Mice deficient for Purkinje cell-specific Mof display impaired motor coordination, ataxia, a backward-walking phenotype, and a reduced life span, similar to A-T. Treatment of Mof(F/F)/Pcp2-Cre(+) mice with HDAC inhibitors modestly prolongs Purkinje cell survival and delays death.52 Increased trimethylation of histone H3 on Lysine at position 27 (Lys27) (H3K27me3) mediated by polycomb repressive complex 2 is also important in the A-T phenotype. Enhancer of zeste homolog 2 (EZH2), a core catalytic component of polycomb repressive complex 2, is described as a new ATM kinase target, and ATM-mediated phosphorylation of EZH2 on Serine residue (Ser)734 reduces protein stability. Thus, formation of polycomb repressive complex 2 is elevated along with H3K27me3 in ATM deficiency. Chromatin immunoprecipitation and sequencing showed an increase in H3K27me3 “marks” and a dramatic shift in their location. The change in H3K27me3 chromatin-binding pattern is directly related to cell cycle re-entry and cell death of ATM-deficient neurons. Lentiviral knockdown of EZH2 rescued Purkinje cell degeneration and behavioral abnormalities in ATM-/- mice, demonstrating that EZH2 hyperactivity is another key factor in A-T neurodegeneration.53 Use of HDAC inhibitors and manipulation of EZH2 are exciting new options to be explored further.
Glucocorticoids have been shown to protect post-mitotic neurons from apoptosis through a mechanism involving the cyclin-dependent kinase inhibitor p21Wafl/Cip1 molecule.54 Of note, ATM signaling appears to function predominantly in immature recently post-mitotic neurons to trigger apoptosis of cells that have experienced excess DNA damage during brain development.55 Even though steroids are not curative and cannot be proposed for long-term therapies due to their side effects, it is suggested that during the clinical course there is a phase when neurological impairment can be rescued to some extent.56 In a multicenter, double-blind, randomized, placebo-controlled, crossover trial, oral betamethasone use showed significant reduction in ataxia scores.57 Dexamethasone has been shown to induce a splicing event, which leads to the skipping of mutations upstream of nucleotide residue 8450 of the ATM coding sequence. The resulting transcript provides an alternative open reading frame translated in a new ATM variant with the complete kinase domain. This partly restores ATM activity in A-T cells by a new molecular mechanism that overcomes most of the mutations so far described within this gene.58 This is a very exciting new area for further research. In order to minimize the frequent side effects of chronic corticosteroid treatment, a Phase II trial has recently showed the efficacy and safety of encapsulation of dexamethasone sodium phosphate into autologous erythrocytes, allowing slow release of dexamethasone for up to 1 month after dosing.59
Role of ATM in immunity
A-T patients are more susceptible to infections, in part because of varying degrees of immunodeficiency. A number of important roles for ATM have been identified in both adaptive and innate immunity, as well as inflammatory responses, that may underlie many aspects of pathology in A-T. ATM plays an important role in the development of both T-cells and B-cells, and A-T patients often exhibit abnormal T and B lymphocyte counts and deficient antibody responses.
Lymphocytes are the main cellular component of the adaptive immune system, which counteract infections and participate in tumor surveillance. To react to a wide range of antigens, lymphocytes generate a diverse repertoire of antigen-specific receptors that depend on programmed chromosomal rearrangements initiated by enzymes that introduce DNA breaks. These processes, ie, V(D)J recombination (T and B lymphocytes) and class switch recombination (in B lymphocytes), are both dependent on intact ATM function. The ability of ATM to monitor the development of lymphocytes through regulation of both DNA repair and apoptosis plays a critical role in tumor suppression.13 ATM null mice have reduced numbers of mature single positive cluster of differentiation (CD)4 or CD8 T-cells and increased numbers of immature double-positive thymocytes.60,61 During the development of T and B lymphocytes, V(D)J recombination is required for the assembly of antigen receptor genes. The recombination activating genes 1 and 2 (RAG1 and RAG2) constitute the RAG recombinase that generates DNA DSBs to catalyze recombination between the V, D, and J gene fragments in order to define the binding properties of the receptor.62 ATM colocalizes with RAG at endogenous recombination loci and RAG-induced DNA breaks persist in ATM-deficient cells.63–65 Persistent breaks in the immunoglobulin (Ig) heavy chain locus promote translocations, including those with Myc, that are known to promote genesis of lymphoma and occur at higher rates in ATM-deficient cells.66
In response to cytokine secretion or infection, B-cells initiate antibody class switching.62 This stimulation leads to rearrangements of the switch regions, thus altering the effector function of the antibody. Class switching is catalyzed by the activation-induced deaminase that promotes strand breakage. Human A-T patients frequently have impaired development of immunoglobulin subtypes IgA, IgG2, IgG4, and IgE in serum, and ATM-deficient mice show strong defects in class switch recombination.67–70
Role of ATM in metabolism
Impaired glucose metabolism and type 2 diabetes are more frequently seen in A-T than in the general population. Molecular roles of ATM in metabolism are suggested by insulin signaling through phosphorylation of translator regulator elF-4E-BP1 by ATM, and the increased reactive oxygen species seen in ATM-deficient animals.48,71,72 The use of antioxidants in ATM null mice has been shown to delay tumor formation.73–76 Reactive oxygen species can directly activate ATM, which in turn promotes antioxidant responses through stimulation of the pentose phosphate pathway, and ATM plays a role in monitoring mitochondrial quality control, again pointing to a central function of ATM in controlling cellular metabolism of reactive oxygen species.47,77 ATM may directly regulate mitochondrial homeostasis through responding to reactive oxygen species or by regulating mitochondrial quality control genes, such as PINK1 or Parkin.46 As these proteins are mutated in Parkinson’s disease, which is also characterized by degeneration of the central nervous system, understanding the regulation of mitochondrial integrity by ATM may shed light on the etiology of neurodegeneration in A-T. Recently, a multicenter study showed an association between the glycemic response to metformin and the rs11212617 single nucleotide polymorphism at a locus that includes the ATM gene.78,79
Role of ATM in longevity
Lifespan is attributable to genetic factors, and some studies have attempted to identify putative genes implicated in human longevity. Apart from the apolipoprotein E gene, not many genes have been consistently associated with longevity. There is some evidence that extreme longevity could be associated with increased resistance to oxidative stress.80,81 There is an association between a genetic variant (rs189037) in the ATM gene promoter and longevity through an effect on ATM transcription in Chinese nonagenarians/centenarians. The relevant study showed that the C/T genotype is associated with moderate levels of ATM gene expression, suggesting that the best way to prolong the lifespan is via a moderate level of ATM.82,83 The encoded protein is important in resistance to oxidative stress through its role in detection of reactive oxygen species lesions and DNA repair defects.84,85
Role of ATM role in fertility
Fertility as a clinical issue is often overlooked in A-T because affected patients are young and die early. ATM has a crucial role in completion of mouse gametogenesis since both male and female ATM-deficient mice are sterile with gonadal dysgenesis as a result of a defect in meiotic recombination.60 Meiosis is the special cell division that ensures formation of haploid cells, spermatozoa and ova from diploid progenitor germ cells. Germ cells undergo two rounds of chromosome segregations after replicating their genomes only once. ATM is essential in DSB repair during meiotic recombination.86,87 Testes and ovaries from ATM-/- animals display massive germ cell loss. While ATM-/- spermatocytes arrest at the pachytene stage of meiotic prophase, ATM-/- females lose all oocytes during the first days of life, before completing meiotic prophase.88–90 Recently, a particular variant in the promoter region of the ATM gene, rs189037 (G>A), was identified to be associated with idiopathic nonobstructive azoospermia.91
Future perspectives and conclusion
Although there is no definitive treatment or cure at present, gene therapy could be possible in the future. Some of the studies done in mouse models are promising.92 One of the main problems with this modality is the relatively large size of the gene, making it difficult for it to be delivered to cells, especially across the blood–brain barrier. Until the advancements in gene therapy make it possible in future we will be exploring diverse ATM substrates and their functions. Gene editing technologies, such as zinc-finger nucleases, should allow manipulation of ATM and associated genes in higher organisms, which may more faithfully recapitulate the human condition, particularly central nervous system pathology. Reprogramming of somatic cells into induced pluripotent stem cells provides an opportunity to gain insight into the molecular and cellular basis of disease. Because the cellular DNA damage response poses a barrier to reprogramming, generation of induced pluripotent stem cells from patients with chromosomal instability syndromes has proven to be difficult. A-T induced pluripotent stem cells, recently obtained in this way, can be differentiated into functional neurons and thus represent a suitable model system in which to investigate A-T-associated neurodegeneration.93 The response to glucocorticoids through a novel mechanism provides new possibilities for its’ role as possible disease-modifying agents. HDAC inhibitors targeting different enzymes and manipulation of EZH2 are exciting new options to be explored further.
Disclosure
The authors report no conflicts of interest in this work.
References
Syllaba L, Henner K. [Contributions to the independence of idiopathic and congenital double athetosis]. Rev Neurol. 1926;1:541–562. French. | |
Boder E, Sedgwick RP. Ataxia-telangiectasia; a familial syndrome of progressive cerebellar ataxia, oculocutaneous telangiectasia and frequent pulmonary infection. Pediatrics. 1958;21:526–554. | |
Chun HH, Gatti RA. Ataxia-telangiectasia, an evolving phenotype. DNA Repair. 2004;3:1187–1196. | |
Savitsky K, Sfez S, Tagle DA, et al. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum Mol Genet. 1995;4:2025–2032. | |
Taylor AM, Byrd PJ. Molecular pathology of ataxia telangiectasia. J Clin Pathol. 2005;58:1009–1015. | |
Becker-Catania SG, Chen G, Hwang MJ, et al. Ataxia-telangiectasia: phenotype/genotype studies of ATM protein expression, mutations, and radio sensitivity. Mol Genet Metab. 2000;70:122–133. | |
Verhagen MM, Abdo WF, Willemsen MA, et al. Clinical spectrum of ataxia-telangiectasia in adulthood. Neurology. 2009;73:430–437. | |
Atamna H, Cheung I, Ames BN. A method for detecting basic sites in living cells: age dependent changes in base excision repair. Proc Natl Acad Sci U S A. 2000;97:686–691. | |
Coppede F, Migliore L. DNA repair in premature aging disorders and neurodegeneration. Curr Aging Sci. 2010;3:3–19. | |
Wang S, Okun MS, Suslov O, et al. Neurogenic potential of progenitor cells isolated from postmortem human Parkinsonian brains. Brain Res. 2012;1464:61–72. | |
Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst.) 2009;8:1004–1008. | |
Stracker TH, Petrini JH. The MRE11complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. | |
Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. | |
Sasaki M, Lange J, Keeney S. Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol. 2010;11:182–195. | |
Tsantoulis PK, Kotsinas A, Sfikakis PP, et al. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27: 3256–3264. | |
Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. | |
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8: 193–204. | |
Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28:3067–3073. | |
Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. | |
Kozlov SV, Graham ME, Jakob B, et al. Autophosphorylation and ATM activation: additional sites add to the complexity. J Biol Chem. 2011;286:9107–9119. | |
Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006;25:3504–3514. | |
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51:6304–6311. | |
Kastan MB, Zhan Q, El-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia telangiectasia. Cell. 1992;71:587–597. | |
Danska JS, Guidos CJ. Essential and perilous: V(D)J recombination and DNA damage checkpoints in lymphocyte precursors. Semin Immunol. 1997;9:199–206. | |
Difilippantonio S, Celeste A, Kruhlak MJ, et al. Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J Exp Med. 2007;204:1003–1011. | |
Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH. Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Mol Cell. 2008;31:21–32. | |
Niida H, Murata K, Shimada M, et al. Cooperative functions of Chk1 and Chk2 reduce tumor susceptibility in vivo. EMBO J. 2010;29:3558–3570. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. | |
Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21:43–48. | |
Eriksson D, Stigbrand T. Radiation-induced cell death mechanisms. Tumor Biol. 2010;31:363–372. | |
Postiglione I, Chiaviello A, Palumbo G. Twilight effect of low dose of ionising radiation on cellular systems: a bird’s eye view on current concepts and research. Med Oncol. 2010;27:495–509. | |
Riesterer O, Matsumoto F, Wang L, et al. A novel Chk inhibitor, XL-844, increases human cancer cell radio sensitivity through promotion of mitotic catastrophe. Invest New Drugs. 2011;29:514–522. | |
Morgan MA, Parsels LA, Zhao L, et al. Mechanism of radiosensitisation by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. | |
Shrivastav M, De Hero LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008;18:134–147. | |
Kuroda S, Urata Y, Fujiwara T. Ataxia-telangiectasia mutated and the Mre11-Rad50-NBS1 complex: promising targets for radio sensitization. Acta Med Okayama. 2012;66:83–92. | |
Hoche F, Seidel K, Theis M, et al. Neurodegeneration in ataxia telangiectasia: what is new? What is evident? Neuropediatrics. 2012;43: 119–129. | |
Yurov YB, Iourov IY, Vorsanova SG. Neurodegeneration mediated by chromosome instability suggests changes in strategy for therapy development in ataxia-telangiectasia. Med Hypotheses. 2009;73: 1075–1076. | |
Lehmann AR, Carr AM. The ataxia-telangiectasia gene: a link between checkpoint controls, neurodegeneration and cancer. Trends Genet. 1995;11:375–377. | |
Shiloh Y, Rotman G. Ataxia-telangiectasia and the ATM gene: linking neurodegeneration, immunodeficiency, and cancer to cell cycle checkpoints. J Clin Immunol. 1996;16:254–260. | |
McKinnon PJ. ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 2012;7:303–321. | |
Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr Biol. 1998;8:1395–1398. | |
Frank KM, Sekiguchi JM, Seidl KJ, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. | |
Gao Y, Sun Y, Frank KM, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95: 891–902. | |
Coufal NG, Garcia-Perez JL, Peng GE, et al. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1(L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci U S A. 2011;108:20382–20387. | |
Valentin-Vega YA, Kastan MB. A new role for ATM: regulating mitochondrial function and mitophagy. Autophagy. 2012;8:840–841. | |
Borghessani PR, Alt FW, Bottaro A, et al. Abnormal development of Purkinje cells and lymphocytes in ATM mutant mice. Proc Natl Acad Sci U S A. 2000;97:3336–3341. | |
Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science. 2010;330:517–521. | |
Barlow C, Dennery PA, Shigenaga MK, et al. Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc Natl Acad Sci U S A. 1999;96:9915–9919. | |
Newman LS, McKeever MO, Okano HJ, et al. Beta-NAP, a cerebellar degeneration antigen, is a neuron-specific vesicle coat protein. Cell. 1995;82:773–783. | |
Herrup K, Li J, Chen J. The role of ATM and DNA damage in neurons: upstream and downstream connections. DNA Repair (Amst). 2013;12:600–604. | |
Li J, Chen J, Ricupero CL, et al. Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat Med. 2012;18:783–790. | |
Kumar R, Hunt CR, Nannepaga S, et al. Purkinje cell-specific males absent on the first (mMof) gene deletion results in an ataxia-telangiectasia-like neurological phenotype and backward walking in mice. Proc Natl Acad Sci U S A. 2011;108:3636–3641. | |
Li J, Hart RP, Mallimo EM, et al. EZH2-mediated H3K27 trimethylation mediates neurodegeneration in ataxia-telangiectasia. Nat Neurosci. 2013;16:1745–1753. | |
Harms C, Albrecht K, Harms U, et al. Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21 (Wafl/Cip 1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci. 2007;27:4562–4571. | |
Biton S, McKinnon PJ, Itsykson P, et al. ATM-mediated response to DNA double stranded breaks in human neurons derived from stem cells. DNA Repair (Amst). 2007;6:128–134. | |
Giardino G, Fusco A, Romano R, et al. Betamethasone therapy in ataxia telangiectasia: unraveling the rationale of this serendipitous observation on the basis of the pathogenesis. Eur J Neurol. 2013;20: 740–747. | |
Zannolli R, Buoni S, Betti G, et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Mov Disord. 2012;27:1312–1316. | |
Menotta M, Biagiotti S, Bianchi M, Chessa L, Magnani M. Dexamethasone partially rescues ataxia telangiectasia-mutated (ATM) deficiency in ataxia telangiectasia by promoting a shortened protein variant retaining kinase activity. J Biol Chem. 2012;287:41352–41363. | |
Chessa L, Leuzzi V, Plebani A, et al. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia-telangiectasia patients: results of a phase 2 trial. Orphanet J Rare Dis. 2014;9:5. | |
Barlow C, Hirotsune S, Paylor R, et al. ATM-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. | |
Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev. 1996;10:2411–2422. | |
Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. | |
Perkins EJ, Nair A, Cowley DO, Van Dyke T, Chang Y, Ramsden DA. Sensing of intermediates in V(D)J recombination by ATM. Genes Dev. 2002;16:159–164. | |
Callen E, Jankovic M, Difilippantonio S, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. | |
Nakamura K, Kato A, Kobayashi J, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell. 2011;41:515–528. | |
Ramiro AR, Jankovic M, Callen E, et al. Role of genomic instability and p53 in AID-induced c-myc-IgH translocations. Nature. 2006;440: 105–109. | |
Lahdesmaki A, Arinbjarnarson K, Arvidsson J, et al. Ataxia-telangiectasia surveyed in Sweden. Lakartidningen. 2000;97:4461–4467. | |
Pan Q, Petit-Frere C, Lahdesmaki A, Gregorek H, Chrzanowska KH, Hammarstrom L. Alternative end joining during switch recombination in patients with ataxia-telangiectasia. Eur J Immunol. 2002;32:1300–1308. | |
Lumsden JM, McCarty T, Petiniot LK, et al. Immunoglobulin class switch recombination is impaired in ATM-deficient mice. J Exp Med. 2004;200:1111–1121. | |
Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–1110. | |
Yang DQ, Kastan MB. Participation of ATM in insulin signaling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol. 2000;2:893–898. | |
Kamsler A, Daily D, Hochman A, et al. Increased oxidative stress in ataxia-telangiectasia evidenced by alterations in redox state of brains from ATM-deficient mice. Cancer Res. 2001;61:1849–1854. | |
Ito K, Hirao A, Arai F, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431:997–1002. | |
Ito K, Takubo K, Arai F, et al. Regulation of reactive oxygen species by ATM is essential for proper response to DNA double-strand breaks in lymphocytes. J Immunol. 2007;178:103–110. | |
Schubert R, Erker L, Barlow C, et al. Cancer chemoprevention by the antioxidant tempol in ATM-deficient mice. Hum Mol Genet. 2004;13:1793–1802. | |
Reliene R, Fleming SM, Chesselet MF, Schiestl RH. Effects of antioxidants on cancer prevention and neuromotor performance in ATM-deficient mice. Food Chem Toxicol. 2008;46:1371–1377. | |
Cosentino C, Grieco D, Costanzo V. ATM activate the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011;30:546–555. | |
Zhou K, Bellenguez C, Spencer CC, et al. Common variants near ATM is associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43:117–120. | |
Leeuwen N, Nijpels G, Becker ML, et al. A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologica. 2012;55:1971–1977. | |
Bostock CV, Soiza RL, Whalley LJ. Genetic determinants of ageing processes and diseases in later life. Maturitas. 2009;62:225–229. | |
Lee HC, Wei YH. Mitochondria and aging. Adv Exp Med Biol. 2012;942:311–327. | |
Chen T, Dong B, Lu Z, et al. A functional single nucleotide polymorphism in promoter of ATM is associated with longevity. Mech Ageing Dev. 2010;131:636–640. | |
Piaceri I, Bagnoli S, Tedde A, Sorbi S, Nacmias B. Ataxia-telangiectasia mutated (ATM) genetic variant in Italian centenarians. Neurol Sci. 2013;34:573–575. | |
Lavin MF, Birrell G, Chen P, et al. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569:123–132. | |
Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. | |
Lichten M. Meiotic recombination: breaking the genome to save it. Curr Biol. 2001;11:R253–R256. | |
Mahadevaiah SK, Turner JM, Baudat F, et al. Recombinational DNA double-stranded breaks in mice precede synapsis. Nat Genet. 2001;27:271–276. | |
Barlow C, Liyanage M, Moens PB, et al. ATM deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development. 1998;125:4007–4017. | |
Hamer G, Kal HB, Westphal CH, Ashley T, DeRooij DG. Ataxia-telangiectasia mutated expression and activation in the testis. Biol Reprod. 2004;70:1206–1212. | |
Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination- defective mouse mutants. Proc Nat Acad Sci U S A. 2005;102:737–742. | |
Li Z, Yu J, Zhang T, et al. rs189037, a functional variant in ATM gene promoter, is associated with idiopathic nonobstructive azoospermia. Fertil Steril. 2013;100:1536–1541. | |
Qi J, Shackelford R, Manuszak R, et al. Functional expression of ATM gene carried by HSV amplicon vector in vitro and in vivo. Gene Ther. 2004;11:25–33. | |
Nayler S, Gatei M, Kozlov S, et al. Induced pluripotent stem cells from ataxia-telangiectasia recapitulate the cellular phenotype. Stem Cells Transl Med. 2012;1:523–535. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.