Back to Journals » Patient Preference and Adherence » Volume 15
Assessing Patient Preferences for Treatment Options for Pediatric Sickle Cell Disease: A Critical Review of Quantitative and Qualitative Studies
Authors Katoch D, Krishnamurti L ![]()
Received 21 May 2021
Accepted for publication 10 August 2021
Published 1 October 2021 Volume 2021:15 Pages 2221—2229
DOI https://doi.org/10.2147/PPA.S264918
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Johnny Chen
Deeksha Katoch,1 Lakshmanan Krishnamurti2
1Department of Pediatrics, SUNY Downstate Medical Center, New York, NY, USA; 2Aflac Cancer and Blood Disorders Center, Department of Pediatrics, Emory University School of Medicine, Atlanta, GA, USA
Correspondence: Lakshmanan Krishnamurti
Aflac Cancer and Blood Disorders Centers, Children’s Healthcare of Atlanta, Emory University, Atlanta, GA, USA
Email [email protected]
Abstract: Sickle cell disease (SCD) resulting from a mutation of the β-globin gene results in sickle deformation of the red blood cell with consequent vaso-occlusion and intravascular hemolysis. SCD results in substantial morbidity, with impaired quality of life and premature mortality. Comprehensive and supportive care, disease modifying therapies and treatments with curative intent are each associated with asymmetrical costs, burden of care, and impact on survival and quality of life. There is thus a considerable decisional dilemma regarding treatment among patients and caregivers. The objective of this review is to evaluate the literature regarding quantitative and qualitative studies of patient preferences in therapy for SCD. Numerous survey-based studies have been performed evaluating SCD patients’ treatment preferences. These studies are limited, however, as they are purely descriptive in nature with limited quantitative information on the relative value of treatment alternatives. Time trade-off and standard gamble studies and health state utility studies have also been utilized to quantify patient utility especially for curative hematopoietic cell transplant. However, these studies suffer from inaccurate assumptions regarding patient preferences. Qualitative studies have garnered the patient and caregiver perspective. Qualitative studies may be limited by selective and purposive sampling, and lack of representativeness due to sample size.
Keywords: anemia, sickle cell, preferences, patient, decision-making, shared
Sickle Cell Disease
Sickle cell disease (SCD) results from a mutation of the β-globin gene which causes the production of abnormal hemoglobin (HbS). The HbS polymerizes in low oxygen tension, resulting in the sickle deformation of red blood cells (RBC).1 The sickled RBCs manifest increased adhesivity, impair laminar flow, and lead to intravascular hemolysis and microvascular occlusion. These pathological changes cause many complications of SCD, including vaso-occlusive crises, acute chest syndrome, stroke, pulmonary hypertension, and splenic auto-infarction, which contribute to substantial morbidity and premature mortality. SCD has significant public health consequences with considerable healthcare utilization and premature mortality. SCD is most prevalent in the Caribbean, Middle East, Africa, India, Central America, and South America. In African Americans, the prevalence of SCD is roughly 1 in 600.2 With universal neonatal screening leading to early detection and intervention, there is a growing population of adults with SCD in the US. Though comprehensive care of patients with SCD has improved survival to adulthood, they frequently suffer from the consequences of the toxic interaction of psychosocial functioning, health behaviors, racial and ethnic disparity, poor socioeconomic status, and healthcare delivery disparities.
Treatment Options in Pediatric Sickle Cell Disease
Since patients with SCD may become functionally asplenic in childhood, they are at risk for overwhelming pneumococcal infections. This provided the rationale for pre-symptomatic diagnosis on newborn screening and early institution of pneumococcal prophylaxis. The institution of universal newborn screening in the US since 1986 has improved the survival of patients with SCD. New diagnoses in adults and children are generally seen in foreign-born individuals. Routine screening for hemoglobinopathy for patients of African, Mediterranean, and Southeast Asian descent is recommended by the American College of Obstetricians and Gynecologists (ACOG).3 Hydroxyurea has been used as a disease-modifying therapy for SCD since its approval by the FDA in 1995 but was limited to adults (Table 1). In 2017, FDA approved the use of Hydroxyurea for children above the age of 2 years, while the National Heart, Lung, or Blood Institute recommends use in children starting at age nine months even in the absence of symptoms. (4) FDA has also approved the use of L-Glutamine, Voxelotor, and Crizanlizumab in older children and adults.4,5 Despite the availability of these disease-modifying therapies, many patients with SCD do not receive adequate treatment.6 Preventative strategies also include chronic blood transfusion for patients with abnormal transcranial Doppler values, which predicts increased risk of stroke. Hematopoietic cell transplantation offers the potential to cure SCD by stabilizing organ damage and improving quality of life. While HCT from HLA identical siblings is associated with excellent survival rates, particularly in young children, emerging alternate donor options and autologous gene therapy offer the potential to expand the applicability of these therapies.7,8 Prenatal diagnosis and preimplantation genetic diagnosis may provide the opportunity for informed reproductive choices with appropriate genetic counseling.9 Alternative medicine and mind-body techniques are being explored, particularly in managing chronic pain in SCD.10 While treatment options for SCD are most applicable in high resource settings such the US and Europe, disease modifying therapies such as hydroxyurea are becoming more available in low resource settings in Africa. We acknowledge that patient preferences are not relevant to treatments to which they do not have access.
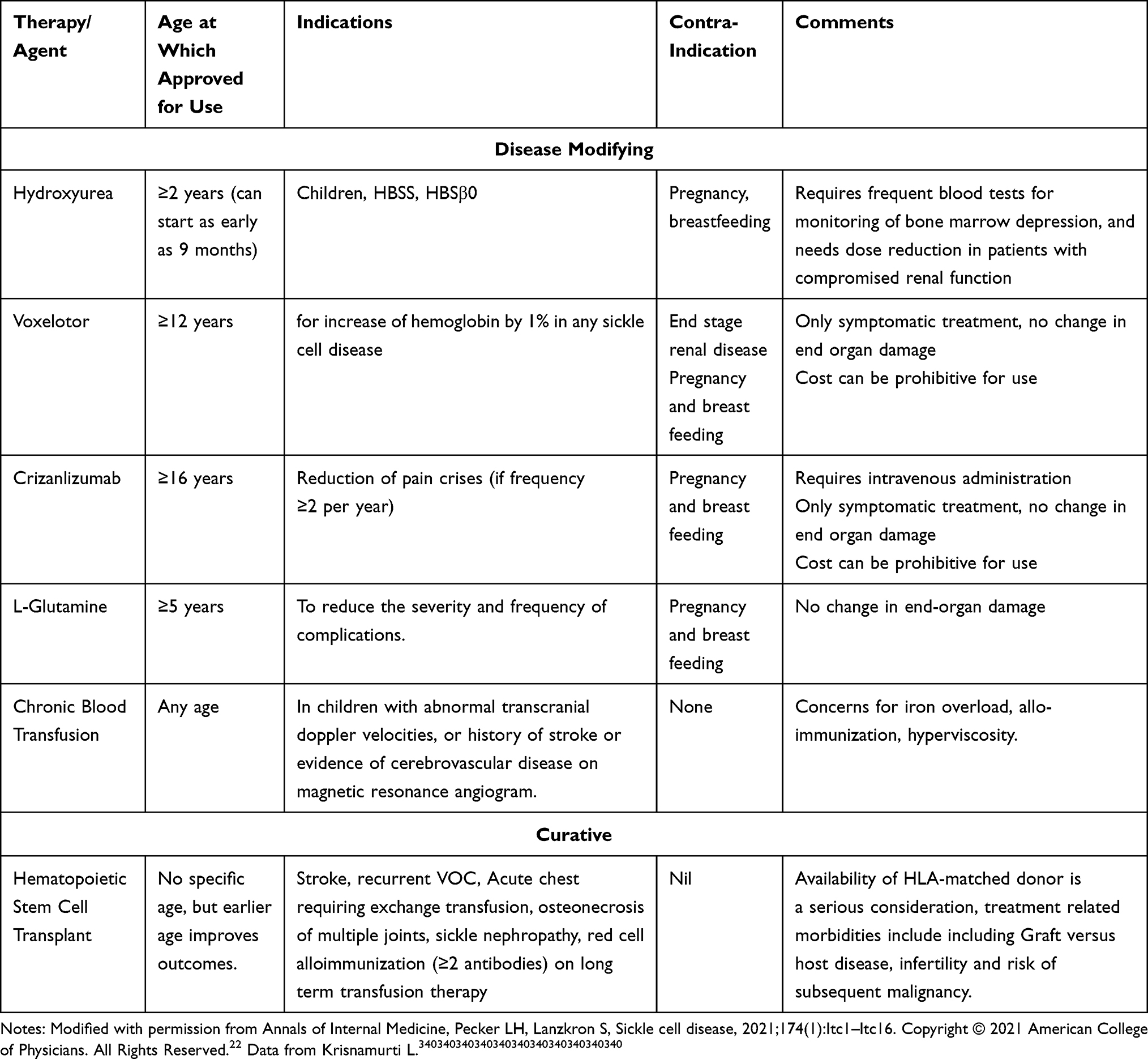 |
Table 1 Treatment Options in Sickle Cell Disease |
Risks of Therapy in Sickle Cell Disease
Newborn screening, pneumococcal prevention, and vaccination in SCD patients have increased survival; however, the quality of life may be severely compromised due to complications like repeated vaso-occlusive crises, chronic pain, and end-organ damage. Silent strokes may also cause increased cognitive deficits. In addition, treatment options available to SCD patients also compound the impaired quality of life. Repeated hospital admissions and therapies like chronic blood transfusion requiring serial often monthly visits to the hospital for transfusions affect productivity and education during school-going years in the pediatric population. Even oral treatments like Hydroxyurea come with risks of myelosuppression which require lifelong monitoring with frequent blood tests to check white and red cell count.2 Curative therapies like HSCT and gene therapy, even when successful, also cause significant acute complications and long-term sequelae. Academic achievement and years of school completed are lower in SCD patients than matched peers, affecting future income-generating potential.11 The toxic interplay of socio-economic disadvantage, psychological behaviors exacerbate the racial and health disparities already faced by SCD patients. Risks of therapy in sickle cell disease (Table 1) contribute to the decisional dilemma faced by patients and caregivers in accepting and adhering to therapies for SCD.
Decision Making in Sickle Cell Disease
Shared decision-making (SDM) involves the collaborative effort by the patient, family, and physician to choose the best-suited management tailored to patient preference and values.12 SDM assumes prime importance in SCD patients who must weigh the tradeoff between serious disease sequelae and treatment-related morbidity. The lack of consensus regarding best outcomes adds to the complexity of decision-making. Both patient and physician attitudes are reflected in the ultimate care received by SCD patients. In studying patient perspectives on shared decision-making, Ross et al found that SCD patients strongly endorse a desire to make an informed decision about their care and prefer that their physician present information to them in an unbiased way.12 Physicians often walk the tight rope of balancing their personal preference of treatment with that of patient expectations. While studying physician perspectives, we found a wide range of physician approaches to shared decision-making. These approaches range in a spectrum from those who are invested in shared decision-making to those that explicitly promote a particular therapeutic plan based on their understanding of patient compliance, socioeconomic status, and the severity of the clinical condition.13
Literature Review
A systematic PubMed search was conducted on April 05, 2021, to identify published studies on patient preferences in SCD. The search used the following keywords and MESH headings: preferences or decision-making or time tradeoff or standard gamble or conjoint analysis or discrete choice combined with sickle cell. This search identified 200 papers published between 1973 and 2021 (Figure 1). Following the application of the limitations “English language” and “human studies,” we remained with 174 papers for analysis. Each study was systematically reviewed to assess for appropriateness for the current review. We excluded studies not specific to sickle cell disease (35), relevant only to sickle cell trait, prenatal screening, or blood donation (57). We also excluded publications that were review or commentary articles that did not evaluate preferences or decision-making or were a methodology analysis (58). Twenty-four articles were analyzed for this review.
 |
Figure 1 Review of literature and identification of studies. |
Survey Studies of SCD Preferences
Badawy et al in their study, surveyed adults and adolescents for their preferred technology-based strategies for improving medication adherence.14 Their study found that daily medication reminder was the most preferred method for improving medication adherence followed by education about SCD and SCD medications, adherence text prompts and medication log. Britto et al studied adolescent patients with SCD and other chronic illnesses to determine what they considered important for health care quality and compared the same with physicians.15,16 They found that patients thought physician trust, autonomy, and the patient–doctor relationship as important metrics for deciding healthcare quality. Adolescent patients preferred to be communicated directly about their medical condition and decisions. Both patients and physicians agreed that pain management was an important determinant of quality of care. The physicians underrated the importance of direct communication with adolescent patients. For patients with sickle cell crisis, Fertleman et al found that parents preferred a fast-track admission policy with direct access to the ward, pre-prescribed analgesia, and administration of pain medication by a nurse on arrival over an emergency department visit.17 In creating an integrative Sickle Cell Wellness Clinic, Junghans-Rutelonis et al interviewed parents. They found that having a group share their experiences and make connections was one of the most critical determinants of “wellness”.18 Patients with SCD often require a central venous catheter (CVC) for exchange transfusion to prevent disease-related complications. The preferred sites for CVC insertion are the internal jugular vein in the neck and femoral vein in the groin. Multiple studies have shown that femoral vein CVCs have higher rates of complications, including catheter-related infections, than internal jugular vein catheterizations.20–23 Park et al interviewed patients who underwent placement of CVC to understand preferences and perceived importance of factors that influenced such choices.24 They found that females preferred femoral vein access site over internal jugular secondary to concerns of scar formation in the neck. They concluded that non-medical problems might guide patient’s preferences, and increased impetus should be provided towards patient education about the risks of procedure to improve patient engagement in shared decision-making.
Methodological Considerations of Survey Based Studies
Survey methods are essential tools to assess attitudes, knowledge, and preferences regarding treatment. The development of patient preference survey questionnaires should follow a psychometric approach, ensuring item generation, pretest and pilot testing, and systematic testing of validity, reliability, and acceptability.25 Survey methodology is limited because few questionnaires meet the standards of psychometric methods and that many studies may not be adequately powered to answer the study question. Further, the results of a survey study may also be influenced by its contents, mode of administration, the burden of administration, the research setting, and the medical literacy of the populations surveyed.
Time Tradeoff and Standard-Gamble Studies in SCD
Kodish et al 1991, using the standard reference-gamble model, assessed a patient preference for hematopoietic stem cell transplant (HSCT) for SCD.26 They assumed that HSCT carried an estimated mortality risk of 15%, of the 67 parents surveyed, 37% accepted short-term mortality of 15% and 12% accepted short-term mortality of 50% or more over the standard care offered at that time. Around 13% of parents would accept both a 15% short-term mortality risk and an additional 15% risk of graft versus host disease (GVHD) for cure with HSCT. The preferences were independent of the disease severity. In the present era of increasing choices of disease-modifying drugs and competing lower mortality and morbidity risks of stem cell transplant for the cure, these preferences may vary. Van Besien et al27 conducted a survey of 100 patients and their health care providers. A reference gamble paradigm was used to assess risk. The study compared the characteristics of those accepting substantial risk vs those not accepting risk. The study also compared agreement on risks recommended by health care providers and taken by patients. Sixty-three of 100 patients indicated a readiness to accept some short-term risk of mortality in exchange for the certainty of a cure. Fifteen patients expressed a willingness to accept a mortality risk of more than 35%. No differences in patient or disease-related variables were identified between those willing to accept risk and those not willing to accept risk. There was a discordance between the recommendations of health care providers and the risk accepted by patients. A substantial proportion of adults with SCD are interested in curative treatment, despite the possibility of considerable risk. However, the decision to accept risk appears to be influenced by individual patient values, which are not easily quantified and often discordant with the health care provider’s assessment. Meier et al assessed parents’ and adolescent patients’ preferences of HSCT as a curative option of SCD; a standard reference- gamble model with at least 5% mortality and 10% risk of significant GVHD was used.28 72% of the 89 respondents were willing to accept 5% or higher mortality, with 56% of respondents willing to accept 10% or higher mortality in exchange for a cure with HSCT. There were no significant differences between adolescents and parents in the acceptable mortality risk. 55% of the respondents were willing to accept a GVHD risk of 10% or more, and no significant difference was found between adolescents and parents. They also illustrated no correlation between disease severity and risk aversion for mortality or GVHD amongst the respondents, a finding similar to that of Kodish et al.26 Adolescent patients were less risk-averse than parents when it came to infertility secondary to HSCT. These three studies conducted more than a decade from each other show increased adoption of HSCT as a treatment modality in SCD as the mortality risk continues to decline.
Methodological Considerations of Time Trade-Off and Standard Gamble Studies
Health state utilities are used to provide quantitative measures of individual values of certain health states, estimating personal preference for an outcome. Utilities are measured on a scale of 0 to 1, in which 0 represents a health state equivalent to death and 1 represents a health state of perfect health.29 These can be assessed using benefit-risk preference methods. Standard gamble is one of the several direct-elicitation methods setting patient preferences. In this, a patient is given an option to choose between his present health state a (standard medical management) and a gamble B (alternate treatment). The gamble B has a probability (p) of success (cure or reversion to perfect health) and probability (1-p) of failure (death). By varying p, the point where the patient is indifferent to the choices is obtained. The utility of the present health state a is equal to the value of p at the point of indifference. Another method assessing benefit-risk preference is time tradeoff, where a patient is asked to choose between his remaining life expectancy in the present health state and shorter lifespan (x years) in normal health. By varying the value of x, we find a point where he is indifferent between the two choices. Hence, x is the time he is willing to tradeoff for good health compared to his present health state.30,31 The direct elicitation methods have their fair share of shortcomings. One of the significant limitations of these methods is that they employ judgments among hypothetical alternatives which do not have the same clinical, emotional, and financial consequences of actual choices.31 Moreover, it has been found that people generally find it challenging to visualize small probabilities and translate population risk to individual risk.
Study of Health State Utilities
Ojelabi et al32 collected data from two SCD outpatient clinics in Ibadan, Nigeria, using a standard algorithm to derive utility scores and measure SF-6D from the SF-36. Socio-demographic, bio-physiological, and psychosocial variables together predicted utility scores. While socio-demographic and bio-physiological factors explained 7.5% and 17.9% of the variance, psychosocial factors explained 4.9% of the variance. Women reported lower utility scores with a small effect size (d = 0.17). Utility score increased with the level of education but decreased with age, anxiety, frequency of pain episodes, and several co-morbidities. The low utility score in SCD suggests a substantial impact of the disease on the Health Related Quality of Life (HRQOL) of patients and the high value they place on their health state due to the limitations they experienced. These observations suggest the need for both clinical care and psychosocial support to help in improving the quality of life of the patients. Oluwole et al in their pilot study in Lagos, Nigeria, explored the feasibility and acceptability of newborn screening of SCD in primary health centers.33 Of the 291 mothers participating in the survey, 86% accepted to get their infants screened for SCD. On inquiring mothers about the correct time for newborn screening, 23% stated soon after birth, and 36% felt within one month is appropriate. Almost all mothers (97.3%) were willing to enroll their infants in SCD clinic immediately if the result came positive. The challenges perceived by mothers for early infant screening included: the cost of affirmative cases, availability of test facilities, delays in testing, accessibility to follow-up care. Obrien et al34 used a decision analysis model to compare no intervention, Hydroxyurea (HU), chronic transfusion, and HSCT. The treatment with the highest average utility (HRQOL) was HSCT (0.85). Average utilities for no therapy, chronic transfusion, and HU were 0.68, 0.71, and 0.80, respectively. The model was quite sensitive to QoL estimates, suggesting that an accurate comparison of HU and HSCT would require robust data gathered on HRQOL in children with SCD following HSCT. Nietert et al35 compared HSCT and chronic blood transfusion outcomes for children with SCD. They have elevated cerebral blood flow velocity with no other marker of disease severity that would make them eligible for HSCT. They used decision analysis to compare the number of quality-adjusted life-years (QALYs) by a population of patients with SCD with an increased risk of stroke because of elevated cerebral blood flow velocity. They constructed Markov models to represent the clinical course of patients with SCD treated with HSCT or chronic blood transfusion. Published results and expert opinion provided risks of stroke and death for different disease states, estimates of transition probabilities from one clinical state to another, and HRQOL. A hypothetical cohort of 100,000 patients was used to conduct an intention-to-treat analysis as well as treatment. Patients with SCD who managed to provide HSCT could expect 16.0 QALYs, compared with 15.7 QALYs for those planning to receive a chronic blood transfusion, of note, the variation around these estimates was considerable. Patients compliant with chronic blood transfusion therapy could expect the best outcomes (19.2 QALYs). Thus, neither HSCT nor chronic transfusion can be considered the “best” treatment for children with SCD who have elevated cerebral blood flow velocity. Therefore, elevated cerebral blood flow is not recommended as the sole criterion for selecting patients with sickle cell for HSCT.
Methodological Considerations of the Study of Health State Utilities
Health state utility-based methods estimate preferences for health states and incorporate values or utilities for health outcomes which can be used in cost-effectiveness analyses to aid resource allocation decisions. Ascertaining the importance people attach to the health improvement they receive from different interventions allows us to determine how best to provide the desired outcomes and avoid those they do not want. Limitations to health state utility-based methods, such as the considerable variation in the measurement scales, sources of preferences, and preference measurement techniques used, are barriers to the widespread acceptance of this methodology.36
Qualitative Studies
Hydroxyurea (HU) utilization in the pediatric age group is affected by caregivers’ concerns regarding HU’s side effects and the belief that their child might not be symptomatic enough to warrant such treatment.37 Creary et al conducted a semi-structured interview of caregivers’ of the patients who were offered HU treatment, including those who agreed and those who refused HU, to understand their preferences regarding HU.38 Caregivers identified the drug’s side effect profile, especially when HU is perceived as a chemotherapy drug, the uncertainty in disease progression, and inability to perceive the severity of SCD in their child as factors swaying them away from the medicine. They also found that even caregivers who chose HU for their children reported being fearful about initiating it. In another qualitative study assessing patient attitude and experience with chronic transfusion therapy (CTT), some parents perceived it as the only option moving forward and wished they had more detailed information about the long-term implication of such treatment.39 They also found that adolescents interviewed were not part of the decision-making process, though all of them had started on CTT at age less than 12 years. Khemani et al interviewed HSCT survivors and their parents regarding their knowledge of HSCT, decision-making process, experience, and impact on their life.40 They found that the major factor leading patients to choose HSCT was the unpredictable onset of disease complications and the constant need to manage such complications. They were also worried about the long-term consequences of the disease and non-curative treatment, especially chronic transfusion. Other factors that positively influenced their decision to pursue HSCT was education imparted by their HSCT physician, strong family support and availability of HLA-identical sibling donor. In a recent study, Schulz explored the decisional conflict in pediatric patients and their families while considering HSCT for SCD.41 They found that families faced decisional conflict at a level that resulted in the decisional delay or feeling unsure. They found that fear of stroke, frequency of hospitalizations, and prevention of SCD complications were most influential in the decision process. Sinha et al examined factors influencing primary caregiver decision-making in HSCT and gene therapy for SCD. They report that patients learned about curative options from social media and news.42 Caregivers sought consultation owing to their child’s diminished quality of life, recent complications, anxiety about future difficulty, or an imminent major medical decision. Gene therapy was seen as a less invasive and more acceptable treatment option as compared to HSCT. Utrankar et al43 conducted a cross-sectional mixed-methods study that included semi-structured interviews, surveys, and focus groups of adolescents and adults with SCD. They evaluated interest, preferences, and anticipated benefits or barriers of a patient-centered adaptation of SCD practice guidelines, prospective technology uses for health, and barriers to technology utilization. Forty-seven individuals completed surveys and interviews, and 39 participated in three separate focus groups. Most participants (91%) were unaware of SCD guidelines. However, almost all (96%) expressed interest in the application of a guideline, identifying benefits (knowledge, activation, individualization, and rewards) and barriers (insufficient information, low motivation, and resource limitations). Current technology health uses included information access, care coordination, and reminders about health-related actions. Prospective technology uses included informational messaging and timely alerts. Barriers to technology use included lack of interest, lack of utility, and preference for direct communication.
Methodological Aspects of Qualitative Studies
Qualitative studies exploring patient preferences in management of SCD have provided several meaningful insights into the decision-making process. These studies have offered insight into reasons people seek treatment, their preferences for treatment options, the desired degree of involvement in decisions, and preferences for healthcare delivery. Qualitative research works inductively from the “ground up” and seeks to build knowledge and experience rather than testing hypotheses.44 Rigor, responsiveness, and reflexivity are components to ensure key quality measures of qualitative research.45 However, the concepts, such as values and priorities, may be imprecise and used interchangeably, under-conceptualized, and under-researched thus limiting the utility of many qualitative studies.46
Conclusion
SCD patients face an increasing number of choices regarding the care of their chronic illness. These choices range from mode, frequency, access to and type of drug or nonpharmacological interventions to preferences regarding treatment with curative intents but potential severe risks such as HSCT and prenatal and preimplantation genetic diagnosis decisions, and newborn screening. As a result of these increased choices, SCD patient’s preferences carry growing importance in the decisions regarding their care, both at a personal and public policy level. Therefore, rigorous methodologies to evaluate patient preferences and willingness to accept risks are needed. Qualitative studies, survey studies, standard gamble, and time tradeoff methods have examined patient preferences in SCD care but have inherent flaws that may bias their results. Further systematic, quantifiable measurement of patient preferences is required to generate insights that can guide physicians and inform public health policy regarding the care of this devastating disease.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017;129(20):2719–2726. doi:10.1182/blood-2017-02-765891
2. Pecker LH, Lanzkron S. Sickle cell disease. Ann Intern Med. 2021;174(1):Itc1–Itc16. doi:10.7326/aitc202101190
3. Pecker LH, Naik RP. The current state of sickle cell trait: implications for reproductive and genetic counseling. Blood. 2018;132(22):2331–2338. doi:10.1182/blood-2018-06-848705
4. Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018;379(3):226–235. doi:10.1056/NEJMoa1715971
5. Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509–519. doi:10.1056/NEJMoa1903212
6. Su ZT, Segal JB, Lanzkron S, Ogunsile FJ. National trends in hydroxyurea and opioid prescribing for sickle cell disease by office-based physicians in the United States, 1997–2017. Pharmacoepidemiol Drug Saf. 2019;28(9):1246–1250. doi:10.1002/pds.4860
7. Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017;129(11):1548–1556. doi:10.1182/blood-2016-10-745711
8. Krishnamurti L. Hematopoietic cell transplantation for sickle cell disease: state of the art. Expert Opin Biol Ther. 2007;7(2):161–172. doi:10.1517/14712598.7.2.161
9. Vrettou C, Kakourou G, Mamas T, Traeger-Synodinos J. Prenatal and preimplantation diagnosis of hemoglobinopathies. Int J Lab Hematol. 2018;40(S1):74–82. doi:10.1111/ijlh.12823
10. Thompson WE, Eriator I. Pain control in sickle cell disease patients: use of complementary and alternative medicine. Pain Med. 2014;15(2):241–246. doi:10.1111/pme.12292
11. Harris KM, Dadekian JN, Abel RA, et al. Increasing educational attainment in adolescents with sickle cell disease. Soc Work Public Health. 2019;34(6):468–482. doi:10.1080/19371918.2019.1629142
12. Ross D, Bakshi N, Khemani K, Sinha C, Loewenstein G, Krishnamurti L. What are the expectations of patients in decision making process for disease modifying therapies for sickle cell disease: do they care about shared decision making? Blood. 2016;128(22):5968. doi:10.1182/blood.V128.22.5968.5968
13. Bakshi N, Sinha CB, Ross D, Khemani K, Loewenstein G, Krishnamurti L. Proponent or collaborative: physician perspectives and approaches to disease modifying therapies in sickle cell disease. PLoS One. 2017;12(7):e0178413. doi:10.1371/journal.pone.0178413
14. Badawy SM, Thompson AA, Liem RI. Technology access and smartphone app preferences for medication adherence in adolescents and young adults with sickle cell disease. Pediatr Blood Cancer. 2016;63(5):848–852. doi:10.1002/pbc.25905
15. Britto MT, DeVellis RF, Hornung RW, DeFriese GH, Atherton HD, Slap GB. Health care preferences and priorities of adolescents with chronic illnesses. Pediatrics. 2004;114(5):1272–1280. doi:10.1542/peds.2003-1134-L
16. Britto MT, Slap GB, DeVellis RF, et al. Specialists understanding of the health care preferences of chronically ill adolescents. J Adolesc Health. 2007;40(4):334–341. doi:10.1016/j.jadohealth.2006.10.020
17. Fertleman CR, Gallagher A, Rossiter MA. Evaluation of fast track admission policy for children with sickle cell crises: questionnaire survey of parents’ preferences. BMJ. 1997;315(7109):650. doi:10.1136/bmj.315.7109.650
18. Junghans-Rutelonis AN, Moquist KL, Blaylark RM, Anderson N, Brown ML. Incorporating integrative medicine and patient preferences into a pilot interdisciplinary sickle cell wellness clinic. Complement Ther Med. 2020;49:102333. doi:10.1016/j.ctim.2020.102333
19. Goetz AM, Wagener MM, Miller JM, Muder RR. Risk of infection due to central venous catheters: effect of site of placement and catheter type. Infect Control Hosp Epidemiol. 1998;19(11):842–845. doi:10.2307/30141562
20. Lorente L, Henry C, Martin MM, Jimenez A, Mora ML. Central venous catheter-related infection in a prospective and observational study of 2595 catheters. Crit Care. 2005;9(6):R631–R635. doi:10.1186/cc3824
21. Trottier SJ, Veremakis C, Obrien J, Auer AI. Femoral deep-vein thrombosis associated with central venous catheterization - results from a prospective, randomized trial. Crit Care Med. 1995;23(1):52–59. doi:10.1097/00003246-199501000-00011
22. Yeral M, Boga C, Oguzkurt L, Asma S, Kasar M, Kozanoglu I. Short-term central venous catheter complications in patients with sickle cell disease who undergo apheresis. J Thromb Thrombolysis. 2014;37(2):97–101. doi:10.1007/s11239-013-0914-z
23. Park PJ, Cole SS, Bercu ZL, Martin J, Newsome J. Understanding patient preferences for femoral central venous catheterization among patients with sickle cell disease. J Vasc Access. 2018;19(2):199–200. doi:10.5301/jva.5000794
24. Kodish E, Lantos J, Stocking C, Singer PA, Siegler M, Johnson FL. Bone marrow transplantation for sickle cell disease. A study of parents’ decisions. N Engl J Med. 1991;325(19):1349–1353. doi:10.1056/NEJM199111073251905
25. Bai F, Ling J, Esoimeme G, Yao L, Wang M, Huang J, et al. A systematic review of questionnaires about patient's values and preferences in clinical practice guidelines. Patient Prefer Adherence. 2018;12:2309–23. doi:10.2147/PPA.S177540
26. van Besien K, Koshy M, Anderson-Shaw L, et al. Allogeneic stem cell transplantation for sickle cell disease. A study of patients’ decisions. Bone Marrow Transplant. 2001;28(6):545–549. doi:10.1038/sj.bmt.1703208
27. Meier ER, Dioguardi JV, Kamani N. Current attitudes of parents and patients toward hematopoietic stem cell transplantation for sickle cell anemia. Pediatr Blood Cancer. 2015;62(7):1277–1284. doi:10.1002/pbc.25446
28. Chang EM, Saigal CS, Raldow AC. Explaining health state utility assessment. JAMA. 2020;323(11):1085–1086. doi:10.1001/jama.2020.0656
29. Stiggelbout AM, de Haes JC. Patient preference for cancer therapy: an overview of measurement approaches. J Clin Oncol. 2001;19(1):220–230. doi:10.1200/jco.2001.19.1.220
30. Brett Hauber A, Fairchild AO, Reed johnson F. Quantifying benefit-risk preferences for medical interventions: an overview of a growing empirical literature. Appl Health Econ Health Policy. 2013;11(4):319–329. doi:10.1007/s40258-013-0028-y
31. Ojelabi AO, Bamgboye AE, Ling J, Stillwaggon E. Preference-based measure of health-related quality of life and its determinants in sickle cell disease in Nigeria. PLoS One. 2019;14(11):e0223043. doi:10.1371/journal.pone.0223043
32. Oluwole EO, Adeyemo TA, Osanyin GE, Odukoya OO, Kanki PJ, Afolabi BB. Feasibility and acceptability of early infant screening for sickle cell disease in Lagos, Nigeria-a pilot study. PLoS One. 2020;15(12):e0242861. doi:10.1371/journal.pone.0242861
33. O’Brien SH, Hankins JS. Decision analysis of treatment strategies in children with severe sickle cell disease. J Pediatr Hematol Oncol. 2009;31(11):873–878. doi:10.1097/MPH.0b013e3181b83cab
34. Nietert PJ, Abboud MR, Silverstein MD, Jackson SM. Bone marrow transplantation versus periodic prophylactic blood transfusion in sickle cell patients at high risk of ischemic stroke: a decision analysis. Blood. 2000;95(10):3057–3064. doi:10.1182/blood.V95.10.3057
35. Neumann PJ, Goldie SJ, Weinstein MC. Preference-based measures in economic evaluation in health care. Annu Rev Public Health. 2000;21:587–611. doi:10.1146/annurev.publhealth.21.1.587
36. Oyeku SO, Driscoll MC, Cohen HW, et al. Parental and other factors associated with hydroxyurea use for pediatric sickle cell disease. Pediatr Blood Cancer. 2013;60(4):653–658. doi:10.1002/pbc.24381
37. Creary S, Zickmund S, Ross D, Krishnamurti L, Bogen DL. Hydroxyurea therapy for children with sickle cell disease: describing how caregivers make this decision. BMC Res Notes. 2015;8:372. doi:10.1186/s13104-015-1344-0
38. Hawkins LM, Sinha CB, Ross D, et al. Patient and family experience with chronic transfusion therapy for sickle cell disease: a qualitative study. BMC Pediatr. 2020;20(1):172. doi:10.1186/s12887-020-02078-w
39. Khemani K, Ross D, Sinha C, Haight A, Bakshi N, Krishnamurti L. Experiences and decision making in hematopoietic stem cell transplant in sickle cell disease: patients’ and caregivers’ perspectives. Biol Blood Marrow Transplant. 2018;24(5):1041–1048. doi:10.1016/j.bbmt.2017.11.018
40. Schulz GL, Kelly KP, Holtmann M, Armer JM. Navigating decisional conflict as a family when facing the decision of stem cell transplant for a child or adolescent with sickle cell disease. Patient Educ Couns. 2020;104(5):1086–1093. doi:10.1016/j.pec.2020.10.011
41. Krishnamurti L. Hematopoietic CellTransplantation for Sickle Cell Disease. Review. Frontiers in Pediatrics. 2021-January-05 2021;8(846). doi:10.3389/fped.2020.551170
42. Sinha CB, Bakshi N, Ross D, Loewenstein G, Krishnamurti L. Primary caregiver decision-making in hematopoietic cell transplantation and gene therapy for sickle cell disease. Pediatr Blood Cancer. 2021;68(1):e28749. doi:10.1002/pbc.28749
43. Utrankar A, Mayo-Gamble TL, Allen W, et al. Technology use and preferences to support clinical practice guideline awareness and adherence in individuals with sickle cell disease. J Am Med Inform Assoc. 2018;25(8):976–988. doi:10.1093/jamia/ocy036
44. Gooberman-Hill R. Qualitative approaches to understanding patient preferences. Patient. 2012;5(4):215–223. doi:10.2165/11633720-000000000-00000
45. Rolfe DE, Ramsden VR, Banner D, Graham ID. Using qualitative health research methods to improve patient and public involvement and engagement in research. Res Invol Engage. 2018;4:49. doi:10.1186/s40900-018-0129-8
46. Bastemeijer CM, Voogt L, van Ewijk JP, Hazelzet JA. What do patient values and preferences mean? A taxonomy based on a systematic review of qualitative papers. Patient Educ Couns. 2017;100(5):871–881. doi:10.1016/j.pec.2016.12.019
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.