Back to Journals » International Medical Case Reports Journal » Volume 19
Apical Hypertrophic Cardiomyopathy Misdiagnosed as Hypertensive Heart Disease Due to Anchoring Bias in an African Man: A Case Report
Authors Okema JN ![]() , Chukwuocha C, Danvictor E, Uche CN
, Chukwuocha C, Danvictor E, Uche CN ![]() , Ekengwu CC, Egharevba JE, Ebunoluwa EO, Bongomin F
, Ekengwu CC, Egharevba JE, Ebunoluwa EO, Bongomin F ![]()
Received 27 February 2026
Accepted for publication 10 June 2026
Published 12 June 2026 Volume 2026:19 605653
DOI https://doi.org/10.2147/IMCRJ.S605653
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Thomas E Hutson
James Nelson Okema,1,2 Chigozie Chukwuocha,3 Ebirim Danvictor,3 Clinton Nwanaga Uche,4 Chigozie Charles Ekengwu,3,5 Jovita Eghe Egharevba,6 Esther Oniagba Ebunoluwa,7 Felix Bongomin1,8,9
1Faculty of Medicine, Gulu University, Gulu, Uganda; 2Department of Internal Medicine, St. Mary’s Hospital Lacor, Gulu, Uganda; 3Cardiology Unit, Department of Internal Medicine, Federal Teaching Hospital, Owerri, Nigeria; 4College of Medicine, Imo State University, Owerri, Imo State, Nigeria; 5Cardiology Unit, Department of Internal Medicine, University College Hospital, Ibadan, Nigeria; 6Department of Human Physiology, College of Medicine, Federal University of Technology, Owerri, Imo State, Nigeria; 7Department of Human Physiology, Olabisi Onabanjo University, Ago Iwoye, Ogun State, Nigeria; 8Department of Medical Microbiology and Immunology, Gulu University, Gulu, Uganda; 9Department of Internal Medicine, Gulu Regional Referral Hospital, Gulu, Uganda
Correspondence: James Nelson Okema, Department of Internal Medicine, St. Mary’s Hospital Lacor, P.O. Box 180, Gulu, Uganda, Email [email protected]
Background: Apical hypertrophic cardiomyopathy (ApHCM) is an uncommon variant of hypertrophic cardiomyopathy, accounting for approximately 5– 10% of all cases. It is frequently misdiagnosed as hypertensive heart disease in African patients with longstanding hypertension, particularly in resource-limited settings where cardiac magnetic resonance imaging (CMR) is unavailable. Anchoring bias, the tendency to adhere to an initial diagnosis despite contradictory evidence is a well-recognized contributor to this diagnostic error.
Case Presentation: A 60-year-old African man with a 15-year history of hypertension presented with exertional dyspnea and palpitations. Initial differentials included hypertensive heart disease, coronary artery disease, and dilated cardiomyopathy. Troponin I was negative. Electrocardiography revealed deep symmetric T-wave inversions (7– 9 mm) in anterolateral leads without voltage criteria for left ventricular hypertrophy. Transthoracic echocardiography demonstrated apical-predominant hypertrophy (apical wall 18 mm, posterior wall 11 mm; ratio 1.6), preserved ejection fraction (55– 60%), and classic systolic apical cavity obliteration with an ace-of-spades configuration. Speckle-tracking echocardiography showed selective apical longitudinal strain reduction (− 12%) with preserved basal strain (− 18%). These findings established ApHCM. Dual renin-angiotensin system blockade and unindicated clopidogrel were discontinued, bisoprolol was optimized, and symptoms resolved completely within two weeks.
Conclusion: Deep T-wave inversions without voltage criteria for left ventricular hypertrophy should prompt focused apical echocardiographic assessment, even in patients with longstanding hypertension. An apical-to-posterior wall thickness ratio of at least 1.5 with systolic apical cavity obliteration permits ApHCM diagnosis without CMR. Selective apical strain reduction distinguishes ApHCM from hypertensive heart disease. Recognizing anchoring bias is essential to avoid diagnostic delay when electrocardiographic and echocardiographic findings are discordant.
Keywords: apical hypertrophic cardiomyopathy, anchoring bias, hypertensive heart disease, echocardiography, strain imaging, Africa, resource-limited settings
Introduction
Apical hypertrophic cardiomyopathy (ApHCM) accounts for approximately 5 to 10% of hypertrophic cardiomyopathy cases worldwide and is an uncommon variant of HCM that remains infrequently reported in African populations, likely reflecting under-recognition rather than true absence.1,2 Hypertrophic cardiomyopathy is primarily an autosomal dominant genetic disorder caused by mutations in sarcomere protein-encoding genes, most commonly MYH7 (beta-myosin heavy chain) and MYBPC3 (myosin-binding protein C), though pathogenic variants in TNNI3, TNNT2, TPM1, and ACTC1 have also been identified.3 ApHCM is defined by apical wall thickness of at least 15 mm and an apical-to-posterior wall thickness ratio of at least 1.5, typically without left ventricular outflow tract obstruction.4,5
In settings where cardiac magnetic resonance imaging is unavailable, ApHCM commonly mimics hypertensive heart disease, particularly in patients with longstanding hypertension. Deep symmetric T-wave inversions without electrocardiographic voltage criteria for left ventricular hypertrophy represent an important diagnostic clue that may be overlooked.6 Failure to reconcile such discordant electrocardiographic and echocardiographic findings can delay recognition of apical cavity obliteration and characteristic strain abnormalities.7
The key differential diagnoses to consider in a patient presenting with exertional symptoms, deep T-wave inversions, and left ventricular hypertrophy include: hypertensive heart disease, apical hypertrophic cardiomyopathy, coronary artery disease with apical ischemia, cardiac amyloidosis, Fabry disease, athlete’s heart, and Takotsubo cardiomyopathy. Distinguishing features including wall thickness distribution, cavity morphology, strain patterns, biomarkers, and clinical context allow systematic exclusion of these conditions.
In many African centers, echocardiography remains the primary cardiac imaging modality. Systematic assessment of relative apical hypertrophy using simple wall thickness ratios can permit accurate diagnosis without advanced imaging.8 Speckle-tracking echocardiography has emerged as a valuable diagnostic tool that can demonstrate selective apical strain impairment, distinguishing ApHCM from hypertensive remodeling.9,10
This case illustrates how reliance on a prior diagnosis of hypertensive heart disease can obscure ApHCM despite emerging features that are inconsistent with hypertensive remodeling, highlighting the role of anchoring bias in diagnostic delay.10,11
Case Presentation
Patient Information and Clinical Findings
A 60-year-old African man presented to the cardiology clinic with one week of exertional dyspnea, generalized fatigue, and intermittent palpitations. He denied chest pain, syncope, orthopnea, or paroxysmal nocturnal dyspnea. His height was 172 cm and weight 78 kg, giving a body mass index of 26.4 kg/m2, blood pressure was 148/92 mmHg and heart rate were 76 beats per minute with a regular rhythm. No cardiac murmurs were audible. There was no peripheral edema, and respiratory examination was normal.
He had a 15-year history of hypertension, with antihypertensive therapy initiated eight years earlier and inconsistent blood pressure control (Table 1). He had been labeled with hypertensive heart disease based on prior investigations. A transthoracic echocardiogram performed several years earlier reportedly showed left ventricular hypertrophy, but no measurements or images were available for review. A chest radiograph at that time demonstrated cardiomegaly.
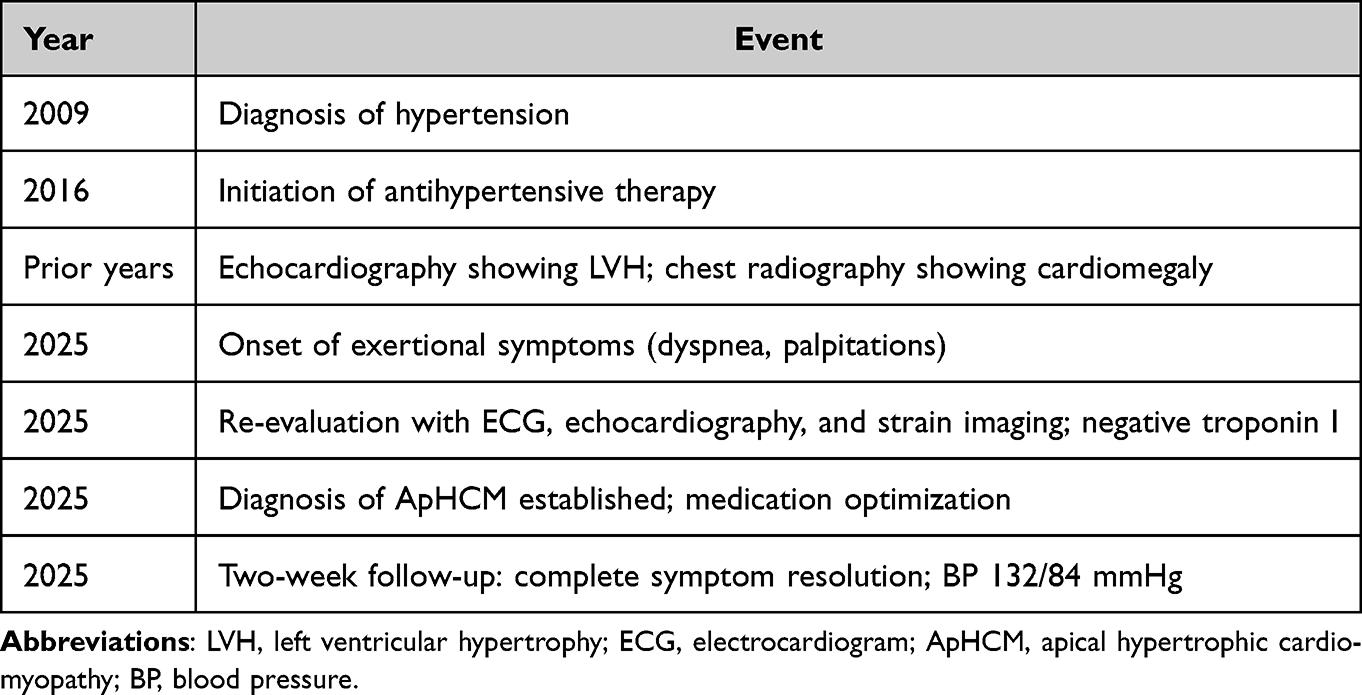 |
Table 1 Diagnostic Timeline Showing Progression From Initial Diagnosis of Hypertension in 2009 to Recognition of Apical Hypertrophic Cardiomyopathy in 2025 |
Past Medical History
The patient had a longstanding diagnosis of systemic hypertension (15 years), managed with antihypertensive therapy for the last eight years. He was on amlodipine, bisoprolol, losartan, telmisartan-hydrochlorothiazide, and clopidogrel. Clopidogrel had been initiated years earlier for undocumented indications.
There was no prior documented history of diabetes mellitus, chronic kidney disease, coronary artery disease, cerebrovascular accident, peripheral arterial disease, or prior cardiac procedures. He had no known history of rheumatic heart disease or thyroid disorders. His renal function at presentation showed mild impairment with estimated glomerular filtration rate (eGFR) of 45 mL/min/1.73 m2, with no prior documentation of renal disease.
Family History
There was no family history of cardiomyopathy, sudden unexplained cardiac death, relatives diagnosed with an enlarged heart, or early-onset heart failure in first-degree relatives under 50 years of age. While a detailed multigenerational family history was not formally elicited at first presentation, a recognized limitation of this case, no spontaneously reported family history of cardiac disease was identified. Genetic counseling and formal family screening were recommended following the ApHCM diagnosis, given the autosomal dominant inheritance pattern. He did not smoke or consume alcohol.
Diagnostic Assessment
Twelve-lead electrocardiography showed sinus rhythm with normal PR, QRS, and QT intervals. Sokolow-Lyon and Cornell voltage criteria for left ventricular hypertrophy were not met. Deep symmetric giant T-wave inversions measuring seven to nine mm were present in leads V4 to V6, I, and aVL, consistent with the characteristic electrocardiographic pattern of apical hypertrophic cardiomyopathy without voltage criteria for left ventricular hypertrophy (Figure 1).
 |
Figure 1 Twelve-lead electrocardiogram showing sinus rhythm with deep symmetric T-wave inversions (7–9 mm) in leads V2–V6, I, and aVL. Voltage criteria for left ventricular hypertrophy (Sokolow-Lyon and Cornell) are not met. The red circle indicates the deep symmetric T-wave inversions in precordial leads V4–V6 and similar T-wave changes in limb leads I and aVL. This voltage-negative T-wave pattern is characteristic of ApHCM. |
Transthoracic echocardiography demonstrated marked apical-predominant hypertrophy with a maximal apical wall thickness of 18 mm and posterior wall thickness of 11 mm, yielding an apical-to-posterior wall thickness ratio of 1.6. Classic systolic apical cavity obliteration with an ace-of-spades configuration was present (Figure 2). Left ventricular ejection fraction was preserved at 55 to 60%. There was no left ventricular outflow tract obstruction at rest or with Valsalva maneuver.
 |
Figure 2 Transthoracic echocardiogram (parasternal long-axis view, end-diastolic and end-systolic frames) showing apical-predominant hypertrophy (apical wall thickness 18 mm, posterior wall thickness 11 mm; apical-to-posterior ratio 1.6). The red circle identifies the classic “ace-of-spades” systolic apical cavity obliteration, a pathognomonic feature of ApHCM distinguishing it from the concentric hypertensive remodeling pattern. |
Speckle-tracking strain imaging (Figure 3) showed selective reduction in apical longitudinal strain at minus 12%, with preserved basal strain at minus 18%. This pattern contrasts with the diffuse strain impairment typically seen in hypertensive heart disease. Strain imaging was available locally as part of routine echocardiographic assessment and did not require cardiac magnetic resonance imaging. Diastolic assessment showed impaired relaxation with mild left atrial enlargement.
 |
Figure 3 Speckle-tracking longitudinal strain bull’s-eye plot demonstrating selective reduction in apical longitudinal strain (−12%) with preserved basal strain (−18%). The Orange-shaded apical segments indicate impaired strain, while blue-shaded basal segments indicate preserved deformation. This selective apical strain reduction pattern is characteristic of ApHCM and contrasts with the diffuse strain impairment typically observed in hypertensive heart disease. |
Twenty-four-hour Holter monitoring showed predominant sinus rhythm with occasional premature atrial contractions and no non-sustained ventricular tachycardia. Laboratory tests showed hemoglobin of 13.2 g/dL, serum creatinine of 1.4 mg/dL corresponding to an estimated glomerular filtration rate of 45 mL/min/1.73 m2, and negative troponin I, effectively excluding acute myocardial injury and reducing the likelihood of acute coronary syndrome as a competing diagnosis.
Chest radiography (Figure 4) demonstrated mild cardiomegaly with a cardiothoracic ratio of 0.53 on the posteroanterior view, with clear lung fields and no evidence of focal consolidation, pleural effusion, or pulmonary vascular congestion; the lateral view confirmed borderline cardiac enlargement without retrosternal mass or significant hilar prominence. Taken together, the radiographic appearances were interpreted as nonspecific cardiomegaly consistent with hypertensive heart disease, a conclusion that initially obscured the underlying diagnosis of apical hypertrophic cardiomyopathy.
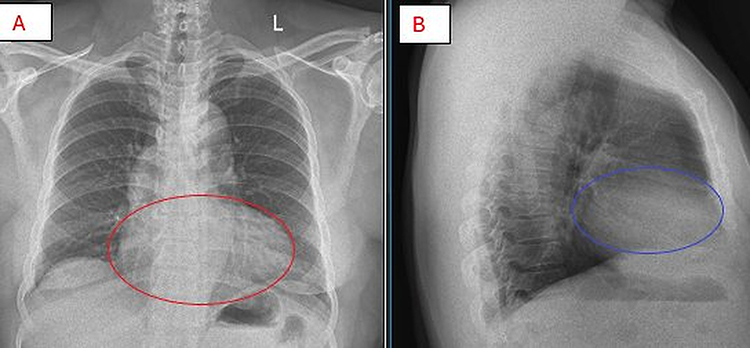 |
Figure 4 Chest radiography in posteroanterior (PA) view (A) and lateral view (B), where the red circle in (A) outlines mild cardiomegaly with a cardiothoracic ratio of 0.53 amid otherwise clear lung fields without focal consolidation, pleural effusion, or pulmonary vascular congestion, the blue circle in (B) demarcates borderline cardiac enlargement on lateral view without retrosternal mass or significant hilar prominence, with these combined findings consistent with nonspecific cardiomegaly that initially reinforced the hypertensive heart disease diagnosis rather than suggesting apical hypertrophic cardiomyopathy. |
Differential Diagnosis
The primary differential was hypertensive heart disease, given 15 years of suboptimal blood pressure control with documented left ventricular hypertrophy and cardiomegaly. However, several key discordant features collectively favored apical hypertrophic cardiomyopathy. Deep anterolateral T-wave inversions measuring 7–9 mm was present without voltage criteria for left ventricular hypertrophy, a pattern atypical for hypertensive remodeling. Echocardiography revealed apical-predominant hypertrophy with an apical-to-posterior wall thickness ratio of 1.6, contrasting with the concentric remodeling expected in hypertensive heart disease, and classic “ace-of-spades” systolic apical cavity obliteration, a finding absents in uncomplicated hypertension. Speckle-tracking strain imaging demonstrated selective apical strain impairment (apical strain −12%, basal strain −18%), in contrast to the diffuse strain reduction typically observed in hypertensive heart disease. Troponin I was negative, effectively excluding acute myocardial injury and arguing against acute coronary syndrome or apical ischemia as the primary diagnosis. Cardiac amyloidosis was considered but rendered unlikely by the absence of biventricular thickening, restrictive filling, pericardial effusion, or characteristic sparkling echotexture. Fabry disease was similarly excluded on clinical grounds, given the absence of corneal verticillata, angiokeratomas, or a family history consistent with X-linked inheritance. Taken together, these pathognomonic features established ApHCM despite anchoring bias from the longstanding hypertensive diagnosis.
Therapeutic Intervention
Bisoprolol was continued as first-line therapy for symptomatic non-obstructive ApHCM, consistent with 2024 hypertrophic cardiomyopathy guidelines.12 Dual renin-angiotensin system blockade was rationalized by discontinuing telmisartan-hydrochlorothiazide while retaining losartan with renal function monitoring (baseline eGFR 45 mL/min/1.73 m2). Clopidogrel was stopped due to undocumented indication, eliminating iatrogenic bleeding risk. Systolic blood pressure target was set at 130–140 mmHg to balance hypertension control against excessive afterload reduction. Cardiac magnetic resonance imaging with late gadolinium enhancement was recommended for comprehensive sudden cardiac death risk stratification.13,14
Follow-Up and Outcomes
At two-week follow-up, the patient reported complete resolution of exertional dyspnea and palpitations. Blood pressure was controlled at 132/84 mmHg with no adverse drug effects or interim arrhythmic events. Repeat echocardiography has not been performed. Sudden cardiac death risk stratification remains pending cardiac magnetic resonance imaging to assess late gadolinium enhancement burden and scar distribution. The patient was counseled regarding ApHCM prognosis, activity restrictions, and arrhythmia symptoms. Ongoing cardiology follow-up was arranged with beta-blocker optimization and blood pressure monitoring.
Discussion
Hypertensive heart disease typically produces concentric left ventricular hypertrophy, electrocardiographic voltage criteria for hypertrophy, and diffuse or basal-predominant longitudinal strain impairment. In contrast, this patient showed apical-predominant hypertrophy, systolic apical cavity obliteration, absence of voltage-defined hypertrophy, and selective apical strain reduction. This constellation is characteristic of apical hypertrophic cardiomyopathy and is not explained by hypertensive remodeling alone.15
ApHCM, like other HCM phenotypes, arises predominantly from autosomal dominant mutations in sarcomere protein-encoding genes. The most commonly implicated genes are MYH7 and MYBPC3, with additional contributions from TNNI3, TNNT2, TPM1, and ACTC1.3 Notably, ApHCM exhibits considerable clinical heterogeneity. The clinical presentation ranges from asymptomatic incidental discovery to exertional dyspnea, palpitations, chest pain, or less commonly, sudden cardiac death. Some patients experience progressive disease with diastolic dysfunction, atrial fibrillation, or apical aneurysm formation, while others maintain a benign long-term course.4,5 This phenotypic variability, combined with the frequent co-existence of hypertension (a common comorbidity in older adults), substantially increases the risk of misdiagnosis in clinical practice. The variability of genetic penetrance and expression means that even family members carrying the same pathogenic variant may present with markedly different clinical phenotypes, further complicating recognition in index cases.3
The presence of giant T-wave inversions without electrocardiographic voltage criteria for left ventricular hypertrophy is a critical diagnostic clue for ApHCM, even in patients with longstanding hypertension.6,16 In this case, the non-obstructive phenotype and absence of a cardiac murmur may have falsely reassured clinicians and reinforced the existing diagnosis of hypertensive heart disease. Such discordant findings should prompt diagnostic reassessment rather than confirmation of the initial label.
Relative apical hypertrophy assessed by simple wall thickness ratios has emerged as a practical diagnostic approach when cardiac magnetic resonance imaging is unavailable. An apical-to-posterior wall thickness ratio of at least 1.5, combined with systolic apical cavity obliteration, permits confident diagnosis of ApHCM using transthoracic echocardiography alone.4,5,17 Although basal septal thickness is often used as the reference, the posterior wall was selected because septal hypertrophy was not disproportionate and the posterior wall provided a stable non-apical comparator.17 Recent studies have refined diagnostic criteria using indexed apical wall thickness thresholds, improving diagnostic accuracy to 92% compared with 69% using conventional unindexed thresholds.18
Speckle-tracking echocardiography further strengthens diagnostic confidence by demonstrating selective apical strain impairment, a pattern that contrasts with the diffuse strain reduction seen in hypertensive heart disease.9,10,19 Regional strain analysis using two-dimensional speckle tracking has demonstrated decreased deformation mainly confined to the mesocardium in apical HCM, while endocardial deformation may be preserved.20 Importantly, strain imaging can be performed during routine echocardiography and does not require advanced imaging infrastructure.
ApHCM is generally associated with lower sudden cardiac death risk than septal phenotypes, although risk stratification remains essential.12–14 The 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guidelines recommend 24–48-hour ambulatory electrocardiographic monitoring for sudden cardiac death risk stratification in all hypertrophic cardiomyopathy patients.12 Late gadolinium enhancement on cardiac magnetic resonance imaging provides additional prognostic information regarding fibrosis burden and sudden cardiac death risk.21
Anchoring bias has been well described as a cause of diagnostic delay when clinicians adhere to an initial diagnosis despite emerging contradictory data.11,22,23 Croskerry identified anchoring as one of the most common cognitive biases in clinical reasoning, occurring when initial diagnostic impressions persist despite accumulating evidence to the contrary.24 Norman et al further elaborated that such cognitive biases, combined with atypical presentations and systemic factors, account for the majority of diagnostic errors in clinical practice.25
In this case, prolonged reliance on a prior diagnosis of hypertensive heart disease delayed recognition of ApHCM despite clear electrocardiographic and echocardiographic discordance. This case demonstrates that cognitive bias can delay diagnosis even when diagnostic tools are available. Similar cases of ApHCM misdiagnosis have been reported in African patients, emphasizing the need for heightened diagnostic vigilance in this population.2,26
Systematic reassessment of established diagnoses in the presence of discordant findings is essential, particularly in resource-limited settings where access to advanced imaging is constrained and clinical vigilance is critical.24 Educational interventions focused on cognitive bias recognition and systematic diagnostic reassessment have shown promise in reducing diagnostic errors.27
Limitations
This case report has several limitations that should be acknowledged. First, cardiac magnetic resonance imaging with late gadolinium enhancement was not performed, precluding definitive tissue characterization and comprehensive sudden cardiac death risk stratification. Second, genetic testing was not undertaken, so a causative sarcomere gene mutation could not be confirmed; the coincident presence of longstanding hypertension means that a contribution of hypertensive remodeling to the observed phenotype cannot be entirely excluded. Third, a formal multigenerational family history was not systematically elicited at the initial consultation, limiting cascade screening evaluation. Fourth, the absence of prior echocardiographic measurements made it impossible to determine the timeline of hypertrophy development. Fifth, the short follow-up period (two weeks) precludes assessment of long-term outcomes. These limitations are common in resource-limited settings and reflect the real-world diagnostic challenges this case is intended to highlight.
Conclusion
This case highlights a clinically important and underrecognized diagnostic pitfall: the misdiagnosis of ApHCM as hypertensive heart disease driven by anchoring bias. The key clinical implication is that deep symmetric T-wave inversions without electrocardiographic voltage criteria for left ventricular hypertrophy should immediately prompt focused apical echocardiographic assessment, even in patients with longstanding hypertension. An apical-to-posterior wall thickness ratio of at least 1.5 combined with systolic apical cavity obliteration is sufficient to establish the diagnosis of ApHCM without CMR in resource-limited settings. Selective apical longitudinal strain reduction with preserved basal strain, demonstrated by speckle-tracking echocardiography, provides an additional discriminating feature. Clinicians should maintain awareness that HCM is a genetic sarcomere disorder that can coexist with hypertension and that recognition requires systematic re-evaluation of discordant findings rather than anchoring to an established diagnosis. Teaching cognitive bias recognition alongside pattern recognition of ApHCM may reduce a meaningful proportion of diagnostic errors that no additional imaging alone would prevent.
Patient Perspective
The patient expressed relief at receiving a definitive diagnosis after years of suboptimal symptom control. He appreciated understanding that his condition was distinct from simple hypertensive heart disease and required different management strategies. He was motivated to maintain regular cardiology follow-up and medication adherence.
Ethics Approval and Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the consent is available for review by the Editor-in-Chief. This is a case report. Formal ethics committee approval was not required according to institutional policy.
Acknowledgments
The authors thank the staffs of the Cardiology Unit, Department of Internal Medicine, Federal Teaching Hospital, Owerri, Nigeria for their support in patient care.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, or publication of this article.
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Paluszkiewicz J, Krasinska B, Milting H, et al. Apical hypertrophic cardiomyopathy: diagnosis, medical and surgical treatment. Kardiochirurgia I Torakochirurgia Polska. 2018;15(4):246–9. doi:10.5114/kitp.2018.80922
2. Rugbeer Y, Ismail M, Nadar S, et al. The ace of spades: apical hypertrophic cardiomyopathy in an African Male Patient. Cureus. 2022;14(2):e22514. doi:10.7759/cureus.22514
3. Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation. 1995;92(4):785–789. doi:10.1161/01.cir.92.4.785
4. Rouskas P, Zegkos T, Ntelios D, et al. Prevalence, characteristics, and natural history of apical phenotype in a large cohort of patients with hypertrophic cardiomyopathy. Hellenic J Cardiol. 2023;73:8–15. doi:10.1016/j.hjc.2023.02.004
5. Eriksson MJ, Sonnenberg B, Woo A, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39(4):638–645. doi:10.1016/s0735-1097(01)01778-8
6. Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381(9862):242–255. doi:10.1016/S0140-6736(12)60397-3
7. Moon JC, Fisher NG, McKenna WJ, Pennell DJ. Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart. 2004;90(6):645–649. doi:10.1136/hrt.2003.014969
8. Kitaoka H, Doi Y, Casey SA, Hitomi N, Furuno T, Maron BJ. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003;92(10):1183–1186. doi:10.1016/j.amjcard.2003.07.027
9. Carasso S, Yang H, Woo A, et al. Systolic myocardial mechanics in hypertrophic cardiomyopathy: novel concepts and implications for clinical status. J Am Soc Echocardiogr. 2008;21(6):675–683. doi:10.1016/j.echo.2007.10.021
10. Maron MS, Maron BJ, Harrigan C, et al. Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009;54(3):220–228. doi:10.1016/j.jacc.2009.05.006
11. Croskerry P. The importance of cognitive errors in diagnosis and strategies to minimize them. Acad Med. 2003;78(8):775–780. doi:10.1097/00001888-200308000-00003
12. Ommen SR, Mital S, Burke MA, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy. Circulation. 2024;149(23):e1239–e1311. doi:10.1161/CIR.0000000000001250
13. O’Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM Risk-SCD). Eur Heart J. 2014;35(30):2010–2020. doi:10.1093/eurheartj/eht439
14. Chan RH, Maron BJ, Olivotto I, et al. Prognostic value of quantitative contrast-enhanced cardiovascular magnetic resonance for the evaluation of sudden death risk in patients with hypertrophic cardiomyopathy. Circulation. 2014;130(6):484–495. doi:10.1161/CIRCULATIONAHA.113.007094
15. Nagueh SF, Bierig SM, Budoff MJ, et al. American Society of Echocardiography clinical recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy. J Am Soc Echocardiogr. 2011;24(5):473–498. doi:10.1016/j.echo.2011.03.006
16. Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol. 1979;44(3):401–412. doi:10.1016/0002-9149(79)90388-6
17. Kubo T, Gimeno JR, Bahl A, et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J Am Coll Cardiol. 2007;49(25):2419–2426. doi:10.1016/j.jacc.2007.02.061
18. Hughes RK, Shiwani H, Rosmini S, et al. Improved diagnostic criteria for apical hypertrophic cardiomyopathy. JACC Cardiovasc Imaging. 2024;17(5):501–512. doi:10.1016/j.jcmg.2023.07.012
19. Saito M, Okayama H, Yoshii T, et al. Clinical significance of global two-dimensional strain as a surrogate parameter of myocardial fibrosis and cardiac events in patients with hypertrophic cardiomyopathy. Eur Heart J Cardiovasc Imaging. 2012;13(7):617–623. doi:10.1093/ejechocard/jer318
20. Saccheri MC, Cianciulli TF, Morita LA, et al. Speckle tracking echocardiography to assess regional ventricular function in patients with apical hypertrophic cardiomyopathy. World J Cardiol. 2017;9(4):363–370. doi:10.4330/wjc.v9.i4.363
21. Maron MS, Rowin EJ, Maron BJ. How to image hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2017;10(7):e005372. doi:10.1161/CIRCIMAGING.116.005372
22. Graber ML, Franklin N, Gordon R. Diagnostic error in internal medicine. Arch Intern Med. 2005;165(13):1493–1499. doi:10.1001/archinte.165.13.1493
23. Hussain I, Rather SH, Yatoo GN. Enhancing diagnostic reasoning through cognitive bias awareness in medical students: a case-based learning initiative. Cureus. 2025;17(6):e65403. doi:10.7759/cureus.65403
24. Croskerry P. Achieving quality in clinical decision making: cognitive strategies and detection of bias. Acad Emerg Med. 2002;9(11):1184–1204. doi:10.1111/j.1553-2712.2002.tb01574.x
25. Norman GR, Monteiro SD, Sherbino J, Ilgen JS, Schmidt HG, Mamede S. The causes of errors in clinical reasoning: cognitive biases, knowledge deficits, and dual process thinking. Acad Med. 2017;92(1):23–29. doi:10.1097/ACM.0000000000001421
26. Adedeji AT, Gqunta KM, Thienemann F, Chin A, Herbst PG, Ntusi NA. Apical hypertrophic cardiomyopathy: a case report from South Africa highlighting diagnostic challenges. Cardiovasc J Afr. 2019;30(5):e1–e4. doi:10.5830/CVJA-2019-017
27. Graber ML, Kissam S, Payne VL, et al. Cognitive interventions to reduce diagnostic error: a narrative review. BMJ Qual Saf. 2012;21(7):535–557. doi:10.1136/bmjqs-2011-000149
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.