Back to Journals » Drug Design, Development and Therapy » Volume 20
Artificial Intelligence in Selected Domains of Drug Discovery: A Critical Narrative Review
Authors Bin Abdulqader MI, Alsaqabi AA, Mohammed AE, Alghamdi SS ![]()
Received 5 March 2026
Accepted for publication 22 May 2026
Published 19 June 2026 Volume 2026:20 607228
DOI https://doi.org/10.2147/DDDT.S607228
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Marwah I Bin Abdulqader,1 Abdulrhman A Alsaqabi,1 Afrah E Mohammed,2,3 Sahar S Alghamdi1,4,5
1Department of Pharmaceutical Sciences, College of Pharmacy, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia; 2Department of Biology, College of Science, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia; 3Microbiology and Immunology Unit, Natural and Health Sciences Research Center, Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia; 4Medical Research Core Facility and Platforms, King Abdullah International Medical Research Center (KAIMRC), Ministry of the National Guard Health Affairs, Riyadh, Saudi Arabia; 5King Abdulaziz Medical City, Ministry of the National Guard Health Affairs, Riyadh, Saudi Arabia
Correspondence: Sahar S Alghamdi, Department of Pharmaceutical Sciences, College of Pharmacy, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia, Tel +966 (11) 4299999, Ext 99516, Email [email protected]
Introduction: Traditional drug discovery is historically characterized by high attrition rates, escalating financial costs, and decades-long development timelines. As global health challenges—particularly antimicrobial resistance and complex malignancies—intensify, the urgent need for innovative and accelerated therapeutic solutions has never been more critical. Artificial Intelligence (AI) has emerged as a supportive computational framework to address these fundamental bottlenecks, offering advanced computational capabilities to navigate vast chemical spaces and optimize molecular design. While AI-based approaches have demonstrated encouraging performance in specific preclinical settings, their practical impact and limitations require careful, objective evaluation. This critical narrative review examines the application of various artificial intelligence technologies in the design and development of antibiotics, anticancer agents, antibodies, and small-molecule drugs, spanning methodologies from conventional machine learning (ML) to advanced deep learning (DL) models.
Methods: A narrative review of studies reporting applications of artificial intelligence in drug discovery and development. It encompassed articles published between 2000 and 2026 and was informed by literature retrieved from multiple electronic databases. The selected studies focused on AI applications in antibiotics, anticancer agents, antibodies, and small-molecule discovery and development. Studies published before 2000, incomplete reports, or those not directly related to pharmaceutical applications of AI were not considered. Review or meta-analysis articles were also excluded from the primary results, though utilized for background context. Although the inclusion criteria covered studies from 2000 to 2026, one earlier study published before 2000 was also included to provide historical context for the early development of neural network applications in molecular biology.
Results and conclusion: The reviewed literature demonstrates that AI has transitioned from a theoretical concept to a useful framework in early-stage drug discovery, particularly in virtual screening and lead optimization. However, this review identifies a significant “translational gap”; most AI applications remain confined to computational settings, facing challenges in data quality, model interpretability, and a lack of prospective clinical validation. We conclude that while AI significantly accelerates computational efficiency and hypothesis generation, realizing its full potential to combat pressing global health threats requires rigorous experimental integration, standardized data governance, and continuous human expertise to ensure therapeutic efficacy and safety.
Keywords: artificial intelligence, AI in drug discovery, anticancer, antibiotics, antibodies, small molecules
Introduction
The use of technological tools based on theoretical foundations to address complex problems has been increasingly explored in recent years. As the world continues to witness rapid technological growth, newly developed computational tools designed to achieve defined goals have received significant attention. Specifically, artificial intelligence (AI) has emerged as a key framework to support and accelerate drug discovery processes.1 This interest has been driven by advances in understanding human intelligence and learning capabilities, and by efforts to translate aspects of this knowledge into computational systems aimed at supporting and accelerating complex processes. To achieve these objectives, a wide range of computer-controlled technologies capable of enhancing data-driven reasoning and task execution have been developed and collectively referred to as artificial intelligence (AI). The implementation of AI across various fields has contributed to improvements in efficiency and development, supporting more advanced analytical outcomes. The healthcare sector, in particular, has experienced growing integration of AI technologies across multiple applications, ranging from drug discovery to procedural implementation. This increasing adoption of AI reflects its ability to acquire, structure, and apply knowledge to support development and assist in addressing complex problems.2
Over the years, the discovery and development of therapeutic agents has become an increasingly active area for computational approaches, reflecting the growing complexity of modern drug research. Drug discovery is a multidisciplinary field focused on identifying new therapeutic molecules for specific biological targets.3 It is widely recognized as being associated with high costs, extended development timelines, and relatively low success rates, which ensure the need for more efficient and supportive strategies.4 Despite many years of scientific advancement, drug discovery remains a demanding process. In many cases, it may take more than a decade for a promising molecule to progress from early laboratory testing to clinical evaluation, and only a limited number of candidates ultimately succeed. The combination of long development timelines, substantial financial investment, and increasing biological complexity has highlighted the inherent limitations of traditional computational approaches. While conventional statistical models such as classical QSAR have provided valuable support in early-stage screening, they are often challenged when attempting to capture multifactorial biological behavior, including efficacy, safety, and pharmacokinetic performance. Within this context, artificial intelligence—particularly deep learning—has emerged not as a replacement for experimental research, but as a complementary framework capable of integrating complex chemical and biological data to support more informed and efficient decision-making throughout both drug discovery and subsequent development stages.5
Traditionally, large-scale chemical libraries are screened to identify a limited number of promising candidates. Estimates suggest that the drug discovery and development pipeline prior to clinical testing may require many years and substantial financial investment, in which billions of dollars are spent to accomplish a result.6 However, artificial intelligence has been applied primarily as a methodological and developmental support tool, with the aim of improving efficiency in selected stages of the drug discovery process rather than replacing conventional workflows. AI-based software systems enable the evaluation of large and complex datasets to support the identification and optimization of potential drug candidates. Additionally, several areas within drug discovery, including drug design, have attracted increasing interest for AI-assisted approaches. Structure-based drug design, for example, has been introduced to reduce extensive screening requirements and support more informed decision-making, potentially lowering time and cost through advanced computational techniques.7
The advancement of AI techniques applied in drug discovery primarily involves ML and deep learning (DL). ML, a subset of AI, enables computational systems to learn patterns from data and improve predictive performance, often with limited direct human intervention. However, ML-based models may exhibit limited ability to capture complex non-linear relationships, which is counteracted by the development of DL approaches. DL, a specialized subset of ML, applies deep neural networks to capture more complex patterns within data. Compared with conventional ML methods, DL models often demonstrate reduced generalization error and support more detailed feature representation, although their performance remains dependent on data quality and model design. By utilizing neural networks inspired by the structure of the human brain, DL approaches can enhance pattern recognition capabilities in complex biological and chemical datasets.
As shown in Figure 1, early progress in AI was limited prior to the mid-20th century due to constraints in computational power and theoretical frameworks. The formal emergence of AI in the 1950s marked a shift toward computational systems designed to assist analytical tasks, followed by the introduction of ML techniques in the late 1980s. DL experienced substantial advancement after 2012, driven by improvements in data availability and computational resources, which broadened its application within the drug discovery field. More recently, the introduction of large-scale language models such as ChatGPT in 2022 has further increased interest in AI-driven methodologies. In addition, the incorporation of automated input processing in DL models can reduce reliance on manually engineered features, resulting in less complex model development workflows compared with traditional ML approaches.4,8
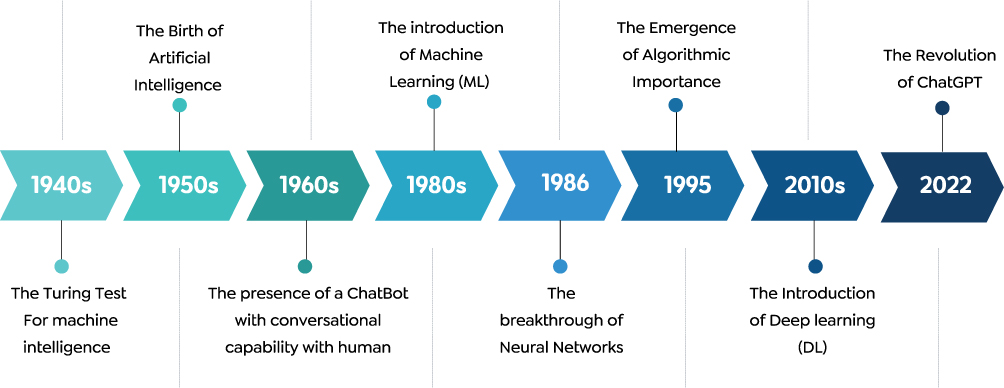 |
Figure 1 A chronological timeline illustrating the major historical milestones in the evolution of artificial intelligence. The sequential chevron-shaped arrows denote the continuous progression of time from the 1940s to 2022. The vertical lines with circular nodes connect specific technological breakthroughs and events to their corresponding decades or exact years. Abbreviations: ML, Machine Learning; DL, Deep Learning. |
In the area of drug design and development, DL algorithms demonstrate considerable prospects via numerous models such as Convolutional Neural Networks (CNN), Deep Neural Networks (DNN), and Recurrent Neural Networks (RNN).9–11 Nevertheless, ML applications can be utilized to quickly identify the active biological compound in a shorter period. Some of those techniques include Genetic Optimization for Ligand Docking (GOLD), Deep Learning-Based Protein Variant Prediction (deep-PVP), and Support Vector Machine (SVM).12 Many of these examples of ML and DL could be employed to assist drug discovery and development workflow across multiple areas, such as quantitative structure-based virtual systems (QSAR) modeling, ligand-based virtual screening (LBVS), ligand-based drug design (LBDD), drug repurposing, and lead identification with reported applications across many studies.13 The nested relationship between artificial intelligence (AI), machine learning (ML), deep learning (DL), and neural networks is illustrated in Figure 2.
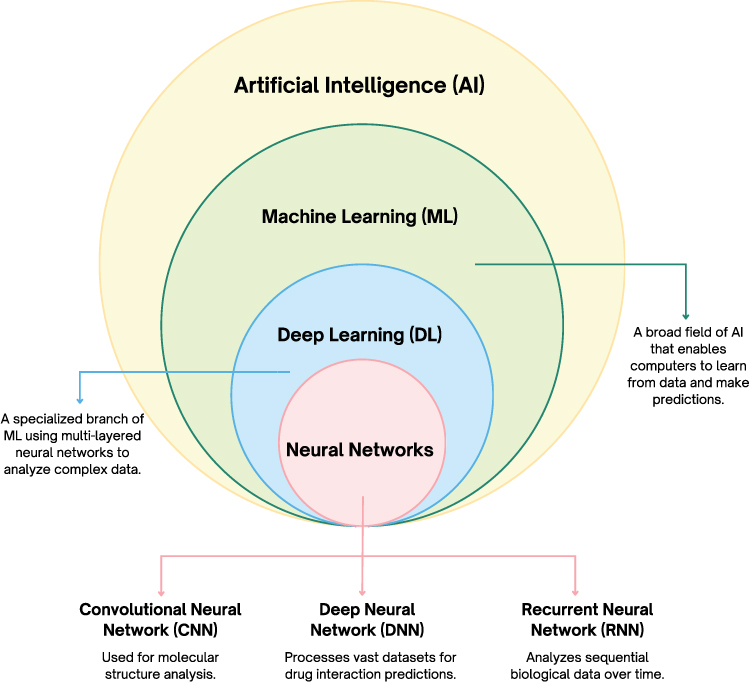 |
Figure 2 A figure representation illustrating the hierarchical relationship between Artificial Intelligence and its subfields. The concentric circles denote that Machine Learning (ML) is a subset of Artificial Intelligence (AI), Deep Learning (DL) is a subset of ML, and Neural Networks form the core of DL. Explanatory text boxes connected by directional lines provide specific definitions for these domains, while the downward arrows branch into specialized neural network architectures. Bold text is utilized to emphasize the names of the core technological fields and the specific network models. Abbreviations: AI, Artificial Intelligence; ML, Machine Learning; DL, Deep Learning; CNN, Convolutional Neural Network; DNN, Deep Neural Network; RNN, Recurrent Neural Network. |
To reflect the expanding application of artificial intelligence (AI) in drug discovery and development, this review provides a critical narrative overview of current AI methodologies and their reported applications in the discovery of therapeutic agents, including antibiotics, anticancer agents, antibodies, and small-molecule drugs. Rather than offering an exhaustive or quantitative analysis, the review focuses on representative approaches that illustrate how AI techniques have been integrated into drug discovery workflows. Compared with conventional computational methods such as classical QSAR models, which rely on predefined molecular descriptors and relatively simple statistical relationships, AI-based approaches enable direct learning from data, allowing for improved capture of complex biological interactions and the ability to handle larger and more heterogeneous datasets. However, these advantages are accompanied by challenges related to model interpretability, data quality, and generalizability. This review further emphasizes these limitations alongside implementation challenges, including validation constraints and dataset representativeness. Finally, potential future directions are discussed, highlighting the importance of integrating AI tools in a cautious and complementary manner to support, rather than replace, established drug discovery processes.
Methodology
This review examined published literature that was identified by an initial screening process, focusing on studies published between 2000 and 2026 that addressed AI applications related to drug discovery and development. Studies that did not meet the thematic scope of this review were not chosen, such as incomplete studies, studies lacking clear or valid conclusions, AI applications in non-pharmaceutical fields, or studies published before 2000. Additionally, review articles and meta-analyses were excluded from the primary data analysis and results, although some were cited to provide background context in the introduction and discussion sections. The included studies were further classified based on different techniques, targets, and areas of drug discovery, while supporting each domain with representative examples discussed in the relevant sections. Moreover, the studies discussed across multiple sections were drawn from different scientific databases, including PubMed, Web of Science, and Google Scholar. This review was conducted as a critical narrative review and therefore did not follow a formal systematic review protocol.
Comparison Between Traditional and AI-Driven Drug Discovery
Historically, Traditional Drug Discovery has mainly depended on a linear trial-and-error approach. This process usually involves physical High-Throughput Screening (HTS) of chemical libraries, extensive in vitro experiments, and repeated chemical optimization by medicinal chemists. Although this strategy has contributed to the development of many important medications, it is associated with high financial costs, long development timelines, and high attrition rates. In many cases, problems related to toxicity or poor pharmacokinetic properties are identified only during the later stages of clinical trials.
In contrast, the use of AI has introduced a more predictive and data-driven approach to drug discovery. AI-Driven Drug Discovery uses computational methods to explore large chemical spaces without the immediate need for physical synthesis. Machine learning and deep learning models can support rapid virtual screening, de novo molecule design, and early prediction of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties. As a result, potentially unsuitable compounds may be identified earlier in the development process. This can help reduce development time and lower overall costs. Despite the growing role of AI in drug discovery, traditional experimental approaches remain essential for validating the biological activity, safety, and therapeutic potential of candidate compounds. Computational predictions can support and accelerate the discovery process; however, in vitro, in vivo, and clinical studies are still required to confirm the actual effectiveness and safety of potential drugs. Methodological differences between Traditional and AI-Driven Drug Discovery are summarized in Table 1.
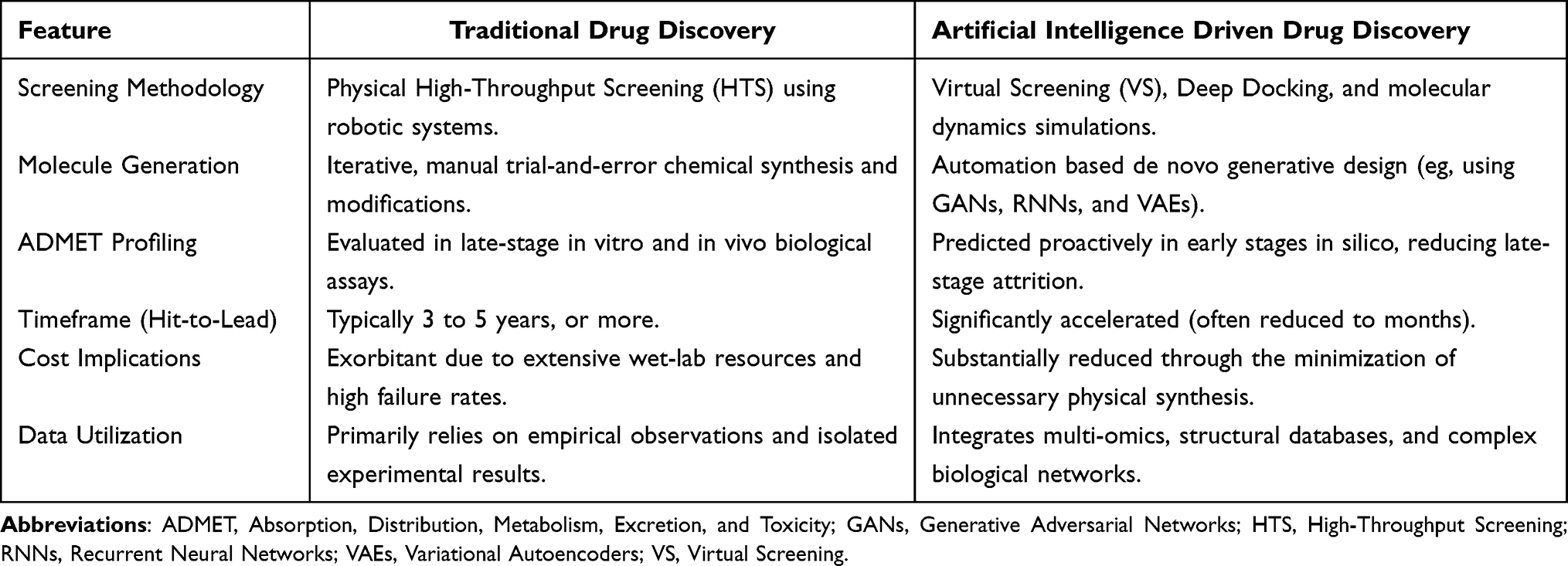 |
Table 1 Key Methodological Differences Between Traditional and AI-Driven Drug Discovery |
The Transformation into AI in Drug Discovery and Development
The use of AI was first introduced in chemistry and molecular biology to help in protein secondary structure estimation.14 However, in the field of drug discovery, early AI applications focused on modeling structure–activity relationships due to AI’s strong pattern recognition capabilities.4 Medicinal chemists face the challenge of evaluating several compound structures through processes that may take months to years.15 The integration of AI technologies can facilitate and support the screening and evaluation process, potentially accelerating selected stages of drug discovery. Both ML and DL methods recognize patterns and shared structural similarities. As researchers aim to improve drug discovery and development processes while maintaining accuracy, AI technologies have continued to develop to support more detailed chemical structure prediction and modeling of complex biological activities. The advancement in AI has been applied not only to predict, categorize, or hypothesize from large datasets, but also to assist in addressing complex research questions and prioritizing potential therapeutic candidates, rather than independently generating therapeutic solutions.16
AI in Antibiotics Discovery and Development
Between 2014 and 2019, 14 new antibiotics were approved, a number considered limited due to the increasing burden of antimicrobial resistance, which has been associated with substantial global mortality.16 This landscape has motivated the exploration of artificial intelligence (AI)–based approaches as supportive tools in antibacterial discovery. Rather than serving as standalone solutions to antimicrobial resistance, AI methodologies are primarily applied to assist in screening, prioritization, and early-stage evaluation of candidate compounds. Table 2 provides a summarized overview of representative AI applications in antibiotic drug discovery and development.
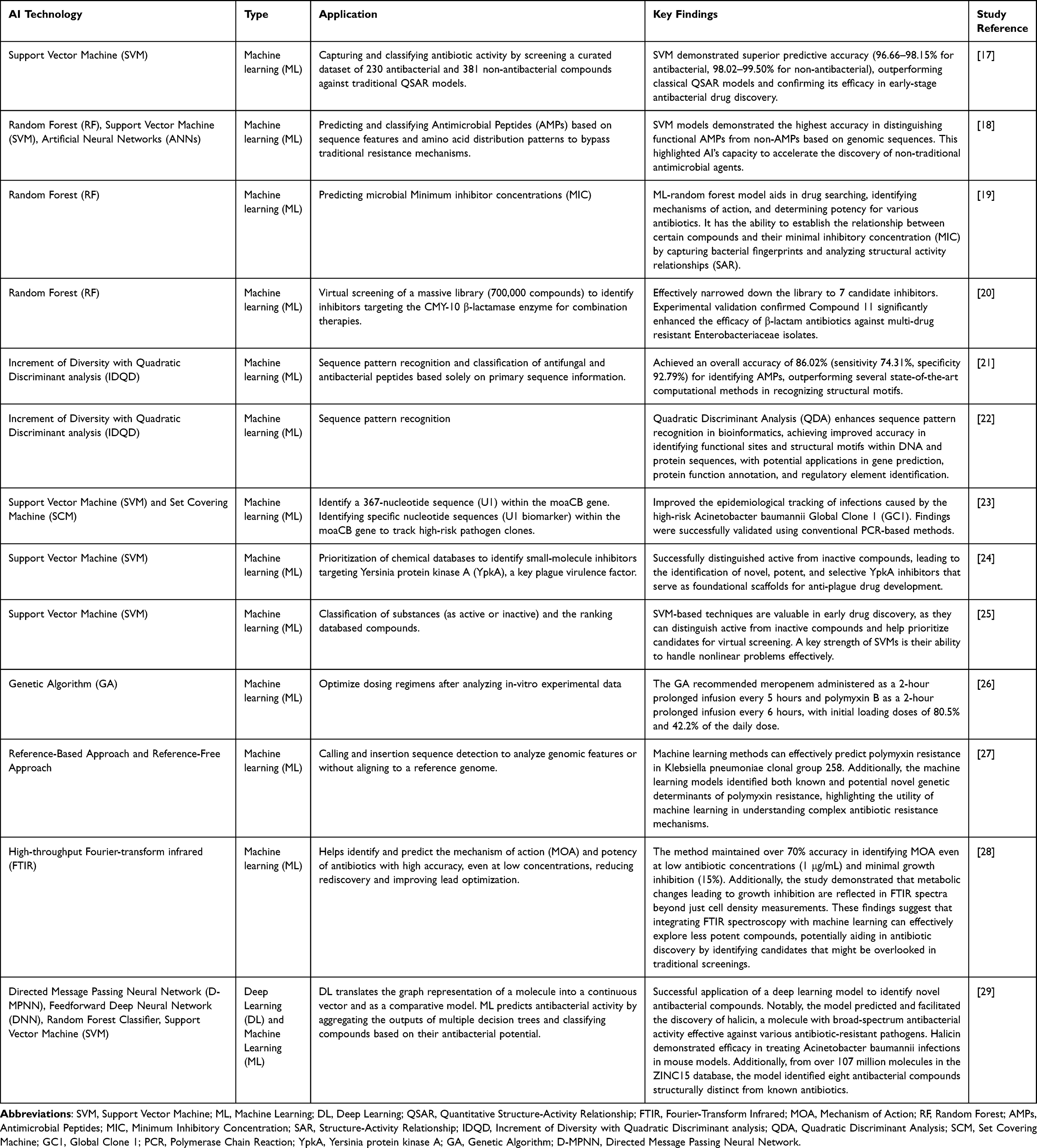 |
Table 2 Summary of Studies Implementing AI Approaches in Antibiotics Discovery and Development |
Machine Learning Approaches
Machine learning (ML) techniques have increasingly been applied to support in improving the efficiency of identifying potential antibacterial agents within large chemical and biological datasets. Support vector machine (SVM) models and other supervised learning approaches have been reported to achieve high predictive performance in classifying active versus inactive compounds;17 however, reported accuracy values depend strongly on dataset composition, feature representation, and validation strategies.26 Random forest (RF) models have been used in structure–activity relationship (SAR) analysis, mechanism-of-action prediction, and potency estimation in antibiotic research.19 In several studies, RF-based approaches were applied to evaluate compound activity using minimum inhibitory concentration (MIC) data and bacterial fingerprint information.18,21,22 These models have also supported compound prioritization following virtual screening campaigns.24 Other ML approaches, including Increment of Diversity with Quadratic Discriminant (IDQD), have been employed to classify antibacterial peptides and analyze genomic sequence patterns relevant to antimicrobial activity.21,22 Antimicrobial peptides (AMPs) are considered promising alternatives to conventional antimicrobial agents. However, there is still a lack of portable, easy-to-use, and efficient tools for identifying AMP sequences from genome-scale datasets.30
Large-scale ML-driven screening platforms have enabled the categorization of compounds as active or inactive against organisms such as Escherichia coli, facilitating the identification of promising candidates for subsequent experimental validation.27 Additionally, machine learning algorithms have been utilized to identify molecular patterns linked to antibiotic permeability in Gram-negative bacteria, forming an “antibiotic vocabulary” useful for distinguishing antibacterial compounds from inactive molecules.31 ML methods have additionally been applied to identify inhibitors of specific bacterial targets. For example, predictive modeling combined with virtual screening has been used to prioritize inhibitors of Yersinia protein kinase A (YpkA), which were then subjected to experimental testing.24 In studies targeting FabI in Staphylococcus aureus, multiple algorithms—including decision trees, random forests, multilayer perceptrons, k-nearest neighbors, Naive Bayes, and SVM—were compared, with some models demonstrating improved classification performance within the evaluated datasets.28 Recently, a deep learning-guided screening approach successfully identified a novel structural class of antibiotics specifically targeting the multidrug-resistant pathogen Acinetobacter baumannii, highlighting the potential utility of AI-assisted approaches in identifying pathogen-specific therapeutic candidates.32 Beyond compound identification, ML has been explored for optimizing antibiotic dosing regimens and predicting resistance phenotypes, including polymyxin resistance in Klebsiella pneumoniae, through genomic feature analysis.27 ML-based models have also been developed to assess the potency and toxicity of antimicrobial peptides, although predictive performance remains influenced by dataset size and representativeness. ML has further been integrated with high-throughput analytical techniques, such as Fourier-transform infrared spectroscopy, to infer potential mechanisms of action through metabolic fingerprint analysis.28
Deep Learning Approaches
Deep learning (DL) models have expanded the capacity to explore large chemical spaces and prioritize structurally diverse antibiotic candidates. In a prominent example, a DL framework was used to identify novel antibacterial compounds with reported activity against multidrug-resistant organisms, including Mycobacterium tuberculosis and Acinetobacter baumannii.29 The proposed mechanism of action in this context involved disruption of bacterial metabolic processes and iron sequestration. Nonetheless, these findings were primarily demonstrated in preclinical and in vitro settings, and their broader clinical applicability requires further investigation. Overall, both ML and DL approaches have contributed to improving the efficiency of candidate identification and prioritization in antibiotic discovery. However, model performance remains dependent on data quality, external validation, and careful interpretation, particularly when addressing the multifactorial challenge of antimicrobial resistance.
AI in Anticancer Drug Discovery
Discovering newer anticancer agents remains a major priority due to the high mortality rates and the emergence of therapeutic resistance. AI-based methods have increasingly been explored to support target identification, genomic data interpretation, and compound prioritization. Rather than directly accelerating drug approval or guaranteeing higher success rates, AI technologies are primarily applied in early-stage discovery to assist in pattern recognition, target prediction, and molecular screening. Table 3 provides a summary of selected studies applying AI in anticancer drug discovery and development.
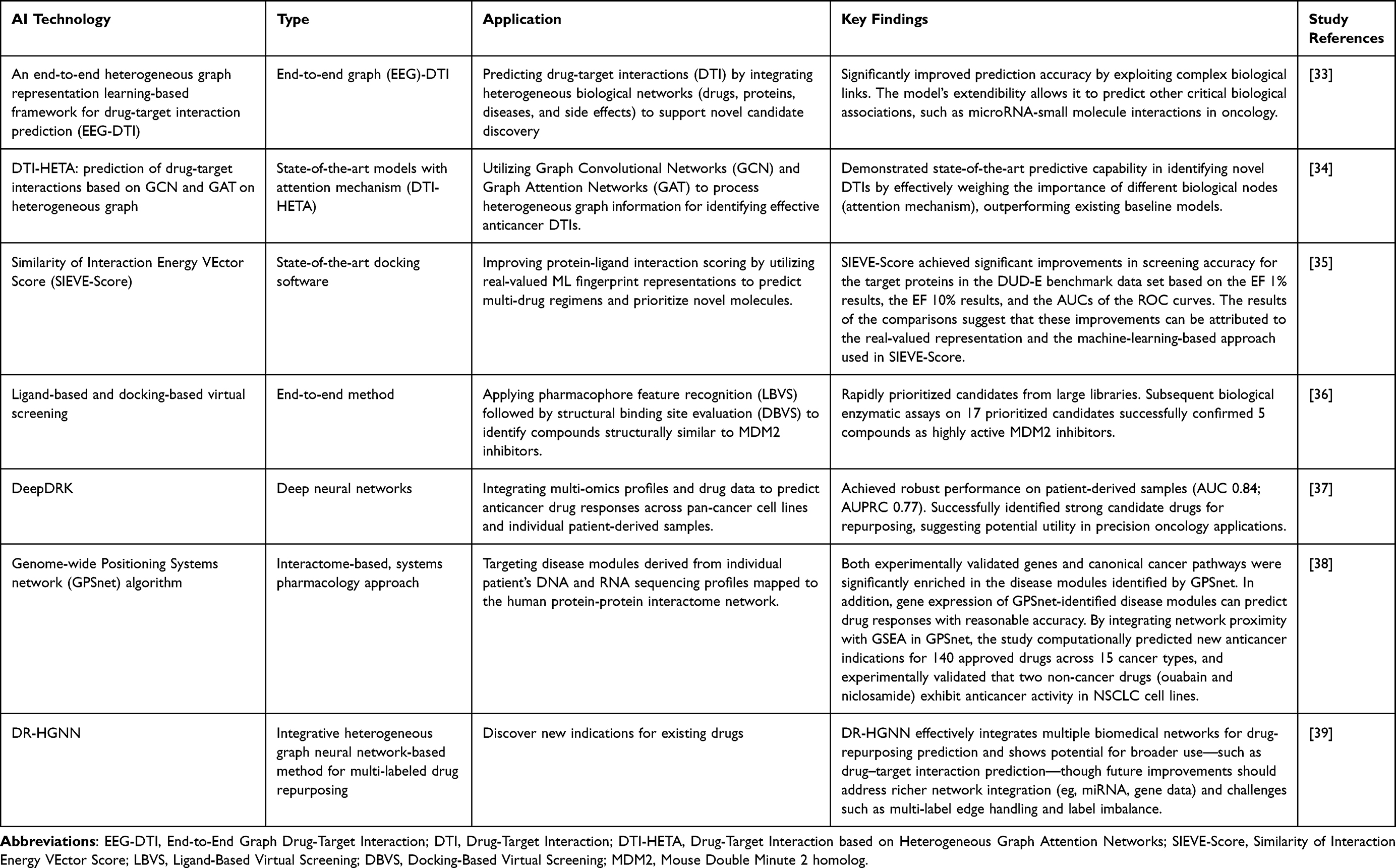 |
Table 3 Summary of Studies Implementing AI Approaches in Anticancer Drug Discovery and Development |
Computer-aided drug design (CADD) approaches integrate machine learning (ML) and deep learning (DL) models for drug-target interaction (DTI) prediction, screening, and drug repositioning. Traditional DTI prediction often relies on molecular docking and structural modeling, which can be computationally intensive and dependent on available 3D protein structures. Graph-based neural network models, such as EEG-DTI and DTI-HETA, were developed to represent drug–target interactions as heterogeneous graphs and to improve interaction prediction using convolutional and attention mechanisms.33,34 These models were evaluated on benchmark datasets and demonstrated improved predictive performance compared with selected baseline models; however, their results remain dependent on dataset composition and internal validation frameworks. The SIEVE-Score was developed to improve protein–ligand interaction scoring using ML-based fingerprint representations.35 Ligand-based and docking-based virtual screening is an approach that supports the efficient discovery of potential anticancer leads. Notably, one identified compound showed promising antiproliferative activity and may serve as a scaffold for further optimization in the development of MDM2-targeted anticancer therapies.36 Network-based methods such as GPSnet utilize patient-specific gene expression data and drug–target interaction networks to identify repositioning candidates.37,38 In the reported case of ouabain as a potential candidate in lung adenocarcinoma, findings were derived from network inference and preclinical validation rather than confirmed therapeutic efficacy. Thus, GPSnet provides hypothesis-generating associations rather than direct clinical recommendations. Other frameworks, including MTRD and graph-based models such as DR-HGNN and GDRnet, integrate drug–protein–disease interaction networks to predict drug–disease associations.39 While these models report strong predictive metrics within curated datasets, their performance remains influenced by training data distribution, similarity assumptions, and limited external validation across independent clinical cohorts.
Overall, AI approaches in anticancer drug discovery have expanded the capacity to integrate multi-omics datasets, prioritize candidates, and model drug–target interactions. Nevertheless, challenges such as dataset bias, ground-truth uncertainty, overfitting risk, and the limited translation of computational predictions into clinically validated therapies remain significant. Consequently, AI currently functions primarily as a decision-support tool in early-stage oncology research rather than as a standalone determinant of therapeutic development outcomes. The overarching AI-driven computational workflows and screening strategies employed for the discovery of both antibacterial and anticancer therapeutic agents are comprehensively illustrated in Figure 3.
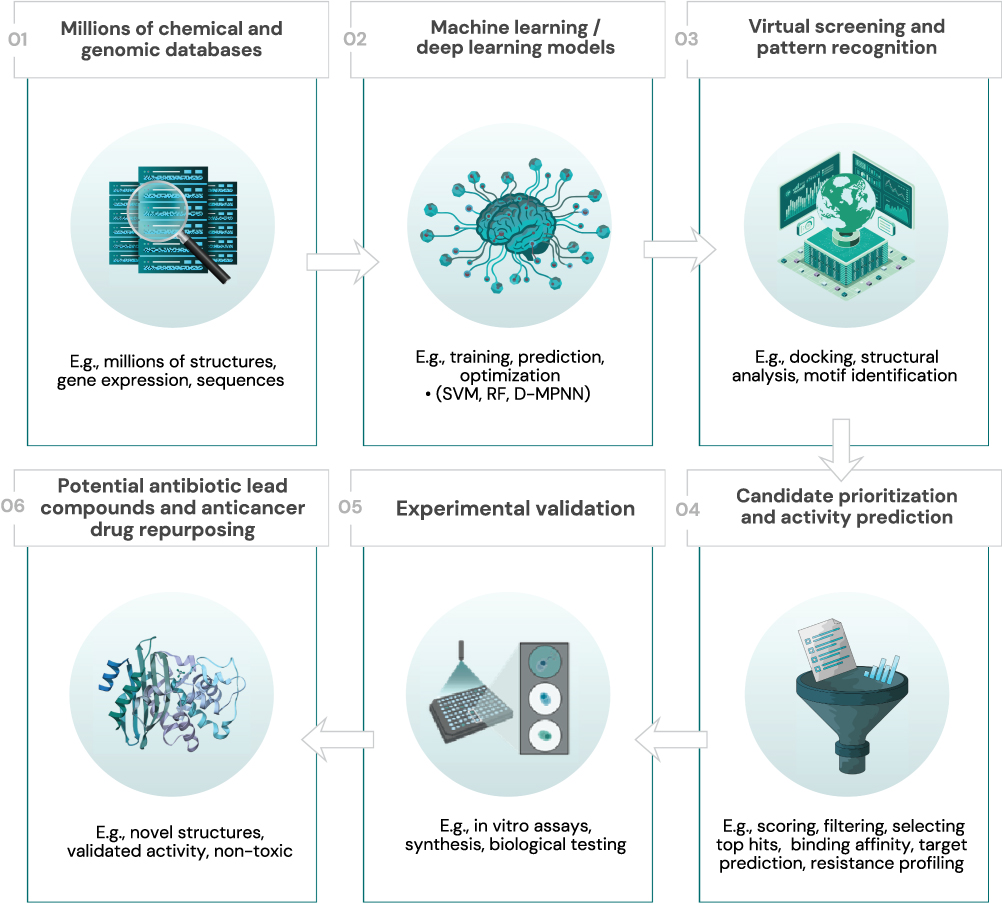 |
Figure 3 A schematic workflow illustrating the integration of artificial intelligence in drug discovery, specifically highlighting the pipeline for identifying antibiotic leads and repurposing anticancer drugs. The sequential numerical indicators (01–06) represent the chronological progression of the computational and experimental phases. Bold text is used to denote the primary stages of the workflow. Abbreviations: SVM, Support Vector Machine; RF, Random Forest; D-MPNN, Directed Message Passing Neural Network. |
AI in Antibodies Discovery
Antibodies are commonly used therapeutic agents in chronic inflammatory diseases, cancer, and autoimmune disorders. These serious human illnesses continue to receive significant attention, particularly given the complexity of biologic development and the relatively limited annual number of newly approved antibody therapeutics.40,41 Most approved agents are IgG-based, despite the availability of alternative formats such as antibody–drug conjugates (ADCs), bispecific antibodies, and antibody fragments. Accordingly, the development of newer antibody therapeutics has relied heavily on iterative engineering strategies rather than a purely accelerated discovery process. The unpredictable and sometimes unfavorable properties of antibodies may influence immunogenic responses in patients. However, it is essential to distinguish between computational prediction of anti-drug antibody (ADA) risk and actual clinical immunogenicity outcomes. AI and ML-based techniques have been explored to rank clinical candidate antibodies according to predicted immunogenic features and to guide engineering efforts aimed at reducing theoretical T-cell epitope content. In the case of emicizumab, epitope modification strategies were applied to reduce predicted T-cell activation. Nevertheless, a reduction in in silico epitope burden does not directly equate to reduced clinical ADA incidence, as immunogenicity is multifactorial and influenced by patient-related and formulation-specific variables.42,43 Reports of moderate-to-severe ADA responses in certain bispecific antibodies have raised further discussion regarding format-related immunogenicity. AI-driven workflow for the design and optimization of therapeutic antibodies is outlined in Figure 4. Current evidence does not conclusively demonstrate that bispecific antibodies are inherently more immunogenic than monospecific antibodies; rather, immunogenicity appears to depend on molecular structure, target biology, dosing strategy, and patient characteristics.
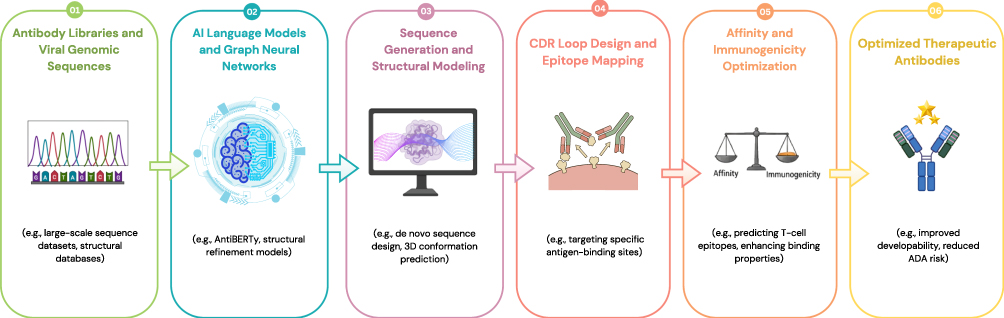 |
Figure 4 A sequential workflow showing the application of AI in the design and optimization of therapeutic antibodies. The numbered steps (01–06) and block arrows illustrate the progression from initial genomic sequences to optimized antibodies. Abbreviations: CDR, Complementarity-Determining Regions; ADA, Anti-Drug Antibody. |
AI Applications in SARS-CoV-2 Antibody Research
The emergence of SARS-CoV-2 stimulated rapid integration of AI-based tools into antibody research. However, several distinct tasks were involved, including viral genomic sequence analysis, structural modeling of antibody–antigen complexes, CDR design, and epitope mapping. Deep learning approaches such as convolutional neural networks (CNNs) were applied to analyze viral genomic sequences and distinguish SARS-CoV-2 variants from related viruses.44,45 More broadly, the integration of machine learning with structural and computational biology has increasingly supported antibody engineering workflows, including sequence analysis, structural prediction, and antigen–antibody interaction modeling.46 These models primarily support viral classification and sequence analysis rather than direct antibody binding prediction. Structural and graph-based neural network models, including REFINEGNN, have been applied to antibody structure refinement and CDR conformational modeling.47 REFINEGNN performs graph-based refinement of predicted antibody structural conformations to improve modeling accuracy. Although improvements in structural prediction performance have been reported relative to selected computational baselines, these metrics reflect modeling benchmarks rather than experimentally validated therapeutic enhancement.
Autoregressive and generative graph neural networks have been explored to design CDR sequences and three-dimensional conformations aimed at improving predicted antigen binding. Claims regarding docking success rates or substantial increases in neutralization capability should be interpreted cautiously, as computational docking scores do not guarantee in vitro or clinical efficacy. Language model–based approaches such as ProtGPT and AntiBERTy have been utilized for antibody sequence generation.48–50 These models expand sequence diversity and provide high-throughput design capabilities; however, sequence generation quality remains dependent on training data representation and does not ensure functional binding without experimental validation. Bayesian neural networks and related generative ML strategies have been proposed to identify tight-binding antibodies and explore sequence spaces not directly present in screening libraries.51 While these approaches broaden exploration capacity, their predictions rely on modeled binding probabilities rather than experimentally confirmed affinity. RosettaAntibodyDesign (RAbD) integrates structural and sequence features to guide CDR optimization.52 Reported improvements in binding affinity following CDR replacement are derived from controlled experimental or computational settings and should not be generalized beyond the evaluated systems.
Critical Considerations
Overall, AI has supported antibody engineering by enabling epitope prediction, structural modeling, developability assessment, and generative sequence design. Yet challenges such as dataset bias, limited ground-truth structural data, overfitting in benchmark evaluations, and the gap between computational prediction and clinical validation remain substantial. Consequently, AI currently functions as a decision-support and hypothesis-generating tool in antibody discovery rather than as an independent determinant of clinical immunogenicity or therapeutic success.
AI in Small Molecules Design
Small-molecule agents are characterized by their relatively low molecular weight, allowing intracellular penetration and oral bioavailability in many cases. Since approximately 2015, AI-assisted approaches have increasingly been incorporated into small-molecule discovery workflows.53,54 Rather than independently discovering drugs, AI methods typically assist in candidate prioritization, optimization, and screening efficiency. Examples such as DSP-1181, EXS21546, and DSP-0038 entered early clinical phases following AI-assisted optimization processes.55 Importantly, these compounds were not “discovered solely by AI”, but resulted from integrated computational modeling combined with medicinal chemistry and experimental validation. In addition to its expanding role in small-molecule drug discovery, machine learning has shown promising utility in the prediction of adverse drug events, supporting improved medication safety and patient care.56
Classical Virtual Screening (VS)
Classical virtual screening strategies include structure-based virtual screening (SBVS) and ligand-based virtual screening (LBVS). SBVS relies on knowledge of a target’s binding site structure, whereas LBVS identifies compounds based on similarity to known active ligands without requiring explicit structural information. Traditional docking and scoring approaches remain computationally intensive and are subject to scoring-function limitations. AI has been integrated into these pipelines primarily to improve ranking efficiency rather than to replace classical methods. For example, ML-assisted workflows were applied to identify Cyclin-Dependent Kinase 5 (CDK5) inhibitors using algorithms such as random forest and k-nearest neighbors.57 Reported hit identification was based on in silico prioritization followed by experimental testing, and computational hit rates should not be interpreted as confirmed biological efficacy.
ML-Based Virtual Screening
Machine learning models have been used to improve compound prioritization within SBVS and LBVS frameworks. Platforms such as AtomNet apply deep learning to predict protein–ligand binding probability from structural features.58 In one study, AtomNet screened millions of compounds and prioritized candidates for synthesis and biological evaluation. While several compounds demonstrated activity in subsequent assays, these results reflect a multi-step pipeline combining DL prediction, experimental testing, and biological validation. Statements implying that AI alone “identified tumor-inhibiting compounds” oversimplify the development process. Furthermore, performance metrics such as IC50 ranking are meaningful only after experimental confirmation, not at the purely computational stage. Similarly, Deep Docking frameworks were used in ligand-based screening contexts.59 Reported improvements in screening speed or ranking accuracy should be interpreted within dataset and validation constraints.
Generative Models in Molecular Design
Deep learning–based generative models, including recurrent neural networks (RNN), variational autoencoders (VAE), and generative adversarial networks (GAN), have been applied to propose new molecular structures.60 These models generate candidate structures using string-based (eg, SMILES) or graph-based representations. Tools such as MolAICal and DeepLigBuilder were developed to generate molecules predicted to bind specific targets.61,62 These systems prioritize structural compatibility and docking feasibility, but predicted binding does not equate to confirmed biological potency. Deep Generative Models for 3D Linker Design applies graph-based modeling to generate linker fragments while maintaining pharmacophoric features.63 Claims such as “300% similarity” require contextual clarification and should not be interpreted as guaranteed structural improvement beyond computational benchmarks. Generative RNN models with long short-term memory (LSTM) architectures have also been used in de novo design workflows, producing candidate compounds subsequently evaluated experimentally.64 Although some generated molecules progressed to hit-to-lead optimization, synthesis feasibility and stability were confirmed through experimental validation rather than AI prediction alone. The application of AI in therapeutic antibody design and optimization is summarized in Figure 5.
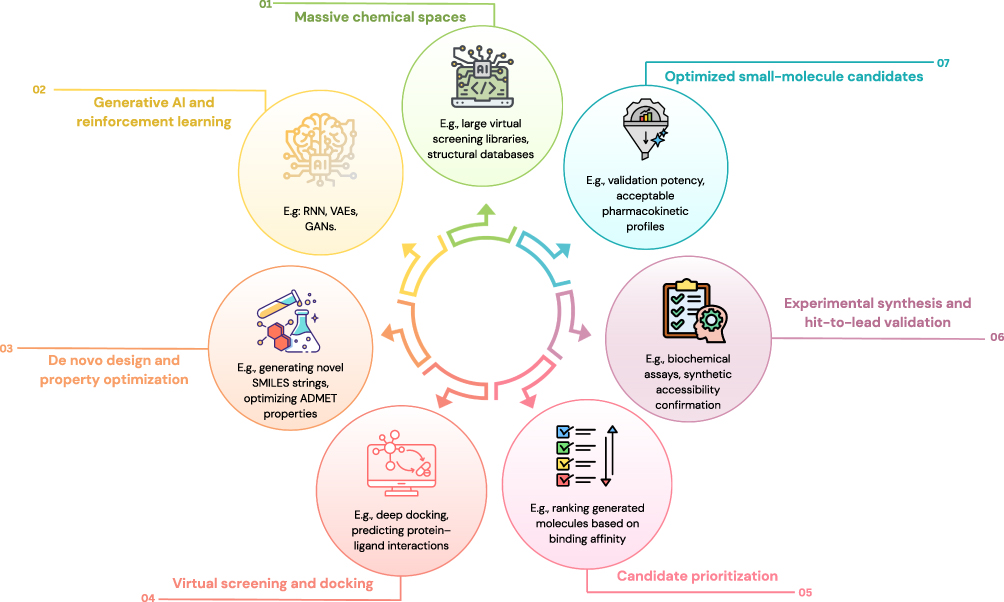 |
Figure 5 A schematic workflow illustrating the small molecules AI-driven drug discovery cycle. The numbered steps (01–07) indicate the sequential progression of the pipeline. Abbreviations: RNN, Recurrent Neural Network; VAEs, Variational Autoencoders; GANs, Generative Adversarial Networks; SMILES, Simplified Molecular-Input Line-Entry System; ADMET, Absorption, Distribution, Metabolism, Excretion, and Toxicity. |
Reinforcement Learning (RL) and Property Optimization
Reinforcement learning (RL) has been integrated with generative architectures to optimize desired properties such as binding affinity or physicochemical constraints. Approaches including Mol-CycleGAN apply transformation-based learning to modify molecular structures toward predefined objectives.65 These models operate within constrained chemical spaces defined by training data, and their performance depends strongly on reward function design and dataset bias. While RL approaches may accelerate lead refinement, they remain hypothesis-generating tools.
AI in Lead Optimization and Development
Neural network models such as LSTM architectures have been applied to predict activity profiles across large compound libraries.66,67 Some reports describe the identification of potent inhibitors (eg, p300 or CDK8 inhibitors) following AI-assisted optimization. However, reported IC50 values reflect experimentally measured outcomes after medicinal chemistry refinement, not purely computational predictions. AI contributes to prioritization and design suggestions, while potency confirmation relies on biochemical assays.
Critical Considerations
Despite promising integration of AI into small-molecule workflows, several limitations remain. Many reported results are derived from retrospective benchmark datasets with limited prospective validation. Dataset bias, imbalanced active/inactive examples, and overfitting risk may inflate apparent performance. Furthermore, synthetic accessibility, off-target effects, and in vivo pharmacokinetics remain challenges not fully captured by current AI models. Consequently, AI functions primarily as a supportive decision-making tool within multidisciplinary drug discovery pipelines rather than as an autonomous discovery engine.
Discussion of Persistent Challenges in AI Implementation
Across the diverse therapeutic areas reviewed—spanning antibiotics, anticancer agents, antibodies, and small molecules—AI has consistently demonstrated the capacity to enhance early-stage drug discovery, particularly through improved virtual screening and pattern recognition (as summarized in Table 1 and Table 2). However, a comprehensive evaluation of the literature indicates that computational advancements do not entirely eliminate the fundamental hurdles of drug development. Rather than viewing AI as a standalone solution, it is essential to discuss the practical limitations it faces. As summarized in Figure 6, several challenges continue to hinder the implementation of AI in drug discovery and development. While AI provides robust supportive tools for molecular modeling and candidate prioritization, its implementation introduces specific methodological, technical, and data-related constraints that must be objectively considered. Integrating these technologies is a complex process that encounters several persistent challenges, which are outlined below:
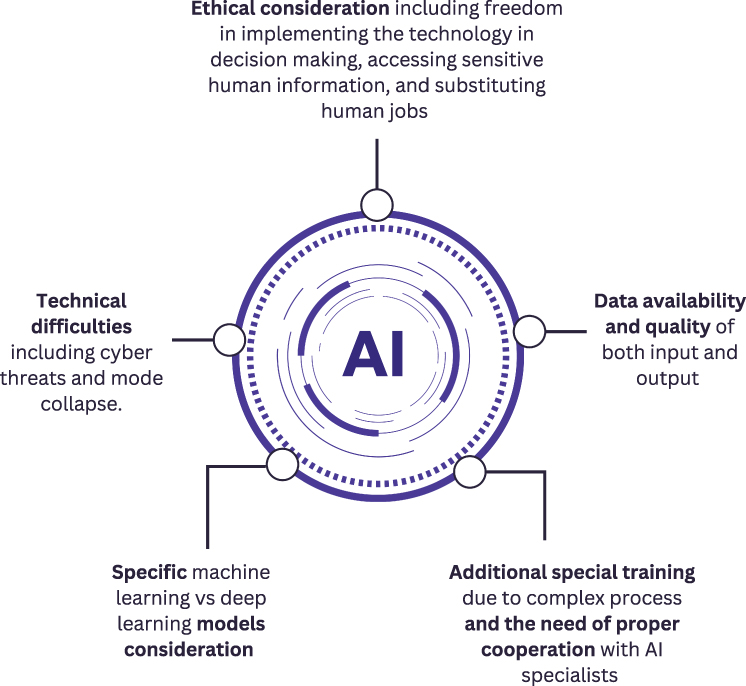 |
Figure 6 A figure summarizing the primary challenges and considerations associated with the implementation of Artificial Intelligence. |
Ethical and Data Governance Considerations
AI systems in drug discovery primarily function as decision-support tools rather than autonomous decision-makers. Therefore, statements suggesting that AI independently “makes decisions vital for human health” overstate its current role. Instead, AI outputs influence prioritization of compounds, targets, or experimental strategies within human-led workflows. A major concern in drug discovery is data bias and representativeness. Incomplete or unbalanced datasets may lead to skewed predictions and misleading compound prioritization.68 In biomedical contexts, experimental data are often heterogeneous, derived from different assay conditions and population subsets, which complicates model generalization. The use of proprietary datasets and sensitive patient-derived information also raises concerns regarding confidentiality and data protection.69–71 However, these issues relate specifically to clinical and translational datasets rather than to AI autonomy itself. Consequently, governance frameworks in drug discovery must emphasize responsible data curation and controlled access rather than generalized concerns about machine “freedom” in healthcare.
Technical Expertise and Model Interpretability
AI implementation in drug discovery requires interdisciplinary collaboration between data scientists, computational chemists, and experimental researchers. Rather than requiring “special training due to data loss”, the primary challenge lies in ensuring correct model interpretation and appropriate feature engineering. Machine learning models are highly sensitive to input representation and feature selection. For example, the use of SMILES strings as textual molecular representations may influence how models interpret chemical space.72 Improper featurization or biased training data can affect interpretability and downstream decision-making. Furthermore, misunderstanding model limitations may lead to overconfidence in predictive accuracy.73,74 Thus, expertise is required not only for technical implementation but also for critical evaluation of model outputs within experimental pipelines.
Data Availability, Quality, and Metadata
AI models in drug discovery are highly dependent on data volume and quality.13 Limited availability of well-annotated experimental datasets can constrain model performance, particularly in early-stage target discovery. Metadata—such as assay conditions, cell line context, dose ranges, and experimental protocols—significantly influence model reliability. Inaccurate or incomplete metadata may impair model generalization, not because AI “fails”, but because underlying biological context is inconsistently captured. Moreover, generative models may propose chemically valid structures that are synthetically inaccessible or biologically unstable This gap between computational feasibility and experimental applicability represents a practical limitation rather than a failure of algorithmic design.73
Infrastructure and Model Deployment Constraints
Some AI and deep learning models require significant computational resources and technical support. High-performance computing infrastructure, data storage capacity, and secure access systems may not be uniformly available across research settings. However, increased computational demand reflects model complexity rather than inherent inefficiency, and it does not automatically translate into poor project insight. Instead, organizations must balance infrastructure investment with projected research benefits.13
Technical Risks in Generative Modeling
In generative AI systems, particularly generative adversarial networks (GANs), technical issues such as mode collapse may arise. Mode collapse refers to a phenomenon in which the generator produces limited structural diversity, repeatedly sampling a narrow subset of outputs despite broader training distributions. In the context of small-molecule design, this may result in structurally similar compounds being proposed repeatedly, potentially limiting chemical space exploration. This issue reflects training instability and reward-function imbalance rather than “data loss” or stacking in a local minimum. Additionally, cybersecurity risks exist when integrating AI into healthcare data ecosystems, especially when large biological or patient-derived datasets are involved. These concerns relate primarily to data protection infrastructure rather than AI-specific decision-making mechanisms.69
Summary of Practical Limitations
Overall, the principal challenges of AI implementation in drug discovery include:
- Dataset heterogeneity and bias
- Dependence on high-quality annotated metadata
- Risk of overfitting and limited external validation
- Computational resource demands
- Gap between computational prediction and experimental verification
AI technologies, therefore, function as supportive analytical tools within multidisciplinary drug discovery pipelines, rather than as autonomous systems capable of independently overcoming biological complexity. Recent reviews further emphasize that AI-driven antimicrobial discovery continues to rely heavily on experimental validation, interdisciplinary integration, and careful interpretation of computational predictions despite rapid advances in generative and predictive modeling approaches.75
The Role of AI in Drug Formulation
Beyond the initial discovery and optimization of lead compounds, AI is increasingly being explored to support pharmaceutical formulation and preparation techniques. Traditionally, drug formulation has been a labor-intensive, trial-and-error process aimed at achieving optimal bioavailability and physical stability. Currently, machine learning models are being deployed to predict the physicochemical properties of Active Pharmaceutical Ingredients (APIs), such as aqueous solubility and dissolution rates, directly from their molecular structures.76 Furthermore, AI algorithms accelerate the formulation process by screening vast databases to optimize excipient selection and predicting potential degradation pathways.77 In the realm of advanced drug delivery, deep learning networks are utilized to optimize the design of nanocarriers and liposomes, precisely tuning their release kinetics for targeted therapies.78 Furthermore, the convergence of AI with emerging manufacturing technologies, particularly 3D printing (additive manufacturing), is contributing to ongoing advancements in personalized medicine. Specifically, ML has been applied to assess the printability of pharmaceutical formulations using selective laser sintering (SLS). A recent study have shown that ML models trained on multi-modal datasets combining formulation composition with analytical characterization data such as Fourier-transform infrared spectroscopy (FT-IR), X-ray powder diffraction (XRPD), and differential scanning calorimetry (DSC) can improve predictive performance, with reported F1 scores reaching approximately 88.9%.79 This multi-modal approach may reduce reliance on traditional trial-and-error experimentation by supporting more data-driven formulation screening and development of 3D-printed drug products. Overall, these findings highlight the potential of artificial intelligence to enhance decision-making and efficiency in SLS-based pharmaceutical manufacturing.
AI in Characterization Techniques
AI may enhance advanced characterization techniques by automating the interpretation of complex analytical data. Traditional physicochemical characterization relies heavily on Nuclear Magnetic Resonance (NMR) spectroscopy, Fourier-transform infrared (FTIR) spectroscopy, and X-ray powder diffraction (XRPD), which generate massive datasets requiring time-consuming manual analysis. Currently, specific AI systems, particularly Convolutional Neural Networks (CNNs) and deep learning algorithms, are employed to rapidly decipher these complex spectra and identify structural features with high precision.80 For instance, specialized AI tools such as DP4-AI have been developed to fully automate the assignment of NMR spectra, facilitating the rapid structural elucidation of complex stereocenters and novel scaffolds.81 In solid-state characterization, machine learning classifiers are increasingly utilized to process XRPD data for real-time polymorph identification, ensuring phase purity during manufacturing. Furthermore, in morphological analysis, AI-based neural networks can enhance the resolution and automated particle reconstruction in systems like Cryo-Electron Microscopy (Cryo-EM).82 By deploying these specific AI-driven analytical tools, researchers can achieve high-throughput quality control and precise physicochemical profiling with minimal human bias.
Future Perspectives in AI-Driven Drug Discovery
Looking ahead, the future perspectives of AI in drug discovery and formulation are promising yet challenging. The transition toward autonomous “self-driving” laboratories, where AI designs, synthesizes, and tests compounds in a closed-loop system, represents a potential future direction. However, several hurdles remain, including the “black box” nature of complex deep learning models and the critical need for high-quality, standardized experimental data. As the field matures, the integration of explainable AI (XAI) and the continuous synergy between computational predictions and wet-lab validation will be essential. Ultimately, AI is not expected to replace pharmaceutical scientists but rather to empower them, shifting the focus from routine screening to strategic innovation and personalized medicine.
Conclusion
This critical narrative review concludes that while AI has significantly accelerated early-stage drug discovery, its current utility is firmly positioned as an advanced decision-support system rather than an autonomous discovery tool. Across the four therapeutic domains examined—antibiotics, anticancer agents, antibodies, and small molecules—AI methodologies ranging from conventional ML to deep generative models, have demonstrated value in target identification, virtual screening, and candidate prioritization. However, a critical evaluation of the literature reveals several ongoing barriers that limit its effective translation into real-world practice. The use of AI is frequently hindered by dataset biases, lack of standardized metadata, and a substantial gap between computational predictions and experimental validation. While AI models can be fast in generating structurally novel and theoretically active compounds, confirming their biological efficacy, synthetic accessibility, and clinical safety remains entirely dependent on conventional experimental workflows. Ultimately, the successful progression of AI in pharmaceutical research relies on a combination approach where computational power is continuously guided and validated by human expertise. Addressing data quality constraints and emphasizing rigorous external validation will be paramount to bridging the gap between in silico capabilities and real-world clinical applications.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Sadybekov AV, Katritch V. Computational approaches streamlining drug discovery. Nature. 2023;616(7958):673–20. doi:10.1038/s41586-023-05905-z
2. Amisha MP, Pathania M, Rathaur VK. Overview of artificial intelligence in medicine. J Family Med Prim Care. 2019;8(7):2328–2331. doi:10.4103/jfmpc.jfmpc_440_19
3. Ali S, Tian X, Chen H, Zhou J. A new era of artificial intelligence (AI): transforming drug discovery and development. J Med Chem. 2025;68(22):23643–23652. doi:10.1021/acs.jmedchem.5c03159
4. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R&D costs. J Health Econ. 2016;47:20–33. doi:10.1016/j.jhealeco.2016.01.012
5. Ali KA, Mohin S, Mondal P, et al. Influence of artificial intelligence in modern pharmaceutical formulation and drug development. Futur J Pharm Sci. 2024;10:53. doi:10.1186/s43094-024-00625-1
6. Paul SM, Mytelka DS, Dunwiddie CT, et al. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9(3):203–214. doi:10.1038/nrd3078
7. Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–949. doi:10.1038/nrd1549
8. Huang D, Ibrahim AK. Deep learning for drug discovery: a study of identifying high efficacy drug compounds using a cascade transfer learning approach. Appl Sci. 2021;11:7772. doi:10.3390/app11177772
9. Veltri D, Kamath U, Shehu A. Deep learning improves antimicrobial peptide recognition. Bioinformatics. 2018;34(16):2740–2747. doi:10.1093/bioinformatics/bty179
10. Müller AT, Hiss JA, Schneider G. Recurrent neural network model for constructive peptide design. J Chem Inf Model. 2018;58(2):472–479. doi:10.1021/acs.jcim.7b00414
11. Chi CT, Lee MH, Weng CF, Leong MK. In silico prediction of PAMPA effective permeability using a two-QSAR approach. Int J Mol Sci. 2019;20(13):3170. doi:10.3390/ijms20133170
12. Niazi SK, Mariam Z. Artificial intelligence in drug development: reshaping the therapeutic landscape. Ther Adv Drug Saf. 2025;16:20420986251321704. doi:10.1177/20420986251321704
13. Paul D, Sanap G, Shenoy S, Kalyane D, Kalia K, Tekade RK. Artificial intelligence in drug discovery and development. Drug Discov Today. 2021;26(1):80–93. doi:10.1016/j.drudis.2020.10.010
14. Qian N, Sejnowski TJ. Predicting the secondary structure of globular proteins using neural network models. J Mol Biol. 1988;202(4):865–884. doi:10.1016/0022-2836(88)90564-5
15. Hughes JP, Rees S, Kalindjian SB, Philpott KL. Principles of early drug discovery. Br J Pharmacol. 2011;162(6):1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
16. Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. London; 2014.
17. Yang XG, Chen D, Wang M, Xue Y, Chen YZ. Prediction of antibacterial compounds by machine learning approaches. J Comput Chem. 2009;30(8):1202–1211. doi:10.1002/jcc.21148
18. Bhadra P, Yan J, Li J, Fong S, Siu SWI. AmPEP: sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci Rep. 2018;8(1):1697. doi:10.1038/s41598-018-19752-w
19. da Cunha B R, Fonseca LP, Calado CRC. Simultaneous elucidation of antibiotic mechanism of action and potency with high-throughput Fourier-transform infrared (FTIR) spectroscopy and machine learning. Appl Microbiol Biotechnol. 2021;105(3):1269–1286. doi:10.1007/s00253-021-11102-7
20. Parvaiz N, Ahmad F, Yu W, MacKerell AD Jr, Azam SS. Discovery of beta-lactamase CMY-10 inhibitors for combination therapy against multi-drug resistant Enterobacteriaceae. PLoS One. 2021;16(1):e0244967. doi:10.1371/journal.pone.0244967
21. Feng P, Wang Z, Yu X. Predicting antimicrobial peptides by using increment of diversity with quadratic discriminant analysis method. IEEE/ACM Trans Comput Biol Bioinform. 2019;16(4):1309–1312. doi:10.1109/TCBB.2017.2669302
22. Lu J, Luo L, Zhang L, Chen W, Zhang Y. Increment of diversity with quadratic discriminant analysis – an efficient tool for sequence pattern recognition in bioinformatics. Open Access Bioinform. 2010;2:89–96. doi:10.2147/OAB.S10782
23. Álvarez VE, Quiroga MP, Centrón D. Identification of a specific biomarker of Acinetobacter baumannii global clone 1 by machine learning and PCR related to metabolic fitness of ESKAPE pathogens. mSystems. 2023;8(3):e0073422. doi:10.1128/msystems.00734-22
24. Hu X, Prehna G, Stebbins CE. Targeting plague virulence factors: a combined machine learning method and multiple conformational virtual screening for the discovery of Yersinia protein kinase A inhibitors. J Med Chem. 2007;50(17):3980–3983. doi:10.1021/jm070645a
25. Maltarollo VG, Kronenberger T, Espinoza GZ, Oliveira PR, Honorio KM. Advances with support vector machines for novel drug discovery. Expert Opin Drug Discov. 2019;14(1):23–33. doi:10.1080/17460441.2019.1549033
26. Smith NM, Lenhard JR, Boissonneault KR, et al. Using machine learning to optimize antibiotic combinations: dosing strategies for meropenem and polymyxin B against carbapenem-resistant Acinetobacter baumannii. Clin Microbiol Infect. 2020;26(9):1207–1213. doi:10.1016/j.cmi.2020.02.004
27. Macesic N, Bear Don’t Walk OJ 4th, Pe’er I, Tatonetti NP, Peleg AY, Uhlemann AC. Predicting phenotypic polymyxin resistance in Klebsiella pneumoniae through machine learning analysis of genomic data. mSystems. 2020;5(3):e00656–19. doi:10.1128/mSystems.00656-19
28. Ribeiro da Cunha B, Fonseca LP, Calado CRC. Metabolic fingerprinting with Fourier-transform infrared (FTIR) spectroscopy: towards a high-throughput screening assay for antibiotic discovery and mechanism-of-action elucidation. Metabolites. 2020;10(4):145. doi:10.3390/metabo10040145
29. Stokes JM, Yang K, Swanson K, et al. A deep learning approach to antibiotic discovery. Cell. 2020;180(4):688–702.e13. doi:10.1016/j.cell.2020.01.021
30. Lawrence TJ, Carper DL, Spangler MK, et al. amPEPpy 1.0: a portable and accurate antimicrobial peptide prediction tool. Bioinformatics. 2021;37(14):2058–2060. doi:10.1093/bioinformatics/btaa917
31. Mansbach RA, Leus IV, Mehla J, et al. Machine learning algorithm identifies an antibiotic vocabulary for permeating Gram-negative bacteria. J Chem Inf Model. 2020;60(6):2838–2847. doi:10.1021/acs.jcim.0c00352
32. Boulaamane Y, Molina Panadero I, Hmadcha A, et al. Antibiotic discovery with artificial intelligence for the treatment of Acinetobacter baumannii infections. mSystems. 2024;9(6):e0032524. doi:10.1128/msystems.00325-24
33. Peng J, Wang Y, Guan J, et al. An end-to-end heterogeneous graph representation learning-based framework for drug-target interaction prediction. Brief Bioinform. 2021;22(5):bbaa430. doi:10.1093/bib/bbaa430
34. Shao K, Zhang Y, Wen Y, Zhang Z, He S, Bo X. DTI-HETA: prediction of drug-target interactions based on GCN and GAT on heterogeneous graph. Brief Bioinform. 2022;23(3):bbac109. doi:10.1093/bib/bbac109
35. Zilian D, Sotriffer CA. SFCscore(RF): a random forest-based scoring function for improved affinity prediction of protein-ligand complexes. J Chem Inf Model. 2013;53(8):1923–1933. doi:10.1021/ci400120b
36. Li BH, Ge JQ, Wang YL, Wang LJ, Zhang Q, Bian C. Ligand-based and docking-based virtual screening of MDM2 inhibitors as potent anticancer agents. Comput Math Methods Med. 2021;2021:3195957. doi:10.1155/2021/3195957
37. Wang Y, Yang Y, Chen S, Wang J. DeepDRK: a deep learning framework for drug repurposing through kernel-based multi-omics integration. Brief Bioinform. 2021;22(5):bbab048. doi:10.1093/bib/bbab048
38. Cheng F, Lu W, Liu C, et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun. 2019;10(1):3476. doi:10.1038/s41467-019-10744-6
39. Sadeghi S, Lu J, Ngom A. An integrative heterogeneous graph neural network-based method for multi-labeled drug repurposing. Front Pharmacol. 2022;13:908549. doi:10.3389/fphar.2022.908549
40. Kaplon H, Chenoweth A, Crescioli S, Reichert JM. Antibodies to watch in 2022. MAbs. 2022;14(1):2014296. doi:10.1080/19420862.2021.2014296
41. Corti D, Purcell LA, Snell G, Veesler D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell. 2021;184(12):3086–3108. doi:10.1016/j.cell.2021.05.005
42. Sampei Z, Igawa T, Soeda T, et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8(2):e57479. doi:10.1371/journal.pone.0057479
43. Akpalu DE, Frederick B, Nnane IP, et al. Pharmacokinetics, pharmacodynamics, immunogenicity, safety, and tolerability of JNJ-61178104, a novel tumor necrosis factor-alpha and interleukin-17A bispecific antibody, in healthy subjects. J Clin Pharmacol. 2019;59(7):968–978. doi:10.1002/jcph.1393
44. Lopez-Rincon A, Tonda A, Mendoza-Maldonado L, et al. Classification and specific primer design for accurate detection of SARS-CoV-2 using deep learning. Sci Rep. 2021;11(1):947. doi:10.1038/s41598-020-80363-5
45. Randhawa GS, Hill KA, Kari L. MLDSP-GUI: an alignment-free standalone tool with an interactive graphical user interface for DNA sequence comparison and analysis. Bioinformatics. 2020;36(7):2258–2259. doi:10.1093/bioinformatics/btz918
46. Ching T, Himmelstein DS, Beaulieu-Jones BK, et al. Opportunities and obstacles for deep learning in biology and medicine. J R Soc Interface. 2018;15(141):20170387. doi:10.1098/rsif.2017.0387
47. Jin W, Wohlwend J, Barzilay R, Jaakkola TS. Iterative refinement graph neural network for antibody sequence-structure co-design. ICLR. 2022.
48. Ferruz N, Schmidt S, Höcker B. ProtGPT2 is a deep unsupervised language model for protein design. Nat Commun. 2022;13(1):4348. doi:10.1038/s41467-022-32007-7
49. Ruffolo JA, Gray JJ, Sulam J. Deciphering antibody affinity maturation with language models. 2021. arXiv preprint arXiv:2112.07782.
50. Shin JE, Riesselman AJ, Kollasch AW, et al. Protein design and variant prediction using autoregressive generative models. Nat Commun. 2021;12(1):2403. doi:10.1038/s41467-021-22732-w
51. Olsen TH, Moal IH, Deane CM. AbLang: an antibody language model for completing antibody sequences. Bioinform Adv. 2022;2(1):vbac046. doi:10.1093/bioadv/vbac046
52. Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, et al. RosettaAntibodyDesign (RAbD): a general framework for computational antibody design. PLoS Comput Biol. 2018;14(4):e1006112. doi:10.1371/journal.pcbi.1006112
53. Schneider G. Automating drug discovery. Nat Rev Drug Discov. 2018;17(2):97–113. doi:10.1038/nrd.2017.232
54. Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019;18(6):463–477. doi:10.1038/s41573-019-0024-5
55. Jayatunga MKP, Xie W, Ruder L, Schulze U, Meier C. AI in small-molecule drug discovery: a coming wave? Nat Rev Drug Discov. 2022;21(3):175–176. doi:10.1038/d41573-022-00025-1
56. Hu Q, Chen Y, Zou D, He Z, Xu T. Predicting adverse drug event using machine learning based on electronic health records: a systematic review and meta-analysis. Front Pharmacol. 2024;15:1497397. doi:10.3389/fphar.2024.1497397
57. Di Stefano M, Galati S, Ortore G, et al. Machine learning-based virtual screening for the identification of Cdk5 inhibitors. Int J Mol Sci. 2022;23(18):10653. doi:10.3390/ijms231810653
58. Wallach I, Dzamba M, Heifets A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure-based drug discovery. 2015. arXiv preprint arXiv:1510.02855.
59. Gentile F, Agrawal V, Hsing M, et al. Deep docking: a deep learning platform for augmentation of structure based drug discovery. ACS Cent Sci. 2020;6(6):939–949. doi:10.1021/acscentsci.0c00229
60. Elton DC, Boukouvalas Z, Fuge MD, Chung PW. Deep learning for molecular design—a review of the state of the art. Mol Syst Des Eng. 2019;4(4):828–849. doi:10.1039/C9ME00039A
61. Bai Q, Tan S, Xu T, Liu H, Huang J, Yao X. MolAICal: a soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief Bioinform. 2021;22(3):bbaa161. doi:10.1093/bib/bbaa161
62. Li Y, Pei J, Lai L. Structure-based de novo drug design using 3D deep generative models. Chem Sci. 2021;12(41):13664–13675. doi:10.1039/d1sc04444c
63. Imrie F, Bradley AR, van der Schaar M, Deane CM. Deep generative models for 3D linker design. J Chem Inf Model. 2020;60(4):1983–1995. doi:10.1021/acs.jcim.9b01120
64. Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci. 2018;4(1):120–131. doi:10.1021/acscentsci.7b00512
65. Maziarka Ł, Pocha A, Kaczmarczyk J, et al. Mol-CycleGAN: a generative model for molecular optimization. J Cheminform. 2020;12:2. doi:10.1186/s13321-019-0404-1
66. Merk D, Friedrich L, Grisoni F, Schneider G. De novo design of bioactive small molecules by artificial intelligence. Mol Inform. 2018;37(1–2):1700153. doi:10.1002/minf.201700153
67. Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019;37(9):1038–1040. doi:10.1038/s41587-019-0224-x
68. Bender A, Cortes-Ciriano I. Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 2: a discussion of chemical and biological data. Drug Discov Today. 2021;26(4):1040–1052. doi:10.1016/j.drudis.2020.11.037
69. Vayena E, Blasimme A, Cohen IG. Machine learning in medicine: addressing ethical challenges. PLoS Med. 2018;15(11):e1002689. doi:10.1371/journal.pmed.1002689
70. He J, Baxter SL, Xu J, Xu J, Zhou X, Zhang K. The practical implementation of artificial intelligence technologies in medicine. Nat Med. 2019;25(1):30–36. doi:10.1038/s41591-018-0307-0
71. Torkzadehmahani R, Nasirigerdeh R, Blumenthal DB, et al. Privacy-preserving artificial intelligence techniques in biomedicine. Methods Inf Med. 2022;61(S 01):e12–e27. doi:10.1055/s-0041-1740630
72. David L, Thakkar A, Mercado R, Engkvist O. Molecular representations in AI-driven drug discovery: a review and practical guide. J Cheminform. 2020;12(1):56. doi:10.1186/s13321-020-00460-5
73. Jimenez-Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nat Mach Intell. 2020;2:573–584. doi:10.1038/s42256-020-00236-4
74. Amann J, Blasimme A, Vayena E, Frey D, Madai VI. Explainability for artificial intelligence in healthcare: a multidisciplinary perspective. BMC Med Inform Decis Mak. 2020;20(1):310. doi:10.1186/s12911-020-01332-6
75. Boudza R, Bounou S, Segura-Garcia J, Moukadiri I, Maicas S. Artificial intelligence as a catalyst for antimicrobial discovery: from predictive models to de novo design. Microorganisms. 2026;14(2):394. doi:10.3390/microorganisms14020394
76. Joshi S, Sheth S. Artificial intelligence (AI) in pharmaceutical formulation and dosage calculations. Pharmaceutics. 2025;17(11):1440. doi:10.3390/pharmaceutics17111440
77. Serrano DR, Luciano FC, Anaya BJ, et al. Artificial intelligence (AI) applications in drug discovery and drug delivery: revolutionizing personalized medicine. Pharmaceutics. 2024;16(10):1328. doi:10.3390/pharmaceutics16101328
78. Hathout RM, Metwally AA. Towards better modelling of drug-loading in solid lipid nanoparticles: molecular dynamics, docking experiments and Gaussian processes machine learning. Eur J Pharm Biopharm. 2016;108:262–268. doi:10.1016/j.ejpb.2016.07.019
79. Abdalla Y, Elbadawi M, Ji M, et al. Machine learning using multi-modal data predicts the production of selective laser sintered 3D printed drug products. Int J Pharm. 2023;633:122628. doi:10.1016/j.ijpharm.2023.122628
80. Mater AC, Coote ML. Deep learning in chemistry. J Chem Inf Model. 2019;59(6):2545–2559. doi:10.1021/acs.jcim.9b00266
81. Howarth A, Ermanis K, Goodman JM. DP4-AI automated NMR data analysis: straight from spectrometer to structure. Chem Sci. 2020;11(17):4351–4359. doi:10.1039/d0sc00442a
82. Zhong ED, Bepler T, Berger B, Davis JH. CryoDRGN: reconstruction of heterogeneous cryo-EM structures using neural networks. Nat Meth. 2021;18(2):176–185. doi:10.1038/s41592-020-01049-4
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.