Back to Journals » International Journal of Nanomedicine » Volume 15
ArtinM Grafted Phospholipid Nanoparticles for Enhancing Antibiotic Cellular Uptake Against Intracellular Infection
Authors Suciati T ![]() , Nafisa S
, Nafisa S ![]() , Nareswari TL, Juniatik M, Julianti E, Wibowo MS
, Nareswari TL, Juniatik M, Julianti E, Wibowo MS ![]() , Yudhistira T
, Yudhistira T ![]() , Ihsanawati I
, Ihsanawati I ![]() , Triyani Y
, Triyani Y ![]() , Khairurrijal K
, Khairurrijal K ![]()
Received 7 August 2020
Accepted for publication 25 September 2020
Published 10 November 2020 Volume 2020:15 Pages 8829—8843
DOI https://doi.org/10.2147/IJN.S275449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
Tri Suciati,1 Safira Nafisa,2 Tantri Liris Nareswari,1 Meta Juniatik,1 Elin Julianti,1 Marlia Singgih Wibowo,1 Titah Yudhistira,3 Ihsanawati Ihsanawati,4 Yani Triyani,5 Khairurrijal Khairurrijal4,6
1School of Pharmacy, Bandung Institute of Technology, Bandung, Indonesia; 2Faculty of Pharmacy, Pancasila University, Jakarta, Indonesia; 3Faculty of Industrial Technology, Bandung Institute of Technology, Bandung, Indonesia; 4Faculty of Mathematics and Natural Sciences, Bandung Institute of Technology, Bandung, Indonesia; 5Faculty of Medicine, Bandung Islamic University, Bandung, Indonesia; 6Bioscience and Biotechnology Research Center, Bandung Institute of Technology, Bandung, Indonesia
Correspondence: Tri Suciati
School of Pharmacy, Bandung Institute of Technology, Yusuf Panigoro Building, Ganesa 10, Bandung 40132, Indonesia
Tel/Fax +62 22 2504852
Email [email protected]
Background and Aim: An antimicrobial delivery in the form of surface-modified lectin of lipid nanoparticles was proposed to improve cellular accumulation. ArtinM, an active toll-like receptor 2 (TLR2) agonist lectin isolated from cempedak (Arthocarpus integrifolia) seeds, was selected to induce cellular engulfment of nanoparticles within infected host cells.
Materials and Methods: Lipid nanoparticles were prepared using the emulsification technique before electrostatic adsorption of artinM. The formula comprising of rifampicin, soy phospholipid, and polysorbate 80 was optimized by Box-Behnken design to produce the desired particle size, entrapment efficiency, and drug loading. The optimum formula was characterized for morphology, in vitro release, and cellular transport.
Results and Discussion: Soy phospholipid showed a profound effect on controlling drug loading and entrapment efficiency. Owing to its surface activity, polysorbate 80 contributed significantly to reduce particle size; however, a higher ratio to lipid concentration resulted in a decrease of rifampicin encapsulation. The adsorption of artinM on the surface of nanoparticles was accomplished by electrostatic binding at pH 4, where this process maintained the stability of encapsulated rifampicin. A high proportion of artinM adsorbed on the surface of the nanoparticles shown by haemagglutination assay, zeta potential measurement, and transmission electron microscopy imaging. Cellular uptake revealed by confocal microscopy showed the success in transporting Nile-red labelled nanoparticles across fibroblast cells.
Conclusion: The delivery system of nanoparticles bearing artinM becomes a potential platform technology for antibiotic targeting in the treatment of life-threatening chronic diseases caused by intracellular infections.
Keywords: surface modification, Box–Behnken design, antibiotic, TLR2 agonists, botanical lectin, electrostatic binding
Introduction
Healthcare practitioners are facing a high burden in combating numerous chronic and deadly infection because bacteria evolution forms various virulent factors and/or biofilm resulting in antibiotic tolerant phenotypes.1 Staphylococcus aureus (S. aureus), for example, has for a long time been known to cause a topical nosocomial infection, with the common entry routes are by colonizing nasal tract and skin.2,3 It can be treated by a topical antibiotic in the past,4 but now any efforts to eliminate methicillin-resistant S. aureus (MRSA) and vancomycin intermediate-resistant S. aureus (VISA) local infection have been found to be unsuccessful.1,5,6 A further challenge has emerged by the presence of a more dangerous pathogenic persistent S. aureus recognized as a small colony variant (SCV). These bacteria can enter the human body by invading the skin and mucous membrane through contaminating medical devices used in the surgery, infecting nearly every organ and evading many cell types, both phagocytic and non-phagocytic.6–9 Many research had proven that S. aureus is slowly metabolized by the host which leads to long-term intracellular persistence.8–10
Many antimicrobial drugs are not able to accumulate efficiently within the host cells, resulting in the low or sub-therapeutic intracellular doses and leading to antibiotic resistance and poor therapeutic outcome.11 These emerging researches have been addressed with many approaches in an attempt to increase intracellular concentration, including drug delivery.12 Developing a vehicle for intracellular transport of antibiotics becomes a mandatory option to increase intracellular uptake.13 Rifampicin, which is a broad-spectrum antibacterial with low toxicity and wide applications to latent mycobacterial and non-mycobacterial intracellular infections,14–16 is used as an antibiotic model for this formulation development. Rifampicin used as a first-line tuberculosis therapy is also effective against intracellular and extracellular S. aureus for in vitro and in vivo application.2,14,15,17 However, the single-use of rifampicin is subjected to antimicrobial resistant.18
Following earlier reports, rifampicin is employed in the design and fabrication of the carrier for intracellular targeting.15,16,19 One of the approaches is lipid-based nano-system drug entrapment, which considers the lipid-soluble nature of rifampicin. The structure of phospholipid nanoparticles is similar to mammalian cell wall, hence served as a biocompatible, non-irritant, and biodegradable vehicle for cell permeation.20,21
Having oligosaccharide binding on the cell surface, various proteins or protein domains called lectins have been applied as targeting agents for many drugs.22,23 Lectin grafted nanoparticles are among emerging carriers for enhancing cellular uptake, ie, wheat germ agglutinin (WGA) and Ulex europaeus agglutinin I (UEA1). It was shown that these vehicles result in an effective controlled release of drugs, good physical stability, and increased intracellular trafficking.23,24
ArtinM, a lectin isolated from cempedak (Arthocarpus integrifolia) seeds, is a TLR2 agonist having an activity to activate macrophage through CD14 N-glycan binding.25 The activation of TLR2 mediates intracellular signaling pathway for T-helper activity and cytokine production, such as IL-12 p40, tumor necrosis factor-α (TNF-α), IL-6, and IL-10 which potentially improve phagocytosis and host defense.24 On the other hand, S. aureus enters the host cell via lipoteichoic acid (LTA)-TLR2 interaction, where this interaction causes host immune suppression.26 Thus, artinM surface modification of nanoparticles containing encapsulated-antibiotic is proposed to mimic S. aureus cellular entry pathway, trigger the phagocytosis, and regain immune response. This strategy becomes the hypothesis of our proposed drug delivery as illustrated in Figure 1.
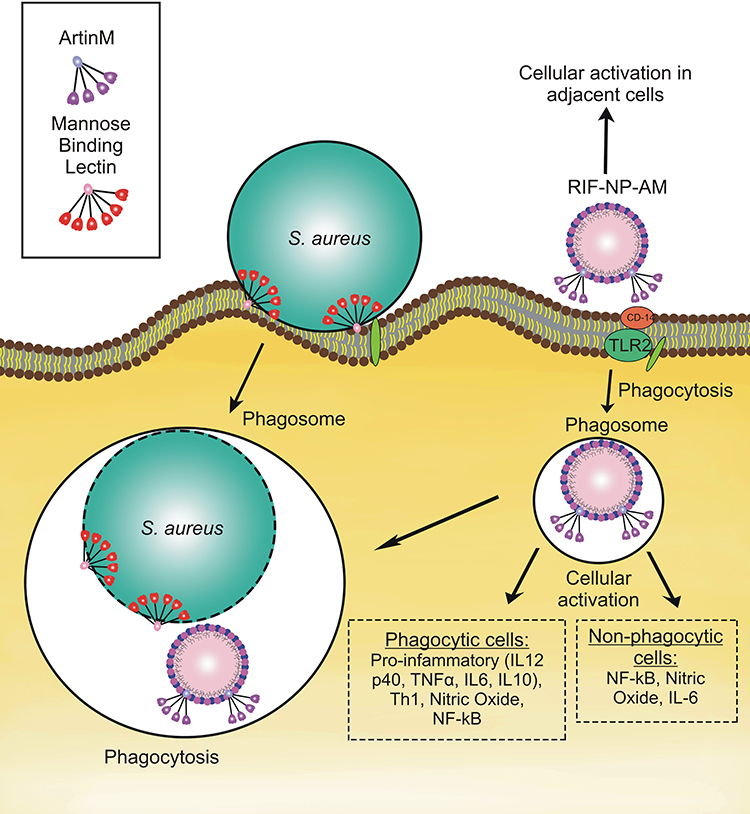 |
Figure 1 Illustration of artinM modified phospholipid nanoparticles as rifampicin carrier against the host cell infected by S. aureus. |
This research aimed at developing surface-modified lipid nanoparticles using lectin as rifampicin vehicles. Rifampicin needs to be encapsulated in lipid-based nano-system such as soy phospholipid (lecithin) due to its lipid solubility. The present of amine and phosphate groups of soy phospholipid is beneficial to adsorb artinM electrostatically for allowing the surface modification of lipid nanoparticles.27
Materials and Methods
Materials
Rifampicin was obtained from PT. Meprofarm (Bandung, Indonesia). Soy lecithin was purchased from Lipoid (Lipoid S 100, Ludwigshafen, Germany). Oleic acid was obtained from Brataco (Bandung, Indonesia). Polysorbate 80, Nile red, D-mannose, and D-galactose were purchased from Sigma-Aldrich (Singapore). Bradford reagent was purchased from Bio-Rad (Biorad®, Hercules, CA, USA). Human erythrocytes type O, Dulbecco’s Modified Eagle Medium (DMEM) and Heat Inactivated Foetal Bovine Serum (FBS) were purchased from GibcoTM of Invitrogen (Gibco BRL, Invitrogen, Carlsbad, CA, USA). Staphylococcus aureus (ATCC 6538) and Balb/3T3 cells were acquired from American Type Culture Collection (USA), and other chemicals found in the laboratory with analytical grade specifications.
ArtinM Isolation
ArtinM was isolated from cempedak (Arthocarpus integrifolia) seeds using a method modified from Roy et al.28 In cempedak seeds, there are two polyspecific lectins with different specificity, namely jacalin and artinM. Considering to the preferential affinity between galactose specific jacalin and mannose specific artinM, jacalin was removed using guar gum-epichlorohydrin beads.29
Twenty-five grams of cempedak seed powder, collected from Subang, Indonesia, were macerated in 250 mL phosphate-buffered saline (PBS) pH 7.4 at 4 °C overnight. The extract was then filtered and centrifuged until crude extract was obtained. The extract was treated with protein fractionation using 40% ammonium sulphate and allowed to stand overnight. This solution was centrifuged, and the supernatant was further precipitated by adding 60% ammonium sulphate. The precipitate was collected and dissolved in PBS pH 7.4, dialyzed against PBS pH 7.4 for three times. Jacalin was separated from protein extract using an affinity matrix with guar gum-epichlorohydrin beads for an hour incubation. The effluent was collected in 5 mL aliquots and measured by ultraviolet-visible (UV-vis) spectrophotometer (Beckman, DU 7500i) at 280 nm. The presence of jacalin in the effluent was evidenced by analyzing jacalin dominant effluent in sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE). Elution was repeated with fresh matrix beads until the constant absorbance was found. Furthermore, complete removal of jacalin trapped on the beads was analyzed from the two latest effluents by SDS-PAGE. The isolated artinM was then characterized, lyophilized, and stored in a desiccator before use. The protein content of artinM isolate was determined by Bradford assay using bovine serum albumin (BSA) as the standard. The absorbance was measured using a UV-vis spectrophotometer (Beckman, DU 7500i) at 595 nm.
ArtinM Characterization
SDS-PAGE analysis was conducted to confirm the identity and molecular weight of artinM using 15% polyacrylamide gel. The solution of lyophilized samples in PBS pH 7.4 were subjected to electrophoresis at 80 V for 120 minutes. The resulting gel was visualized with Coomassie blue staining solution. The SDS-PAGE profile of artinM is recognized as a single band at 14–16 kDa,29 while that of jacalin is as double bands at 12–15 kDa.30
ArtinM activity was analyzed using haemagglutination test adopted from Sano and Ogawa,31 in which a serial dilution of lectin in a 96-well microliter plate with the addition of erythrocyte was used. Approximately 50 µL of 2% erythrocyte suspension in PBS was placed in each well and incubated with artinM solution sample for two hours. ArtinM activity was analyzed by conducting haemagglutination test in PBS at pH 4, 7.4, and 8. The stability of artinM was analyzed upon contact with solvents used in nanoparticle preparation. ArtinM solution in PBS pH 7.4 was incubated with acetate and ethanol 1:1 for 1 hour, subsequently evaporated and reconstituted prior to haemagglutination test. Furthermore, haemagglutination inhibition test was also conducted to confirm artinM identity by supplementing 0.4 M D-mannose and 0.4 M D-galactose to the mixture of erythrocyte and the sample solution.
Formulation of Rifampicin Nanoparticle Surface Modified with ArtinM (Rif-NP-AM)
Rifampicin was encapsulated before the modification process of phospholipid nanoparticles with artinM. The nanoparticle formula was optimized using Box Behnken design. Lecithin nanoparticles were prepared by emulsification method as described by Yanasarn et al with some modifications.32 Lecithin and oleic acid at the ratio of 2:1 was dissolved in ethanol, while rifampicin was dissolved in ethyl acetate. The lecithin-oleic solution was mixed homogeneously with rifampicin solution and the organic solvent was then evaporated. The solid lipid mixture was dispersed in warm water at pH 4 and then added a certain concentration of polysorbate 80 as surfactant. It was stirred at a temperature of 50–55 °C until nanoparticles were formed. The solution of artinM was added to the optimized colloid and mixed until room temperature was achieved.
Box-Behnken design was constructed with 15 runs, 3 factors, and 3 levels. Experimental variables included the lecithin and oleic acid (X1), rifampicin (X2), and polysorbate 80 (X3) concentrations to produce particle size (Y1), entrapment efficiency (EE) (Y2), and drug loading (DL) (Y3) as the responses. The range of variables is presented in Table 1. The rifampicin nanoparticles (Rif-NP) formula was prepared according to the composition shown in Table 2. The results were processed using Minitab 17 and the model is given by Equation (1).
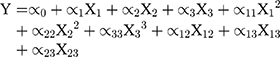
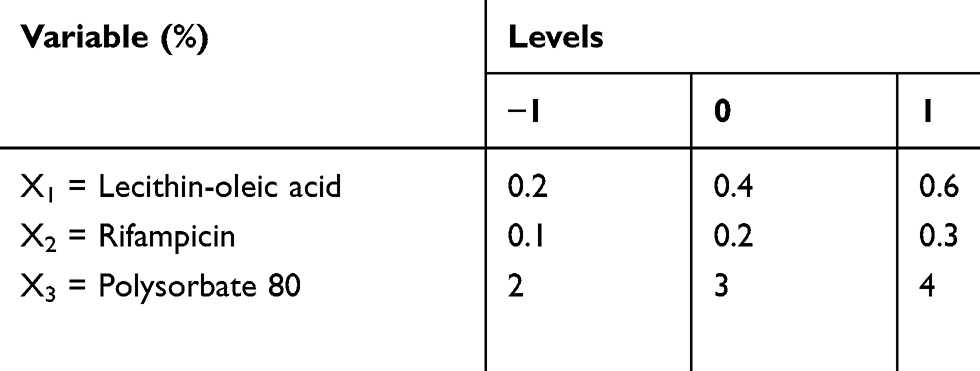 |
Table 1 Variables and Levels Used for Box-Behnken Design |
 |
Table 2 Variables of the Box-Behnken Design for Nanoparticle Formula Optimization |
The particle size was measured as Z-average using a particle size analyzer (PSA) based on photon correlation spectroscopy (DelsaTM Nano C Beckman Coulter) and the distribution as polydispersity index (PDI). The particle surface charge was analyzed using electrophoretic light scattering. The rifampicin entrapment efficiency was determined using an indirect method. The suspended rifampicin-encapsulated nanoparticles were centrifuged at 5000 rpm for 10 minutes to separate the free rifampicin. The supernatant was centrifuged again at 13,000 rpm for 60 minutes. The free rifampicin and the supernatant were tested to determine the amount of the drug that was not entrapped using a UV-visible spectrophotometer (Beckman, DU 7500i) at 475 nm. The percentage of the entrapment efficiency (% EE) and drug loading (% DL) were determined by using Equations (2) and (3), respectively.
(2) (3)
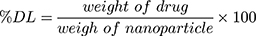
Evaluation and in vitro Release of Rifampicin from Rif-NP-AM
The morphology of Rif-NP-AM nanoparticle sample and lipid nanoparticles with and without rifampicin (NP, rif-NP, and NP-AM) as control was analyzed by transmission electron microscope (TEM Hitachi HT7700) and the presence of artinM on the surface was identified by haemagglutination test. This phenomenon was also supported by the change of nanoparticle electrical charge with zeta potential analyzer (DelsaTM Nano C Beckman Coulter). Furthermore, a diffusion technique using the dialysis membrane was employed to test rifampicin release from the nanoparticles. Free rifampicin, rifampicin loaded in nanoparticles (Rif-NP), and artinM modified nanoparticles (Rif-NP-AM) were each placed inside a dialysis tubing having a cut-off of 10 kDa and incubated in PBS pH 7.4 and phosphate buffer pH 5.2 solution within a water bath shaker at 37 °C. Ascorbic acid was added to the dissolution media to prevent rifampicin degradation.33 Samples were taken at certain intervals and replaced with a fresh medium. The concentration of released rifampicin was determined using a UV-visible spectrophotometer (Beckman, DU 7500i) at 475 nm. The amount of drug released (DR) was determined by using Equation (4).
(4)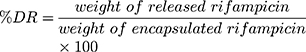
Antimicrobial Activity
S. aureus (ATCC 6538) incubated in Mullet Hinton Agar (MHA) at 37 °C for 18–24 hours were suspended in Muller Hinton Broth (MHB) and diluted until absorbance of 0.08–0.13 at 625 nm was achieved, which is equivalent to 0.5 McFarland standard. The solution was further diluted with MHB at the ratio of 1:20 to get 106 CFU/mL.34 The experimental solutions were composed of nanoparticles without artinM (NP) and Rif-NP-AM. The reference solution was prepared by dissolving rifampicin in DMSO and diluted with MHB.
Approximately 100 µL of MHB was placed in every well of the 96-well microplate. The reference solution and the experimental solution of 100 µL were serially diluted. The negative control (MHB) and positive control (S. aureus suspension in MHB) were also tested. The bacteria suspension was added into the microplate containing the experimental solution and incubated at 35–37 °C for 18–24 hours. The wells resulting in a clear solution were determined as a minimum inhibitory concentration (MIC). The MIC was inoculated onto an agar plate and incubated at 35–37 °C for 18–24 hours. The clear agar plate was recognized as a minimum bactericidal concentration (MBC).
Nanoparticles Internalization into the Cell
Nanoparticles with and without artinM modification were labelled by employing fluorescent Nile red to visualize the cellular entry using a confocal laser scanning microscope (Olympus, Flouview FV10i). Nile red of 0.4% w/w was dissolved in the oil-phase solution containing lipid mixture in ethyl acetate prior to nanoparticle preparation. The Balb/3T3 cells cultured in a T25 flask were split into the confocal dish containing 2 mL of DMEM supplemented with 10% FBS at 2 x 105 cells per dish. The medium was discarded, and the cells were washed with PBS twice. The nanoparticles were diluted with serum-free medium at 50 µg/mL and placed into the confocal dish. The dish was incubated for 30 minutes and observed using a confocal laser scanning microscope (Olympus, Flouview FV10i) at 60× magnification, excitation wavelength of 530 nm, and emission of 553 nm. It was detected using a meta-detector with a scanning range of 595–612 nm.
Statistical Analysis
Analysis of variance (ANOVA) was used to analyze significant factors and their interactions using Minitab 17. A p-value less than 0.05 was considered as statistically significant.
Results and Discussion
Isolation and Characterization of ArtinM
Botanical lectins have long been studied for their function in regulating diverse intracellular signaling in the body.34,35 Here we selected artinM isolated from cempedak seeds to modify cellular transport of lipid nanoparticles carrying antibiotics. The artinM isolate was characterized by SDS-PAGE analysis and haemagglutination test.
ArtinM had been successfully separated from jacalin, which is the identical structure of lectin abundantly produced by cempedak seed. The sequential adsorption using epichlorohydrin cross-linked guar gum beads was selectively retaining jacalin and leaving artinM soluble in the effluent. Figure 2A depicts the absorbance of protein content in the extract eluted on the beads, showing a decrease in absorbance from approximately 3.0 to an almost constant of 0.2 through sequential adsorption. Repeated adsorption of the extract on the matrix was a success key to remove jacalin from the artinM isolate due to a higher amount of jacalin compared to artinM. The endpoint of artinM isolation was at a constant absorbance of the effluent, assuming that jacalin was bound completely onto the matrix beads because of its galactose preferential affinity. Noting that guar gum is a polysaccharide composed of D-galactose side-chain linkage at every two D-mannose units,37 its sidechain, therefore, contributed to jacalin removal from the extract.
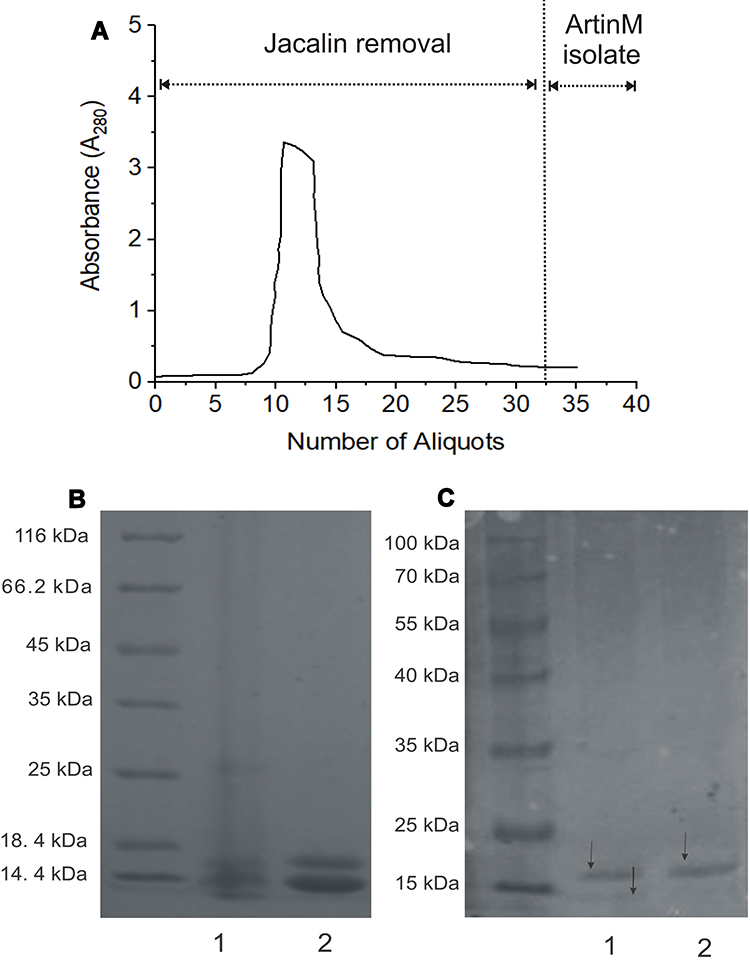 |
Figure 2 ArtinM isolation: (A) Absorbance changes of cempedak seeds aliquot during sequential adsorption of jacalin on guar gum beads, (B) Coomassie-stained SDS–PAGE gel of: crude extract (1) and jacalin dominant effluent (2), (C) Coomassie-stained SDS–PAGE gel of the two latest effluents of artinM isolate. |
This finding is supported by results obtained from the SDS-PAGE analysis given in Figures 2B and 2C. Figure 2B shows distinct two bands representing heavy and light chains at 12–15 kDa in SDS-PAGE electrophoresis gel, which is consistent with the previous report,30 indicating the presence of jacalin in both the crude extract and jacalin dominant effluent. Further adsorption to guar gum beads, a complete artinM separation with jacalin was shown by a single band at about 16 kDa (Figure 2C). This agrees well with previous reports stated that artinM is a homotetramer glycoprotein having a molecular weight of approximately 64–65 kDa.29,38 In the absence of jacalin, excessive adsorption of artinM isolate to the beads should be prevented to avoid strong binding of artinM on the matrix. The concentration of artinM was 14.83 ± 0.7% w/w of freeze-dried isolate powder.
ArtinM was quantified using haemagglutination assay which is generally applied for lectin determination.31 The activity of artinM isolate analyzed by haemagglutination test at pH 7.4 showed a minimum concentration of 1.95 µg/mL. The activity remained stable at pH 4 and 8 (Figure 3A). This characteristic was beneficial from the preparation of lipid nanoparticles where utilizing acidic pH to provide a positive charge of artinM for phospholipid electrostatically binding. On the contrary, the decrease of artinM activity was observed in contact with organic solvents, namely ethyl acetate and ethanol, as shown by an increase of the minimum concentration of haemagglutination reaction to 31.25 µg/mL (Figure 3A). Partial protein denaturation in the solvent-water interface may result in this phenomenon.39 Therefore, the organic solvent used in the preparation process of nanoparticles should be evaporated before artinM adsorption process.
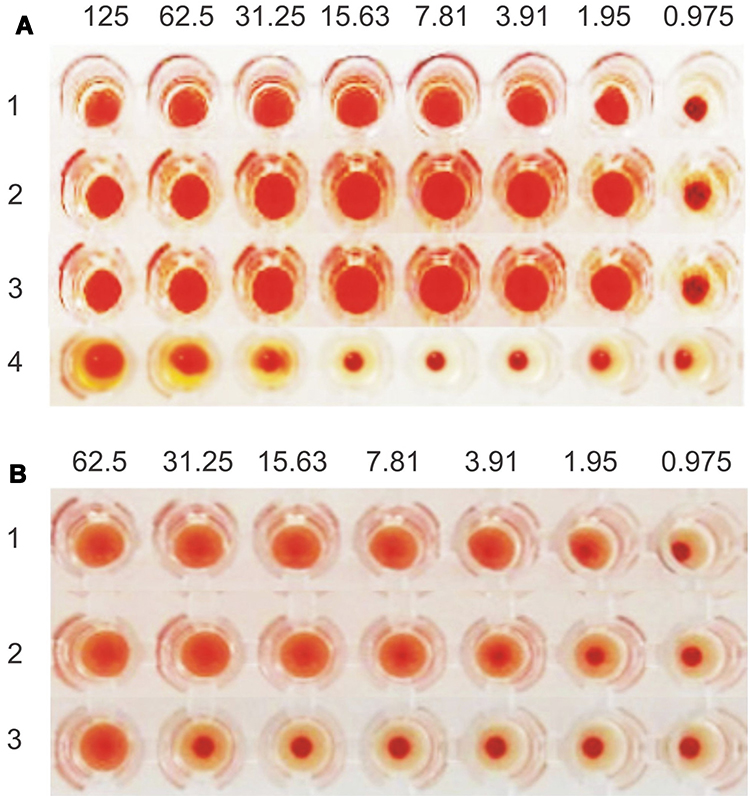 |
Figure 3 ArtinM characterization: (A) Haemagglutination test at pH 7.4 (1), pH 4 (2), pH 8 (3), and ethyl acetate-ethanol solvent mixture (4), and (B) Haemagglutination inhibition test without saccharide (1), with the addition of D-galactose 0.4 M (2) and D-mannose 0.4 M (3). |
Besides the difference in molecular structure, the presence of artinM in the lectin isolate can be distinguished from jacalin by inhibition of haemagglutination test in the presence of a certain saccharide. The binding of artinM to mannose is more prominent compared to jacalin, albeit it polyspecific recognition that also reacts to Glc, Gal, GalNAc, Neu5Ac, and MurNAc.38 ArtinM activity inhibition had been tested by the addition of D-mannose, D-galactose, and alpha-methyl-D-mannoside.39 ArtinM isolate was subjected to haemagglutination inhibition test by adding 0.4 M D-mannose and 0.4 M D-galactose (Figure 3B). It was found that artinM isolate preferentially bound to D-mannose, which is recognized as a dramatic change of the minimum haemagglutination activity from 1.95 µg/mL to 62.5 µg/mL. On another hand, haemagglutination inhibition reaction by D-galactose could not be found, assuring the success in separating artinM from jacalin.
Rifampicin Nanoparticles Surface Modified with ArtinM (Rif-NP-AM) Formula
In the nanoparticle formula, the lecithin structure was modified by adding oleic acid to increase the solubility of rifampicin in the phospholipid core, and hence improving the entrapment efficiency. The same purpose has been conducted by Şenyiğit et al, in which isopropyl myristate was added to solubilize clobetasol within lecithin nanoparticles.41 Our preliminary study found that the solubility of rifampicin in oleic acid was four times higher than other oil, such as isopropyl myristate, castor oil, myritol, olive oil, and virgin coconut oil, which reached ~40 mg/mL.
Therefore, we used a mixture of lecithin and oleic acid at the ratio of 2:1 to produce a homogenous lipid mixture with good physical stability of nanoparticles. In the excess amount of oleic acid, the nanoparticles tend to be creaming due to lipid separation. Since lecithin is insoluble in water, it formed a spherical colloidal dispersion with the addition of polysorbate 80. Surface modification of the nanoparticles were conducted using the electrostatic binding of positively charged of artinM with negatively charged of lecithin phosphate and carboxyl groups of oleic acid. Considering the isoelectric point at 5–6.5,42 the positively charged of artinM was provided by adjusting deionized water at pH 4, while this pH was not detrimental to the encapsulated rifampicin.33
The nanoparticle formula was optimized by employing the surface response methodology.43 Factors determining nanoparticle characteristics included lecithin-oleic acid, rifampicin, and polysorbate 80 concentrations. ArtinM was not included in the optimization because it was adsorbed on the surface after nanoparticle formation. The responses analyzed in this experiment were particle size, entrapment efficiency, and drug loading as shown in Table 2. These nanoparticles were designed to mimic the entry pathway of pathogenic microbes; therefore, we selected the particle size in the range of 200–600 nm to provide cellular uptake.44 The nanoparticles resulted from 15 runs showed a relatively homogeneous size distribution by considering PDI less than 0.7. This finding is in line with that stated in ISO 22,412:2017 that stated a sample with PDI greater than 0.7 has a wide particle size distribution.45 From the Box-Behnken design given by Equation (1), the particle size (Y1) followed Equation (5).
Y1=452 + 133 X1 + 899 X2 – 284 X3 + 1476 X12 – 1158 X22 + 88.8 X32 – 1051 X12 – 462 X13 + 34 X23 (5)
From the ANOVA in Table 3, it is shown that the factor affecting the particle size significantly was polysorbate 80 (X3) as revealed by the p-value less than 0.05. Polysorbate 80 helped to decrease the surface tension of lecithin,46 hence reducing the particle size. This could have resulted from the arrangement of polysorbate 80 molecules on lipid-oil mixture comprising lecithin and oleic acid. Because of its hydrophilicity, polysorbate 80 was on the interface of lipid nanoparticle and formed a denser film packing, preventing the cohesion between the particles. Thus, it explained the mechanism of the addition of polysorbate 80 to prevent the growth of lipid particles.47,48 At a certain concentration of the mixture of lecithin and polysorbate 80, the particle size was reduced almost significantly as indicated by the p-value of 0.058. Lecithin was used as a major component governing the accomplishment of antibiotic delivery. At concentrations less than 5%, lecithin worked synergically with polysorbate 80 in suppressing surface tension to reduce the particle size. On the contrary, lecithin concentrations higher than 5% dominated lipid properties on the interface, hence eliminating polysorbate 80 functions. The particle size characteristics are given in Figures 4A–C. The goal of this study to decrease the particle size had been successfully achieved as shown by the presence of the spherical region in the contour plot in Figure 4B and the desired core particle size in the range of 100–300 nm. Instead of lowering the surface tension, the use of an excessive amount of polysorbate 80 may result in the formation of large particle aggregates, thus, it should be used sufficiently.49
 |
Table 3 ANOVA Analysis of Nanoparticle Formula Optimization Using Box-Behnken Design |
 |
Figure 4 Contour plots of Rif-NP formula using Box-Behnken design for responses: (A–C) particle size (nm), (D–F) % entrapment efficiency, and (G–I) % drug loading. |
In term of entrapment efficiency (Y2), the Box-Behnken design given in Equation (1) resulted in Equation (6):
Y2 =119.3–69.6 X1 – 414 X2 – 30.1 X3 + 27 X12 + 840 X22 + 1.88 X32 + 55 X12 + 21.1 X13 + 41.4 X23 (6)
The entrapment efficiency of rifampicin was significantly influenced by the amount of lecithin-oleic acid (X1) and the amount of rifampicin (X2) with the p-value less than 0.05 as given in Table 3. The contour plots illustrated in Figure 4D-F tell that total lipid was significantly affected by the ability to encapsulate rifampicin ranging from 25% to 50%. A higher amount of lipids resulted in an increase of rifampicin entrapment. This result was in agreement with the finding of increasing progesterone, a lipid-soluble drug, and entrapment in lecithin nanoparticles.20
Polysorbate 80 in combination with lecithin showed concentration-dependent on the entrapment efficiency of rifampicin. At the high concentration of both polysorbate 80 and lecithin, rifampicin was efficiently entrapped in the nanoparticles. Whilst high polysorbate 80 in low lecithin content depressed rifampicin solubility in the nanoparticles, which is shown by the lowest entrapment efficiency in Figure 4E. This is due to rifampicin loss to the water phase at the excess amount of polysorbate 80 hence leading to micelles formation.32 Drug loading represents the ratio of the weight of the drug to the total weight of the formula. For the lipid-soluble rifampicin, the drug loading of nanoparticles (Y3) is proportional to the entrapment efficiency (Y2) as expressed in Equation (7).
Y3 =40.4–73.7 X1- 25.6 X2 – 10.94 X3 + 51.8 X12 + 255.1 X22 + 0.622 X32 – 95.5 X12 + 10.55 X13 + 10.80 X23 (7)
The drug loading was varied up to 26.10%. The capability of lipid nanoparticles to encapsulate rifampicin primarily depended on total lipid and rifampicin concentrations (p<0.05). Polysorbate 80 also contributed to the decrease of entrapment efficiency, as previously mentioned and hence the percentage of drug loading fluctuated. Increasing the concentration of active substances also increases drug loading, but only up to the point which increasing it would yield negative results. Based on the contour plots, it could generally be shown that increasing the concentration of the lecithin-oleic acid mixture and rifampicin, as well as balancing the amount of surfactant, increased the drug loading capacity within the nanoparticles as given in Figure 4G-I.
According to the desired properties, the optimum formula determined by Box-Behnken design consisted of 0.6% lecithin, 0.3% oleic acid, 0.3% rifampicin, 0.006% artinM, and 2% polysorbate 80. Using the optimized formula, the nanoparticles were prepared. The responses, namely the particle size, entrapment efficiency, and drug loading, were then evaluated. They are compared with those predicted from the Box-Behnken design as shown in Table 4. It was found that the deviations of the responses are low (less than 5%) indicating the design is reliable and reproducible.
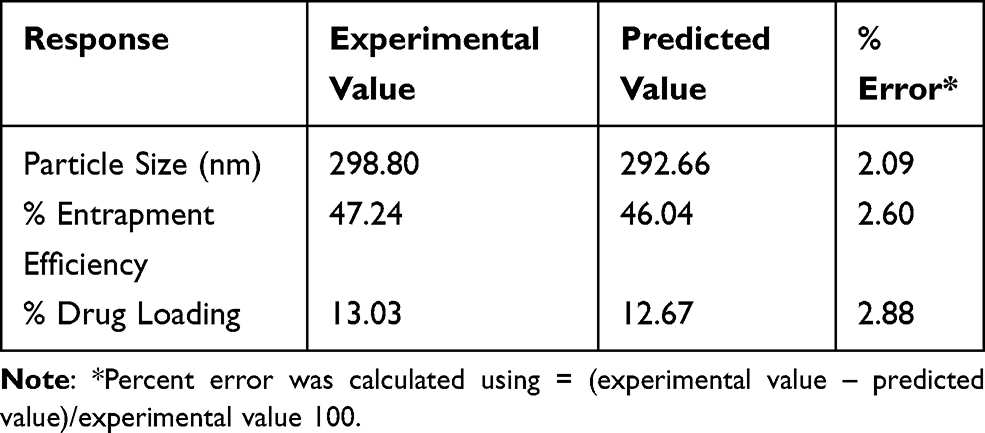 |
Table 4 Comparison of the Experimental and Predicted Value from the Optimized Formulas of Box-Behnken Design |
Rif-NP-AM Evaluation
From the optimized formula, artinM was adsorbed after the nanoparticles formed. To confirm the presence of artinM, the nanoparticles were separated with surfactants and subjected to haemagglutination test. It was shown that the artinM in the nanoparticles had a higher activity, which was demonstrated by the small artinM concentration in the nanoparticles (15.63–3.25 µg/mL) compared to the supernatant (125–250 µg/mL) for blood agglutination. This proved the accomplishment of artinM surface modification, which was revealed by the higher amount of adsorbed artinM. Furthermore, the change of zeta potential of the nanoparticles from −2 mV to 0.8 mV supported the conclusion that the positively charged of artinM at pH 4 was successfully bound to negatively charged of nanoparticles. This is in accordance with the results reported by Liu et al.50
The morphology of rifampicin nanoparticles with and without artinM modification revealed by TEM imaging is shown in Figure 5A-D. The artinM modification of nanoparticles was evidenced by a core-shell particle as illustrated in Figure 5B and D. Similar morphology was found in artinM modified lipid nanoparticles without rifampicin, however, these particles had a smaller size. The size of rifampicin loaded nanoparticles (RIF-NP) was similar to the optimized size in the Box-Behnken design, with a core size domain in the range of 100–300 nm. The grafted artinM indicated from a layered shell surrounding the core nanoparticles resulted in an increase in particle size (Figure 5D and E). The size of Rif-NP-AM was in the designed range of 200–600 nm, suggesting a potential vehicle for cellular engulfment.44
 |
Figure 5 TEM images showing the morphology of (A) lipid nanoparticles (core) (NP), (B) modified with artinM (NP-AM), (C) rifampicin loaded unmodified nanoparticles (Rif-NP), (D) rifampicin loaded modified artinM (Rif-NP-AM), (E) Particle size data from PSA; PI, polydispersity index, (F) Release profiles of rifampicin. |
The successful delivery of rifampicin for treating intracellular infection requires nanoparticles with targeting properties. Endocyted nanoparticle can release rifampicin in intracellular milieu of S. aureus which persist within endosome or escape to the cytoplasm of the host cell.8 The pH value of cytoplasm and endosome of host cells were reported to be 7.4 and 5.2, respectively.51 The in vitro release of rifampicin from nanoparticle grafted and ungrafted artinM is shown in Figure 5F. The percentage of rifampicin release from nanoparticles modified with artinM (Rif-NP-AM) at pH 7.4 and 5.2 showed linear release profiles over time up to 55% and 37% for 30 hours. A similar release profile was found in drug release from unmodified nanoparticles (Rif-NP) at both pHs, indicating that surface modification did not change the release profile. The release rate of free rifampicin as a control was significantly higher at both pH, compared to those from NP and NP-AM, It implied that lipid nanoparticles as the primary diffusion barrier within the nanoparticles leading to the controlled release profile. This finding is in accordance with the in vitro release studied by Guo et al which employed lecithin polymer composite for rifampicin delivery.52 During the preparation of nanoparticles, rifampicin was added to the lipid-surfactant phase where it completely dissolve. Hence, this prolonged and slow release behaviour can be explained by slow diffusion of rifampicin from the lipid core.
Lipid metabolism occurs in the body was not mimicked by the in vitro study, resulting in a slow released of the encapsulated rifampicin in the dissolution media, which may follow a passive diffusion release mechanism. The slow release in dissolution media is advantageous, since it may prevent antibiotic resistance due to a premature release in extracellular. In the body, the nanoparticles release will be enhanced in the intracellular by endosomal degradation. The enzymatic hydrolysis of lipid nanoparticle will optimize the drug release in intracellular pathogenic environments. The enzymatic metabolism of lipids in the in vivo environment will improve the release profile. This result agrees with the findings of John et al, in which the faster release of rifampicin in the enzymatic supplemented media in the in vitro study increased significantly the drug release.53
The significant difference (p < 0.05) of release rate may result in the higher solubility of rifampicin at physiological pH. This finding is in agreement to Rodrigues et al, who mentioned that partially ionized (~40%) of rifampicin at pH 7.4 leads to an increase of solubility.54,55 From these results, higher rifampicin release percentage at pH 7.4 was demonstrated by the feasibility of potential drug delivery in macrophage compartments which was more compatible against cell invaded S. aureus. The inhibition of autophagosome maturing could allow S. aureus to escape to the cytoplasm and lead to the death of the host cell.56 The nature of Rif-NP-AM as a lipid nanoparticle may minimize the chance of drug to release in the general circulation. Moreover, various artinM activities to induce NF-κB and pro-inflammatory production may increase cellular engulfment.24 Thus, it would release rifampicin in the vicinity of intracellular pathogen which may potentiate rifampicin activity against the invaded microbes.
Antimicrobial Activity Test
Rif-NP-AM activity against extracellular S. aureus was conducted using a microdilution method adopted from the Clinical and Laboratory Standards Institute.34 The minimum inhibition concentration (MIC) occurred at 0.781 µg/mL, while the minimum bactericidal concentration (MBC) at 1.563 µg/mL. A similar concentration was also found in the free rifampicin solution. It implied that the process of nanoparticle preparation was not deleterious to the encapsulated rifampicin, even though involving the process of artinM adsorption at pH 4. Furthermore, it also showed that encapsulation did not inhibit rifampicin activity against S. aureus considering that lipase produced by the bacteria may degrade lipid in the formula to liberate the drug.57
Nanoparticle Internalization into the Cell
Lecithin has been purposed for intracellular trafficking of various drugs.20,21,41 For internalization study, a fluorochrome was employed to visualize the Rif-NP-AM using a confocal microscope following previous reports.58–60 Balb/3T3 fibroblast cell selected in this study is among other cells used to evaluate intracellular S. aureus infection.61,62 Fibroblast cell is clinically found invaded by S. aureus in a chronic skin wound.2 The images and emission intensities given in Figure 6 shows that both Rif-NP and Rif-NP-AM were internalized into the cell after 30 minutes of incubation. Interestingly, Rif-NP-AM was internalized more as represented by a higher intensity of fluorescent (Figure 6B). Red dots within the cytoplasm represented the engulfment of Rif-NP-AM and did not penetrate to the nucleus, which was shown by the lowest emission in this area, indicating potential delivery with low toxicity for the host.
 |
Figure 6 Confocal microscope images and emission intensity curves of Nile-red labelled nanoparticles: (A) without artinM (Rif-NP) and (B) with artinM (Rif-NP-AM) incubated in fibroblast cell culture. |
To kill intracellular bacteria, the endosomal released rifampicin can diffuse to the surrounding endosomes and cytoplasm. The soluble rifampicin will serve against the persistent bacteria located in the endosome or escaped in the cytoplasm. Interestingly, ArtinM ability to induce NO and other pro-inflammatory production can potentially increase host cell activity against the invaded pathogens. According to Da Silva et al (2016), artinM has been shown to eradicate Paracoccidioides brasiliensis infected macrophage as an intracellular pathogen, as well as induced the production of T-helper activity and pro-inflammatory cytokine.25 The use of artinM modification in rifampicin loaded nanoparticles is potentially increasing antibiotic targeting and boost host cell immunity for combating intracellular infection.
Internalized S. aureus transforms into a small colony variance (SCV) phenotype, becomes dormant, and inhibits host cellular defense. The SCV is known to invade the phagocytic and non-phagocytic cells such as myoblasts, osteoblasts, cardiomyocytes. Therefore, Rif-NP-AM could be applied as an antibiotic vehicle for emerging intracellular infections causing chronic and deadly diseases, such as endocarditic, diabetic ulcers, and osteomyelitis.
Conclusion
A versatile method for lectin surface modification onto soy phospholipid nanoparticles has been developed. Rifampicin was encapsulated within the lipid nanoparticles prior to the modification process, and thus its stability was maintained. ArtinM was successfully adsorbed after the solidification process of lipid nanoparticles at pH 4. More than 80% of artinM was adsorbed on the nanoparticles, evidenced by the lower haemagglutination concentration of adsorbed fraction compared to unadsorbed fraction, the changes of zeta potential of nanoparticles, and the presence of a layered shell surrounding the core nanoparticles found in TEM images. High emission of Nile-red labeled nanoparticles found in fibroblast cells imaged by a confocal microscope showed success in transporting the particles across the cells. These results imply a promising effect of the surface-modified artinM on phospholipid nanoparticles for traversing host cells in intracellular infection therapy. Research is being conducted to prove the hypothesis of artinM modified nanoparticles in regaining host immune response along with increasing phagocytosis.
Acknowledgments
This research was supported by “Desentralisasi PDUPT” research grant, Directorate of Research and Community Engagement, The Ministry of Research, Technology, and Higher Education, the Republic of Indonesia. We sincerely appreciate Ms. Fatma Ayatiliulil Albab and Ms. Dewi Satwika for assisting confocal microscope imaging.
Disclosure
Prof. Dr. Khairurrijal Khairurrijal reports grants from Institut Teknologi Bandung, during the conduct of the study and grants, personal fees, and non-financial support from Institut Teknologi Bandung, outside the submitted work. The authors report no other potential conflicts of interest in this work.
References
1. Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74(3):417–433. doi:10.1128/MMBR.00016-10
2. Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28(3):603–661. doi:10.1128/CMR.00134-14
3. Sakr A, Brégeon F, Mège J-L, Rolain J-M, Blin O. Staphylococcus aureus Nasal Colonization: an Update on Mechanisms, Epidemiology, Risk Factors, and Subsequent Infections. Front Microbiol. 2018;9:2419. doi:10.3389/fmicb.2018.02419
4. Struthers JK. Clinical Microbiology.
5. Zhang S, Sun X, Chang W, Dai Y, Ma X, de Lencastre H. Systematic review and meta-analysis of the epidemiology of vancomycin-intermediate and heterogeneous vancomycin-intermediate Staphylococcus aureus isolates. PLoS One. 2015;10(8):e0136082. doi:10.1371/journal.pone.0136082
6. Parer S, Lotthé A, Chardon P, Poncet R, Jean-Pierre H, Jumas-Bilak E. An Outbreak of Heterogeneous Glycopeptide-Intermediate Staphylococcus aureus Related to a Device Source in an Intensive Care Unit. Infect Control Hosp Epidemiol. 2012;33(2):167–174. doi:10.1086/663703
7. Huang SS, Singh R, McKinnell JA, et al. Decolonization to Reduce Postdischarge Infection Risk among MRSA Carriers. N Engl J Med. 2019;380(7):638–650. doi:10.1056/NEJMoa1716771
8. Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17(2):59–65.
9. Gunaratnam G, Tuchscherr L, Elhawy MI, et al. ClpC affects the intracellular survival capacity of Staphylococcus aureus in non-professional phagocytic cells. Sci Rep. 2019;9(1):1–2.
10. Proctor RA, Kriegeskorte A, Kahl BC, Becker K, Löffler B, Peters G. Staphylococcus aureus Small Colony Variants (SCVs): a road map for the metabolic pathways involved in persistent infections. Front Cell Infect Microbiol. 2014;4:99.
11. Abed N, Saïd-Hassane F, Zouhiri F, et al. An efficient system for intracellular delivery of beta-lactam antibiotics to overcome bacterial resistance. Sci Rep. 2015;5:13500.
12. Jamaledin R, Yiu CKY, Zare EN, et al. Advances in Antimicrobial Microneedle Patches for Combating Infections. Adv Mater. 2020;32(33):e2002129.
13. Abed N, Couvreur P. Nanocarriers for antibiotics: a promising solution to treat intracellular bacterial infections. Int J Antimicrob Agents. 2014;43(6):485–496.
14. Alt V, Kirchhof K, Seim F, et al. Rifampicin–fosfomycin coating for cementless endoprostheses: antimicrobial effects against methicillin-sensitive Staphylococcus aureus (MSSA) and methicillin-resistant Staphylococcus aureus (MRSA). Acta Biomater. 2014;10(10):4518–4524.
15. Eisen DP, Denholm JS. Recommendations for rifampicin therapy of staphylococcal infection in Infectious Diseases Society of America prosthetic joint infection guidelines are not supported by available literature. Clin Infect Dis. 2013;57(1):159–160.
16. Mohamed W, Sommer U, Sethi S, et al. Intracellular proliferation of Staphylococcus aureus in osteoblasts and effects of rifampicin and gentamicin on S. aureus intracellular proliferation and survival. Eur Cell Mater. 2014;28:258–268.
17. Loss G, Simoes P, Valour F, et al. Staphylococcus aureus small colony variants (SCVs): news from a chronic prosthetic joint infection. Front Cell Infect Microbiol. 2019;9:363.
18. Firsov AA, Golikova MV, Strukova EN, et al. Pharmacokinetically-based prediction of the effects of antibiotic combinations on resistant Staphylococcus aureus mutants: in vitro model studies with linezolid and rifampicin. J Chemother. 2017;29(4):220–226.
19. Chaubey P, Mishra B. Mannose-conjugated chitosan nanoparticles loaded with rifampicin for the treatment of visceral leishmaniasis. Carbohydr Polym. 2014;101:1101–1108.
20. Asprea M, Tatini F, Piazzini V, Rossi F, Bergonzi MC, Bilia AR. Stable, monodisperse, and highly cell-permeating nanocochleates from natural soy lecithin liposomes. Pharmaceutics. 2019;11(1):34.
21. Le NTT, Cao VD, Nguyen TNQ, Le TTH, Tran TT, Thi TTH. Soy lecithin-derived liposomal delivery systems: surface modification and current applications. Int J Mol Sci. 2019;20(19):4706.
22. Martínez-Carmona M, Lozano D, Colilla M, Vallet-Regí M. Lectin-conjugated pH-responsive mesoporous silica nanoparticles for targeted bone cancer treatment. Acta Biomater. 2018;65:393–404.
23. Wang C, Ho PC, Lim LY. Wheat germ agglutinin-conjugated PLGA nanoparticles for enhanced intracellular delivery of paclitaxel to colon cancer cells. Int J Pharm. 2010;400(1–2):201–210.
24. Berger S, Dyugovskaya L, Polyakov A, Lavie L. Short-term fibronectin treatment induces endothelial-like and angiogenic properties in monocyte-derived immature dendritic cells: involvement of intracellular VEGF and MAPK regulation. Eur J Cell Biol. 2012;91(8):640–653.
25. Da Silva TA, Alv Z-F, Cecílio NT, Sardinha-Silva A, Fernandes FF, Roque-Barreira MC. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci Rep. 2017;7(1):7083.
26. Musilova J, Mulcahy ME, Kuijk MM, McLoughlin RM, Bowie AG. Toll-like receptor 2–dependent endosomal signaling by Staphylococcus aureus in monocytes induces type I interferon and promotes intracellular survival. J Biol Chem. 2019;294(45):17031–17042.
27. Umerska A, Matougui N, Groo A-C SP. Understanding the adsorption of salmon calcitonin, antimicrobial peptide AP114 and polymyxin B onto lipid nanocapsules. Int J Pharm. 2016;506(1–2):191–200.
28. Roy I, Sardar M, Gupta MN. Cross-linked alginate–guar gum beads as fluidized bed affinity media for purification of jacalin. Biochem Eng J. 2005;23(3):193–198.
29. Barbosa-Lorenzi VC, Cecilio NT, de Almeida Buranello PA, et al. Recombinant ArtinM activates mast cells. BMC Immunol. 2016;17(1):22.
30. Geraldino TH, Modiano P, Veronez LC, Flória-Santos M, Garcia SB, Jacalin Has P-D-SG. Chemopreventive Effects on Colon Cancer Development. Biomed Res Int. 2017;2017:4614357.
31. Sano K, Ogawa H. Hemagglutination (inhibition) assay. Methods Mol Biol. 2014;1200:47–52.
32. Yanasarn N, Sloat BR, Cui Z. Nanoparticles engineered from lecithin-in-water emulsions as a potential delivery system for docetaxel. Int J Pharm. 2009;379(1):174–180.
33. Rajaram S, Vemuri VD, Natham R. Ascorbic acid improves stability and pharmacokinetics of rifampicin in the presence of isoniazid. J Pharm Biomed Anal. 2014;100:103–108.
34. CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically. M100. Wayne, PA: CLSI; 2020.
35. Cardoso MR, Mota CM, D P R, et al. ArtinM, a D-mannose-binding lectin from Artocarpus integrifolia, plays a potent adjuvant and immunostimulatory role in immunization against Neospora caninum. Vaccine. 2011;29(49):9183–9193.
36. Bidula S, Sexton DW, Yates M, et al. H‐ficolin binds A spergillus fumigatus leading to activation of the lectin complement pathway and modulation of lung epithelial immune responses. Immunology. 2015;146(2):281–291.
37. Pegg AM. Natural Food Additives, Ingredients and Flavourings. Cambridge: Woodhead Publishing; 2012.
38. Cecílio NT, Carvalho FC, Liu Y, et al. Yeast expressed ArtinM shares structure, carbohydrate recognition, and biological effects with native ArtinM. Int J Biol Macromol. 2016;82:22–30.
39. Del Castillo-Santaella T, Peula-García JM, Maldonado-Valderrama J, Jódar-Reyes AB. Interaction of surfactant and protein at the O/W interface and its effect on colloidal and biological properties of polymeric nanocarriers. Colloids Surf B Biointerfaces. 2019;173:295–302.
40. de Almeida Buranello PA, Moulin MR, Jr DA S, Jamur MC, Roque-Barreira MC, Oliver C. The lectin ArtinM induces recruitment of rat mast cells from the bone marrow to the peritoneal cavity. PLoS One. 2010;5(3):e9776.
41. Şenyiğit T, Sonvico F, Barbieri S, Ö Ö, Santi P, Colombo P. Lecithin/chitosan nanoparticles of clobetasol-17-propionate capable of accumulation in pig skin. J Control Release. 2010;142(3):368–373.
42. Misquith S, Rani PG, Surolia A. Carbohydrate binding specificity of the B-cell maturation mitogen from Artocarpus integrifolia seeds. J Biol Chem. 1994;269(48):30393–30401.
43. Myers RH, Montgomery DC, Anderson-Cook CM. Response Surface Methodology: Process and Product Optimization Using Designed Experiments.
44. Oyewumi MO, Kumar A, Cui Z. Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses. Expert Rev Vaccines. 2010;9(9):1095–1107.
45. International standards organizations. ISO 22,412:2017- Particle size analysis — Dynamic light scattering (DLS). 2017.
46. Zhang J, Fan Y, Smith E. Experimental design for the optimization of lipid nanoparticles. J Pharm Sci. 2009;98(5):1813–1819.
47. Riehm DA, Rokke DJ, Paul PG, Lee HS, Vizanko BS, McCormick AV. Dispersion of oil into water using lecithin-Tween 80 blends: the role of spontaneous emulsification. J Colloid Interface Sci. 2017;487:52–59.
48. Nikiforidis CV, Kiosseoglou V. Competitive displacement of oil body surface proteins by Tween 80 – effect on physical stability. Food Hydrocoll. 2011;25(5):1063–1068.
49. Helgason T, Awad TS, Kristbergsson K, McClements DJ, Weiss J. Effect of surfactant surface coverage on formation of solid lipid nanoparticles (SLN). J Colloid Interface Sci. 2009;334(1):75–81.
50. Liu Y, Wang P, Sun C, et al. Wheat germ agglutinin-grafted lipid nanoparticles: preparation and in vitro evaluation of the association with Caco-2 monolayers. Int J Pharm. 2010;397(1–2):155–163.
51. Liu Q, Chen X, Jia J, et al. pH-responsive poly (D, L-lactic-co-glycolic acid) nanoparticles with rapid antigen release behavior promote immune response. ACS Nano. 2015;9(5):4925–4938.
52. Guo P, Xue HY, Buttaro BA, Tran NT, Wong HL. Enhanced eradication of intracellular and biofilm-residing methicillin-resistant Staphylococcus aureus (MRSA) reservoirs with hybrid nanoparticles delivering rifampicin [published online ahead of print, 2020 Aug 30]. Int J Pharm. 2020;589:119784.
53. John JV, Thomas RG, Lee HR, Chen H, Jeong YY, Phospholipid End‐Capped KI. Acid‐Degradable Polyurethane Micelles for Intracellular Delivery of Cancer Therapeutics. Adv Healthc Mater. 2016;5(15):1874–1883.
54. Rodrigues C, Gameiro P, Prieto M, De Castro B. Interaction of rifampicin and isoniazid with large unilamellar liposomes: spectroscopic location studies. Biochim Biophys Acta Gen Subj. 2003;1620(1–3):151–159.
55. Khan MF, Rita SA, Kayser MS, et al. Theoretically Guided Analytical Method Development and Validation for the Estimation of Rifampicin in a Mixture of Isoniazid and Pyrazinamide by UV Spectrophotometer. Front Chem. 2017;5:27.
56. Mestre MB, Colombo MI. Staphylococcus aureus promotes autophagy by decreasing intracellular cAMP levels. Autophagy. 2012;8(12):1865–1867.
57. Hu C, Xiong N, Zhang Y, Rayner S, Chen S. Functional characterization of lipase in the pathogenesis of Staphylococcus aureus. Biochem Biophys Res Commun. 2012;419(4):617–620.
58. Nam HY, Kwon SM, Chung H, et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2009;135(3):259–267.
59. Rima W, Sancey L, Aloy MT, et al. Internalization pathways into cancer cells of gadolinium-based radiosensitizing nanoparticles. Biomaterials. 2013;34(1):181–195.
60. Pridgen EM, Alexis F, Kuo TT, et al. Transepithelial transport of Fc-targeted nanoparticles by the neonatal fc receptor for oral delivery. Sci Transl Med. 2013;5(213):213ra167–213ra167.
61. Löffler B, Tuchscherr L, Niemann S, Peters G. Staphylococcus aureus persistence in non-professional phagocytes. Int J Med Microbiol. 2014;304(2):170–176.
62. Hanses F, Kopp A, Bala M, et al. Intracellular survival of Staphylococcus aureus in adipocyte-like differentiated 3T3-L1 cells is glucose dependent and alters cytokine, chemokine, and adipokine secretion. Endocrinology. 2011;152(11):4148–4157.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.