Back to Journals » Journal of Asthma and Allergy » Volume 9
Aspirin-exacerbated respiratory disease: pathophysiological insights and clinical advances
Received 12 January 2016
Accepted for publication 3 February 2016
Published 10 March 2016 Volume 2016:9 Pages 37—43
DOI https://doi.org/10.2147/JAA.S88739
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
John W Steinke, Jeff M Wilson
Asthma and Allergic Disease Center, Carter Immunology Center, Department of Medicine, University of Virginia Health System, Charlottesville, VA, USA
Abstract: Asthma and chronic rhinosinusitis are heterogeneous airway diseases of the lower and upper airways, respectively. Molecular and cellular studies indicate that these diseases can be categorized into unique endotypes, which have therapeutic implications. One such endotype is aspirin-exacerbated respiratory disease (AERD), which encompasses the triad of asthma, aspirin (or nonsteroidal anti-inflammatory drug) hypersensitivity, and nasal polyposis. AERD has unique pathophysiological features that distinguish it from aspirin-tolerant asthma and other forms of chronic rhinosinusitis. This review details molecular and cellular features of AERD and highlights current and future therapies that are based on these insights.
Keywords: leukotriene, cyclooxygenase, prostaglandin, aspirin-exacerbated respiratory disease, arachidonic acid
Introduction
The constellation of findings of asthma, aspirin (and nonselective nonsteroidal anti-inflammatory drug [NSAID]) hypersensitivity, and nasal polyposis encompasses the phenomena known as aspirin-exacerbated airway disease (AERD). Described by Samter and Beers1 in 1968, and formally called Samter’s triad, the defining feature of this disease is the worsening of respiratory symptoms following ingestion of cyclooxygenase-1 (COX-1) inhibitors such as aspirin and nonselective NSAIDs. Despite being a relatively minor fraction of all asthma and sinusitis cases, aspirin hypersensitivity is associated with increased severity of both upper and lower airway diseases. A recent meta-analysis indicates that 7% of asthmatics are aspirin-sensitive but that the prevalence doubles in severe asthmatics.2 Recent insights into AERD pathophysiology suggest that it represents a unique phenotype of airway disease. Unlike allergic asthma, this disease tends to develop in adulthood, occurs in patients without an atopic history, and displays a slightly higher prevalence in females.3–6 Hallmark features include eosinophilia, expression of Th2 cytokines, and elevated levels of cysteinyl leukotrienes (CysLTs).7–9 Recently, a prominent role for interferon (IFN)-γ in the maturation of eosinophil progenitors in AERD has been proposed.10 Based on these characteristics, it is not surprising that steroids and leukotriene pathway modifiers are the mainstays of therapy. Perhaps paradoxically, aspirin desensitization is a keystone therapy for patients who can tolerate the procedure. This review highlights some recent advances into the cellular and molecular mechanisms involved in AERD.
Role of eicosanoids
Perhaps most central to the underlying pathophysiology of AERD is the dysregulation of pro- and anti-inflammatory lipid mediators. For example, it is well established that proinflammatory CysLTs are markedly upregulated in AERD, whereas the prostanoid prostaglandin E2 (PGE2) is constitutively decreased.9,11–13 The fundamental factors that contribute to this underlying dysregulation remain a source of active investigation, although work discussed later suggest a role for both Th1 and Th2 cytokines.
The enzymes COX and 5-lipoxygenase (5-LO) act as critical switches for regulating downstream production of prostanoids and leukotrienes (Figure 1), respectively, from a common pool of arachidonic acid precursors. Expression and regulation of these and other enzymes involved in eicosanoid metabolism are thought to be important in AERD pathogenesis.
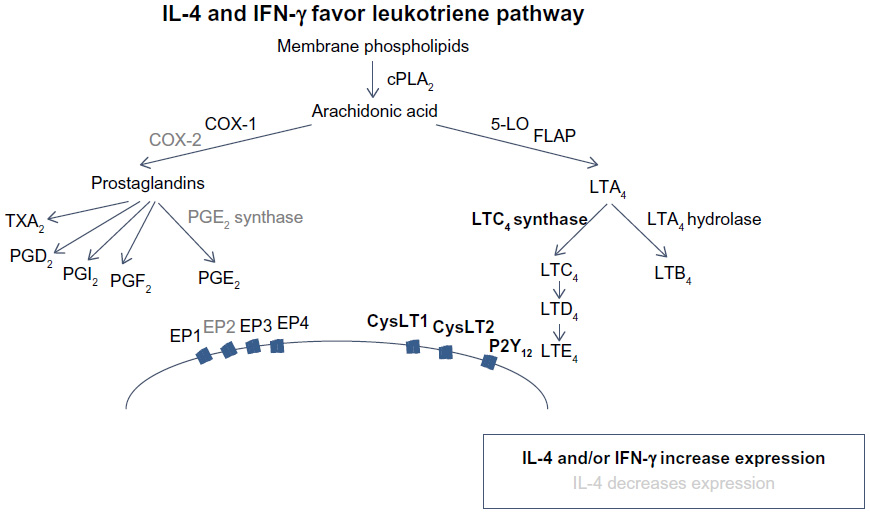 | Figure 1 IL-4 and IFN-γ favor leukotriene pathway. |
COX-1 and COX-2 are prostaglandin synthases that convert arachidonic acid into prostaglandin H2, which acts as a gateway to the synthesis of a number of prostanoids including other prostaglandins, thromboxanes, and prostacyclines (Figure 1). It has been demonstrated that expression of COX-2, but not COX-1, is markedly reduced in AERD.12,14,15 Given that microsomal prostaglandin E2 synthase (mPGES-1), one of the downstream enzymes that generates PGE2, is functionally coupled to COX-2, it is not surprising that PGE2 is decreased in AERD.16 This prostaglandin has pleiotropic effects mediated by four different G-protein-coupled receptors (EP1–EP4); however, acting via the EP2 receptor, it exerts anti-inflammatory effects relevant to AERD by impairing eosinophil activation and mast cell degranulation.17–19 The importance of this pathway has been further demonstrated as PGE2 inhalation into the lung protects against aspirin-induced bronchoconstriction.20,21
In contrast to the impaired prostaglandin production, products of the 5-lipoxygenase pathway are markedly increased in AERD (Figure 1). This reflects increased expression of 5-lipoxygenase itself as well as the downstream enzyme leukotriene C4 synthase (LTC4S). This has been demonstrated in both lung and nasal polyps, largely localizing to mast cell and eosinophil populations.11,12,22 Initially described as the slow-reacting substance of anaphylaxis, studies quickly discerned three distinct lipid mediators that contributed to bronchoconstriction observed in experimental models.23 LTC4 is the first product of this pathway but is readily converted to LTD4, which itself is rapidly metabolized to the more stable LTE4. Levels of LTE4 are elevated in the urine and respiratory secretions of AERD subjects and are further increased upon aspirin provocation.24,25 Signaling is mediated, at least in part, through the G-protein-coupled receptors CysLT1 and CysLT2. Of these, CysLT1 is the high-affinity receptor preferentially binding LTD4, whereas CysLT2 recognizes both LTC4 and LTD4 with equal affinity, albeit at a 10-fold lower affinity than the CysLT1 receptor.26 Despite being able to mediate bronchoconstriction and other proinflammatory activities, LTE4 does not bind avidly to either of these receptors.27–29 CysLT1, but not CysLT2, is upregulated in leukocytes from AERD patients as compared with aspirin-tolerant controls.30,31 A prominent role for CysLT1 in this disease is further supported by its expression on airway smooth muscle and the capacity of CysLT1 receptor antagonists to ameliorate aspirin-induced bronchoconstriction.32–34 The importance of LTE4 has proved challenging, in part, owing to the elusive search for its cognate receptor. Recent reports suggest that LTE4 signals through P2Y12 or GPR99, although additional studies are needed to clarify their role in AERD.35–37
In summary, AERD reflects a state of dysregulated eicosanoid metabolism, with a pathway programmed in favor of enhanced CysLT expression and signaling, against a backdrop of impaired PGE2 expression. Collectively, this action will lead to airway constriction and stimulation of eosinophil and mast cell degranulation, all key factors in AERD pathogenesis. Aspirin or nonselective NSAIDs further contribute to this dysregulation via COX-1 blockade and forcing arachidonic acid into the leukotriene synthesis pathway.
Cells and cytokines
One of the defining features of AERD is the presence of nasal polyps with pronounced eosinophilic infiltrate.7,8,12,22 Additional cellular players include mast cells, macrophages, and T-cells.38,39 Consistent with the eosinophilic predominance is the marked upregulation of eosinophilic cationic protein (ECP), which is a marker of eosinophilic degranulation. A recent study has shown a greater increase in ECP levels, more than eosinophil numbers would predict, suggesting an increased activation state of eosinophils in AERD.40 This is especially interesting as eosinophilia is also pronounced in subjects with certain forms of aspirin-tolerant polyposis. In the same study, the authors reported that chemokines including CCL11 (eotaxin-1) and CCL24 (eotaxin-2), in addition to other mediators involved in proliferation and recruitment (such as interleukin [IL]-5), are not increased in AERD compared with relevant acetyl-salicylic-acid-tolerant polyp subjects.40 Notably though, these mediators are increased in AERD compared with polyp-free sinus disease.
Most studies have shown a cytokine and chemokine milieu consistent with type 2 immunity; however, recent work from our group and others suggests an important role for IFN-γ as well.10,41 We have shown that IL-4 and IFN-γ mRNA, but not IL-5 or IL-13, are upregulated in AERD polyp tissue and that eosinophils are the primary source of these cytokines.10 Interestingly, both IL-4 and IFN-γ increase LTC4S expression on mast cells and eosinophils, providing a mechanistic link for the upregulation of CysLTs seen in AERD.42 For IFN-γ, the effect was not direct but was mediated by promoting the maturation of eosinophils as measured by the upregulation of CCR3 and Siglec-8.6,10 Additionally, IL-4 has also been shown to inhibit COX-2 and mPGES-1.43 Taken together, this suggests that AERD may represent a “mixed” Th1/Th2 disease, where IFN-γ plays an important role in pathogenesis. The mixed nature of cytokine expression fits with the observation that AERD subjects are not atopic as in classical Th2 allergic diseases. The precise role of IFN-γ remains to be determined, because some other studies have not supported this finding, which may reflect the different techniques used to detect expression.40,44
Recent work suggests that IL-33 may be another important cytokine involved in AERD pathogenesis. This alarmin-like cytokine is upregulated in the airway epithelial layer and has been shown to be dependent on CysLT expression in mouse models. It is able to function as a mediator of mast cell activation.45 This is intriguing, because as an innate cytokine, IL-33 represents another mechanism to explain eosinophilic and mast cell involvement in the absence of IgE-mediated atopy.
Additional factors that have recently been shown to be preferentially increased in AERD tissue include granulocyte–macrophage colony-stimulating factor (GM-CSF) and MCP-1, while tissue plasminogen activator (tPA) was decreased.40 Although the pathophysiological relevance of these findings is not clear, prior investigations offer hints. GM-CSF has previously been shown to enhance eosinophil survival in tissues in the absence of IL-5, and GM-CSF-mediated eosinophilic activation has recently been demonstrated in another mucosal inflammatory disease, colitis.46,47 Reduced tPA levels suggest a possible role for the fibrinolytic pathway to contribute to fibrin deposition and thus the remodeling seen in AERD IFN-γ.48 In a similar vein, a role for platelet-associated inflammation has been described in AERD.49 Interestingly, platelet-associated leukocytes were increased in AERD and were shown to enhance LTC4S expression.49
Genetic studies
Microarray and genome-wide association studies have been employed to identify genes relevant to AERD pathogenesis. Stankovic et al50 reported periostin upregulation in chronic rhinosinusitis polyps, although the levels were notably similar in aspirin-tolerant and -sensitive subjects in that study. Follow-up work assessing periostin protein levels with ELISA, however, did show a significant increase in AERD versus aspirin-tolerant asthma and also showed correlation in periostin levels with severity of sinus disease.51 This finding is consistent with recent work establishing periostin as a potential biomarker for Th2 inflammation, congruent with the fact that it is upregulated by IL-4 and IL-13 and is associated with tissue eosinophilia.52 Expression profiling of peripheral blood mononuclear cells from AERD versus aspirin-tolerant asthmatics demonstrated ten genes that exhibited >8-fold change. Interestingly, traditional cytokine and chemokine genes related to type 2 immunity were not represented among these. The two genes that were validated with high sensitivity and specificity were CNKSR family member 3 (CNKSR3) and spectrin β nonerythrocytic 2 (SPTBN2), both of whose functions in AERD remain elusive.53
An association to human leukocyte antigen (HLA) has been described in three separate genome-wide association studies of Korean subjects, with significant association of polymorphisms within the HLA-DPB1 gene being identified.54–56 The prevalence of these polymorphisms in other populations is not clear, although an earlier study in Polish subjects showed a similar result.57 Taken together, these studies have demonstrated novel markers and factors that could be important in AERD pathogenesis, although further investigation will be needed to assess their role in disease pathogenesis and possible utility as biomarkers.
Current and future therapies
In addition to standard asthma therapies such as inhaled corticosteroids and β2-agonists, a keystone of AERD management involves leukotriene blockade. Two classes of medications are in clinical use – the leukotriene receptor antagonists, which include montelukast and zafirlukst, and the 5-LO inhibitor zileuton. Controlled, prospective, placebo-controlled studies with montelukast and zileuton have both shown efficacy in aspirin-sensitive asthma as measured by improved forced expiratory volume in 1 second (FEV1) scores, decreased use of rescue inhalers, and an increase in asthma quality-of-life measures.58,59 Based on their unique mechanism of actions, there is some thought that they may act in an additive way to ameliorate symptoms, although no prospective combined trials, or head-to-head trials, have been conducted in the AERD population. A single head-to-head trial in asthmatics, which did not address aspirin-sensitive asthma, specifically demonstrated modest superiority of zileuton compared to montelukast.60 Leukotriene receptor antagonists are often used as first-line therapy based on practical considerations (as they are less expensive) and have fewer side effects; however, zileuton may have superior efficacy in AERD based on patient survey data.61 Zileuton impairs all leukotriene production by virtue of 5-LO inhibition, whereas the clinically available leukotriene receptor antagonists selectively target CysLT1. As discussed previously, although CysLT1 is the high-affinity receptor for LTD4, leukotrienes also signal via CysLT2 and other LTE4 receptors. Given that CysLT2, like CysLT1, is upregulated in nasal polyps, this provides a mechanistic explanation as to why zileuton would have a broader antileukotriene activity than selective CysLT1 agents.62 Taken together, CysLT2 and other recently described putative leukotriene receptors, such as GPR99 and P2Y12, are potential targets for future research efforts in AERD-directed therapeutics.
On the one hand, while aspirin can trigger acute respiratory symptoms, aspirin desensitization followed by daily aspirin therapy leads to improved long-term symptoms in AERD subjects. The protocol is conducted by starting with small doses of aspirin and gradually achieving doses of 650–1,300 mg daily. Notably, although the approach is similar to traditional allergy desensitization, which addresses IgE-mediated reactions, the pathophysiology is distinct. The most significant improvements with aspirin desensitization relate to upper airway symptoms including smell and decreased polyp formation; however, asthma severity, use of steroids, and hospitalizations are also lessened.63–66 The beneficial mechanisms of aspirin are not entirely clear, though aspirin likely modulates multiple pathways involved in AERD pathogenesis. Our group and others have shown that aspirin blocks IL-4-activated signal transducer and activator of transcription 6 (STAT6), which is a key transcriptional regulator of CysLT1 and has known binding sites in the LTC4S promoter.43,67 This corresponds with an earlier work, which showed downregulation of CysLT1 on leukocytes from nasal mucosa following aspirin desensitization.31 Another study has shown downregulation of IL-4 and MMP-9 levels following desensitization.68
Despite the use of leukotriene pathway inhibitors and aspirin desensitization, AERD remains a disease with high morbidity.61 Advances in AERD pathophysiology, however, offer promising future targets to better address this disease. Multiple monoclonal antibodies targeting immune pathways are in development or have clinical approval for related conditions. Mepolizumab is an anti-IL-5 monoclonal antibody that has been approved for severe eosinophilic asthma and has been shown in a small study to decrease nasal polyposis.69 As previously discussed, IL-5 is not a central mediator of AERD as compared to aspirin-tolerant chronic sinusitis; however, it is clearly elevated in AERD compared with healthy controls or those with chronic sinusitis without polyps, suggesting it may yet be a fruitful target.40 Recently, another anti-IL-5 drug, reslizumab, has been approved and may offer benefit similar to mepolizumab. Dupilumab is an IL-4 α receptor antagonist that blocks both IL-4 and IL-13 signaling. Currently in clinical trials, it has shown benefit in a study of moderate-to-severe eosinophilic asthmatics in decreasing asthma exacerbations and improving FEV1.70 Based on the demonstrated role of IL-4 in AERD, this represents a potentially promising new therapy for aspirin-sensitive asthmatics. Another biologic with proven efficacy in allergic asthma is the anti-IgE monoclonal antibody omalizumab. Interestingly, despite the fact that IgE does not feature prominently in AERD pathogenesis, there have been multiple case reports of omalizumab benefiting AERD patients.71–73 Although the mechanism of action is not clear, these case reports indicate that further prospective studies should be considered. In addition to these biologics already in clinical use or preclinical development, future research efforts targeting novel pathways such as IL-33 or TSLP signaling should also be explored.
Conclusion
AERD is a disease of the upper and lower airways, featuring prominent eosinophilia and eicosanoid dysregulation. In addition to traditional type 2 immune mediators, recent work points to a role for IFN-γ.10 Current therapies include leukotriene modifiers and aspirin desensitization. Although AERD represents a minor constituent of all asthma, it represents a severe subset with high morbidity that warrants ongoing efforts at the level of basic research and clinical trials.
Novel aspects
AERD is a disease characterized by the overexpression and overresponsiveness of the cysteinyl leukotriene pathway. Additionally, there is underproduction of the protective prostaglandin E2. This creates a situation where following ingestion of aspirin or other NSAIDs, there is unconstrained release of inflammatory mediators that lead to symptoms associated with disease. Therapies that target the leukotriene pathway are somewhat effective in providing relief; however, their effect is not complete. Newer biologic therapies aimed at targeting the inflammatory cells themselves may offer enhanced benefit. Two of these that may have an immediate impact are the recently approved drugs mepolizumab and reslizumab, which target eosinophils in uncontrolled asthma. Although not approved for sinusitis, given the asthma component of AERD, these drugs will likely be prescribed, and it will be interesting to see if they improve sinus symptoms and disease severity as eosinophil numbers are decreased.
Disclosure
The authors report that John W Steinke is co-primary investigator on an NIH grant to study sinusitis. The authors report no other conflicts of interest in this work.
References
Samter M, Beers RF Jr. Intolerance to aspirin. Clinical studies and consideration of its pathogenesis. Ann Intern Med. 1968;68(5): 975–983. | |
Rajan JP, Wineinger NE, Stevenson DD, White AA. Prevalence of aspirin-exacerbated respiratory disease among asthmatic patients: a meta-analysis of the literature. J Allergy Clin Immunol. 2015;135(3):676.e1–681.e1. | |
Szczeklik A, Nizankowska E. Clinical features and diagnosis of aspirin induced asthma. Thorax. 2000;55:S42–S44. | |
Vally H, Taylor ML, Thompson PJ. The prevalence of aspirin intolerant asthma (AIA) in Australian asthmatic patients. Thorax. 2002;57:569–574. | |
Mascia K, Borish L, Patrie J, Hunt J, Phillips CD, Steinke JW. Chronic hyperplastic eosiniphilic sinusistis as a predictor of aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2005;94:652–657. | |
Steinke JW, Borish L. Factors driving the aspirin exacerbated respiratory disease phenotype. Am J Rhinol Allergy. 2015;29(1):35–40. | |
Payne SC, Early SB, Huyett P, Han JK, Borish L, Steinke JW. Evidence for distinct histologic profile of nasal polyps with and without eosinophilia. Laryngoscope. 2011;121(10):2262–2267. | |
Bachert C, Wagenmann M, Hauser U, Rudack C. IL-5 synthesis is upregulated in human nasal polyp tissue. J Allergy Clin Immunol. 1997;99:837–842. | |
Sampson AP, Cowburn AS, Sladek K. Profound overexpression of leukotriene C4 synthase in bronchial biopsies from aspirin-intolerant asthmatic patients. Int Arch Allergy Immunol. 1997;113:355–357. | |
Steinke JW, Liu L, Huyett P, Negri J, Payne SC, Borish L. Prominent role of interferon-γ in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. 2013;132:856.e3–865.e3. | |
Cowburn AS, Sladek K, Soja J, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101:834–846. | |
Perez-Novo CA, Watelet JB, Claeys C, van Cauwenberge P, Bachert C. Prostaglandin, leukotiene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol. 2005;115:1189–1196. | |
Schmid M, Gode U, Schafer D, Wigand ME. Arachidonic acid metabolism in nasal tissue and peripheral blood cells in aspirin intolerant asthmatics. Acta Otolaryngol. 1999;119:277–280. | |
Picado C, Fernandez-Morata JC, Juan M, et al. Cyclooxygenase-2 mRNA is down expressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1999;160:291–296. | |
Gosepath J, Brieger J, Mann WJ. New immunohistologic findings on the differential role of cyclooxygenase 1 and cyclooxygenase 2 in nasal polposis. Am J Rhinol. 2005;19:111–116. | |
Murakami M, Nakashima K, Kamei D, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem. 2003;278(39):37937–37947. | |
Sturm EM, Schratl P, Schuligoi R, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol. 2008;181(10):7273–7283. | |
Kay LJ, Yeo WW, Peachell PT. Prostaglandin E2 activated EP2 receptors to inhibit human lung mast degranulation. Br J Pharmacol. 2006;147:707–713. | |
Steinke JW. Editorial: Yin-Yang of EP receptor expression. J Leukoc Biol. 2012;92(6):1129–1131. | |
Sestini P, Armetti L, Gambaro G, et al. Inhaled PgE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153: 572–575. | |
Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood. 2006;107:3243–3250. | |
Steinke JW, Bradley D, Arango P, et al. Cytseinyl leukotriene expression in chronic hyperplastic sinusitis-nasal polyposis: importance to eosinophilia and asthma. J Allergy Clin Immunol. 2003;111:342–349. | |
Brocklehurst WE. The release of histamine and formation of a slow-reacting substance (SRS-A) during anaphylactic shock. J Physiol. 1960;151:416–435. | |
Christie PE, Tagari P, Ford-Hutchinson AW, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis. 1991;143:1025–1029. | |
Sladek K, Szczeklik A. Cysteinyl leukotrienes overproduction and mast cell activation in aspirn-provoked bronchospasm in asthma. Eur Respir J. 1993;6:391–399. | |
Austen KF, Maekawa A, Kanaoka Y, Boyce JA. The leukotriene E4 puzzle: finding the missing pieces and revealing the pathobiologic implications. J Allergy Clin Immunol. 2009;124(3):406–414. | |
Arm JP, O’Hickey S, Spur BW, Lee TH. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am Rev Respir Dis. 1989;140:148–153. | |
Christie PE, Schmitz-Schumann M, Spur BW, Lee TH. Airway responsiveness to leukotriene C4 (LTC4), leukotriene E4 (LTE4) and histamine in aspirin-sensitive asthmatic subjects. Eur Respir J. 1993;6:1468–1473. | |
Laitinen LA, Laitinen A, Haahtela T, Vilkka V, Spur BW, Lee TH. Leukotriene E4 and granulocytic infiltration into asthmatic airways. Lancet. 1993;341:989–990. | |
Corrigan C, Mallett K, Ying S, et al. Expression of the cysteinyl leukotriene receptors cysLT(1) and cysLT(2) in aspirin-sensitive and aspirin-tolerant chronic rhinosinusitis. J Allergy Clin Immunol. 2005;115(2):316–322. | |
Sousa AR, Parikh A, Scadding G, Corrigan CJ, Lee TH. Leukotriene-receptor expression on nasal mucosal inflammatory cells in asprin-sensitive rhinosinusitis. N Engl J Med. 2002;347:1493–1499. | |
Lynch KR, O’Neill GP, Liu Q, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399(6738):789–793. | |
Christie PE, Smith CM, Lee TH. The potent and selective sulfidopeptide leukotriene antagonist, SK&F 104353, inhibits aspirin-induced asthma. Am Rev Respir Dis. 1991;144:957–958. | |
Dahlen B. Treatment of aspirin-intolerant asthma with antileukotrienes. Am J Respir Crit Care Med. 2000;161(Suppl):S137–S141. | |
Paruchuri S, Tashimo H, Feng C, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206(11):2543–2555. | |
Kanaoka Y, Maekawa A, Austen KF. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem. 2013;288(16):10967–10972. | |
Maekawa A, Kanaoka Y, Xing W, Austen KF. Functional recognition of a distinct receptor preferential for leukotriene E4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc Natl Acad Sci U S A. 2008;105(43):16695–16700. | |
Kowalski ML, Grzegorczyk J, Pawliczak R, Kornatowski T, Wagrowska-Danilewicz M, Danilewicz M. Decreased apoptosis and distinct profile of infiltrating cells in the nasal polyps of patients with aspirin hypersensitivity. Allergy. 2002;57(6):493–500. | |
Varga EM, Jacobson MR, Masuyama K, et al. Inflammatory cell populations and cytokine mRNA expression in the nasal mucosa in aspirin-sensitive rhinitis. Eur Respir J. 1999;14(3):610–615. | |
Stevens WW, Ocampo CJ, Berdnikovs S, et al. Cytokines in chronic rhinosinusitis. Role in eosinophilia and aspirin-exacerbated respiratory disease. Am J Respir Crit Care Med. 2015;192(6):682–694. | |
Shome GP, Tarbox J, Shearer M, Kennedy R. Cytokine expression in peripheral blood lymphocytes before and after aspirin desensitization in aspirin-exacerbated respiratory disease. Allergy Asthma Proc. 2007;28(6):706–710. | |
Hsieh FH, Lam BK, Penrose JF, Austen KF, Boyce JA. T helper cell type 2 cytokines coordinately regulate immunoglobulin E-dependent cysteinyl leukotriene production by human cord blood-derived mast cells: profound induction of leukotriene C4 synthase expression by interleukin 4. J Exp Med. 2001;193:123–133. | |
Steinke JW, Culp JA, Kropf E, Borish L. Modulation by aspirin of nuclear phospho-signal transducer and activator of transcription 6 expression: possible role in therapeutic benefit associated with aspirin desensitization. J Allergy Clin Immunol. 2009;124(4):724.e4–730.e4. | |
Zhang N, Van Zele T, Perez-Novo C, et al. Different types of T-effector cells orchestrate mucosal inflammation in chronic sinus disease. J Allergy Clin Immunol. 2008;122(5):961–968. | |
Liu T, Kanaoka Y, Barrett NA, et al. Aspirin-exacerbated respiratory disease involves a cysteinyl leukotriene-driven IL-33-mediated mast cell activation pathway. J Immunol. 2015;195(8):3537–3545. | |
Griseri T, Arnold IC, Pearson C, et al. Granulocyte macrophage colony-stimulating factor-activated eosinophils promote interleukin-23 driven chronic colitis. Immunity. 2015;43(1):187–199. | |
Lamas S, Michel T, Brenner BM, Marsden PA. Nitric oxide synthesis in endothelial cells: evidence for a pathway inducible by TNF-alpha. Am J Physiol. 1991;261(4 Pt 1):C634–C641. | |
Takabayashi T, Kato A, Peters AT, et al. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of tissue plasminogen activator expression. Am J Respir Crit Care Med. 2013;187(1):49–57. | |
Laidlaw TM, Kidder MS, Bhattacharyya N, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. 2012;119(16):3790–3798. | |
Stankovic KM, Goldsztein H, Reh DD, Platt MP, Metson R. Gene expression profiling of nasal polyps associated with chronic sinusitis and aspirin-sensitive asthma. Laryngoscope. 2008;118(5):881–889. | |
Kim MA, Izuhara K, Ohta S, et al. Association of serum periostin with aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. 2014;113(3):314–320. | |
Parulekar AD, Atik MA, Hanania NA. Periostin, a novel biomarker of TH2-driven asthma. Curr Opin Pulm Med. 2014;20(1):60–65. | |
Shin S, Park JS, Kim YJ, Oh T, An S, Park CS. Differential gene expression profile in PBMCs from subjects with AERD and ATA: a gene marker for AERD. Mol Genet Genomics. 2012;287(5):361–371. | |
Kim JH, Park BL, Cheong HS, et al. Genome-wide and follow-up studies identify CEP68 gene variants associated with risk of aspirin-intolerant asthma. PLoS One. 2010;5(11):e13818. | |
Park BL, Kim TH, Kim JH, et al. Genome-wide association study of aspirin-exacerbated respiratory disease in a Korean population. Hum Genet. 2013;132(3):313–321. | |
Kim SH, Cho BY, Choi H, et al. The SNP rs3128965 of HLA-DPB1 as a genetic marker of the AERD phenotype. PLoS One. 2014;9(12):e111220. | |
Dekker JW, Nizankowska E, Schmitz-Schumann M, et al. Aspirin-induced asthma and HLA-DRB1 and HLA-DPB1 genotypes. Clin Exp Allergy. 1997;27(5):574–577. | |
Dahlen SE, Malmstrom K, Nizankowska E, et al. Improvement of aspirin-intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(1):9–14. | |
Dahlen B, Nizankowska E, Szczeklik A, et al. Benefits from adding the 5-lipoxygenase inhibitor zileuton to conventional therapy in aspirin-intolerant asthmatics. Am J Respir Critc Care Med. 1998;157:1187–1194. | |
Kubavat AH, Khippal N, Tak S, et al. A randomized, comparative, multicentric clinical trial to assess the efficacy and safety of zileuton extended-release tablets with montelukast sodium tablets in patients suffering from chronic persistent asthma. Am J Ther. 2013;20(2):154–162. | |
Ta V, White AA. Survey-defined patient experiences with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol Pract. 2015;3(5):711–718. | |
Wu X, Hong H, Zuo K, et al. Expression of leukotriene and its receptors in eosinophilic chronic rhinosinusitis with nasal polyps. Int Forum Allergy Rhinol. 2016;6(1):75–81. | |
Mrowka-Kata K, Czecior E, Kata D, Namyslowski G, Dziechciarz-Werbowska J, Sowa P. Current view on nasal polyps management in Samter’s triad patients. Otolaryngol Pol. 2012;66(6):373–378. | |
Sweet JM, Stevenson DD, Simon RA, Mathison DA. Long-term effects of aspirin desensitization – treatment for aspirin-sensitive rhinosinusitis-asthma. J Allergy Clin Immunol. 1990;85:59–65. | |
Stevenson DD, Simon RA. Selection of patients for aspirin desensitization treatment. J Allergy Clin Immunol. 2006;118:801–804. | |
Stevenson DD, Hankammer MA, Mathison DA, Christiansen SC, Simon RA. Aspirin desensitization treatment of aspirin-sensitive patients with rhinosinusitis-asthma: long-term outcomes. J Allergy Clin Immunol. 1996;98:751–758. | |
Perez GM, Melo M, Keegan AD, Zamorano J. Aspirin and salicylates inhibit the IL-4- and IL-13-induced activation of STAT6. J Immunol. 2002;168(3):1428–1434. | |
Katial RK, Strand M, Prasertsuntarasai T, Leung R, Zheng W, Alam R. The effect of aspirin desensitization on novel biomarkers in aspirin-exacerbated respiratory diseases. J Allergy Clin Immunol. 2010;126(4): 738–744. | |
Gevaert P, Van Bruaene N, Cattaert T, et al. Mepolizumab, a humanized anti-IL-5 mAb, as a treatment option for severe nasal polyposis. J Allergy Clin Immunol. 2011;128(5):989–995.e1–e8. | |
Wenzel S, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Eng J Med. 2013;368(26):2455–2466. | |
Bobolea I, Barranco P, Fiandor A, Cabanas R, Quirce S. Omalizumab: a potential new therapeutic approach for aspirin-exacerbated respiratory disease. J Investig Allergol Clin Immunol. 2010;20(5):448–449. | |
Yalcin AD, Ucar S, Gumuslu S, Strauss LG. Effects of omalizumab on eosinophil cationic peptide, 25-hydroxyvitamin-D, IL-1β and sCD200 in cases of Samter’s syndrome: 36 months follow-up. Immunopharmacol Immunotoxicol. 2013;35(4):524–527. | |
Bergmann KC, Zuberbier T, Church MK. Omalizumab in the treatment of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol Pract. 2015;3(3):459–460. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.