Back to Journals » Nature and Science of Sleep » Volume 18

Obstructive Sleep Apnea Syndrome and Ischemic Stroke: Investigating Inflammatory Mechanisms
Authors Zhang Z ![]() , Zhu Z, Chen Z, Liu X, Yu W, Aqdas T
, Zhu Z, Chen Z, Liu X, Yu W, Aqdas T ![]() , Yuan R
, Yuan R
Received 25 November 2025
Accepted for publication 8 March 2026
Published 23 March 2026 Volume 2026:18 581040
DOI https://doi.org/10.2147/NSS.S581040
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Ahmed BaHammam
Zihao Zhang,1,2 Ziqing Zhu,1,2 Zhiying Chen,2,3 Xiaoqun Liu,1,2 Wenmin Yu,4,5 Toheed Aqdas,6 Rui Yuan7
1Department of Respiratory and Critical Care, School of Clinical Medicine, Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China; 2Jiujiang Clinical Precision Medicine Research Center, Jiujiang, Jiangxi, 332000, People’s Republic of China; 3Department of Neurology, School of Clinical Medicine, Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China; 4Jiangxi Provincial Key Laboratory of Cell Precision Therapy, Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China; 5The School of Basic Medical Science, Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China; 6Department of Otolaryngology, Head and Neck Surgery, Affiliated Hospital of Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China; 7Faculty of International Studies, Jiujiang University, Jiujiang, Jiangxi, 332005, People’s Republic of China
Correspondence: Rui Yuan, Department of Otolaryngology, Head and Neck Surgery, Affiliated Hospital of Jiujiang University, Jiujiang, Jiangxi, 332000, People’s Republic of China, Email [email protected]
Abstract: Obstructive Sleep Apnea Syndrome (OSAS) is characterized by repeated episodes of complete (apnea) or partial (hypopnea) collapse of the upper airway during sleep, leading to chronic intermittent hypoxia and/or hypercapnia, and sleep fragmentation. This review systematically examines the mechanisms linking OSAS and ischemic stroke, with particular emphasis on the roles of the PI3K/Akt signaling pathway, Toll-like receptor signaling pathway, and NLRP3 inflammasome in OSAS-related inflammatory responses, and their contributions to the pathogenesis and progression of ischemic stroke. We also explore how OSAS affects stroke recovery through impaired sleep quality and increased cardiovascular stress, potentially worsening prognosis. This article discusses potential anti-inflammatory therapeutic strategies, underscores the importance of early OSAS intervention in reducing recurrence and improving outcomes, and offers new perspectives on OSAS-related ischemic stroke mechanisms and clinical management.
Keywords: obstructive sleep apnea syndrome, ischemic stroke, inflammatory mechanisms, signalling pathways
Introduction
Obstructive Sleep Apnea Syndrome (OSAS) is a prevalent sleep disorder characterized by recurrent partial or complete upper airway collapse, apnea episodes, and hypoventilation. This condition typically results in intermittent hypoxia, hypercapnia, sleep disruption, and daytime fatigue.1 Epidemiological data indicate that approximately 936 million individuals worldwide aged 30–69 years have moderate to severe OSAS, with about 425 million classified as moderate to severe cases. China reports the highest prevalence rates, followed by the United States, Brazil, and India. The global burden of OSAS continues to rise.2 OSAS significantly impacts daily functioning and overall health, highlighting the need to understand its fundamental mechanisms for improved prevention and treatment strategies. Substantial evidence demonstrates a strong association between OSAS and ischemic stroke (IS).
Ischemic stroke represents a major global health challenge, presenting complex prognostic and therapeutic difficulties. Common manifestations include hemiparesis, speech articulation deficits, aphasia, sensory disturbances, and visual impairment. Currently affecting at least 77.19 million people worldwide, the incidence of IS is projected to increase.3 This review employs current experimental evidence to explore the inflammatory mechanisms connecting OSAS and IS, with the aim of providing valuable insights for future research in this field.
Search Strategy and Selection Criteria
This article is a narrative review focusing on the inflammatory mechanisms linking OSAS and ischemic stroke. We conducted a comprehensive literature search in major databases including PubMed, Web of Science, and Embase up to December 2024. The search strategy employed combinations of the following keywords: “Obstructive Sleep Apnea”, “Ischemic Stroke”, “Inflammation”, “PI3K/Akt signaling”, “Toll-like receptor”, “NLRP3 inflammasome”, and “Intermittent Hypoxia”. We primarily selected original research articles, systematic reviews, and meta-analyses published in English. Studies were included if they provided mechanistic insights into the inflammatory pathways connecting sleep apnea and stroke pathologies. We excluded abstracts without full text, case reports, and studies with insufficient relevance to the specific signaling pathways discussed.
The Pathogenesis of OSAS
The pathogenesis of OSAS involves both anatomical and functional components. Anatomical factors encompass upper airway collapse and morphological abnormalities, while functional elements include nocturnal rostral fluid displacement, reduced respiratory threshold, and increased loop gain.
Anatomic Factors Leading to OSAS
Most OSAS patients present varying degrees of anatomical abnormalities in the upper respiratory tract.4 These structural variations are strongly influenced by BMI, age,5 and gender.6 Respiratory failure in OSAS can be categorized as active or passive. Active collapse relates to impaired neuromuscular control of airway dilator muscles, while passive collapse involves the anatomical structure of the upper airway. Characteristic morphological abnormalities include elongation of the soft palate, mandibular positioning issues, facial structural variations, and increased volume of peripharyngeal soft tissues.7,8 These anatomical alterations compromise upper airway patency and contribute significantly to OSAS development.
Functional Factors Contributing to OSAS
Functional factors affecting OSAS incidence and severity primarily involve upper respiratory tract dynamics. Nocturnal rostral fluid transfer plays a crucial role in OSAS severity, particularly following IS.9 During daytime hours, gravitational forces cause fluid accumulation in the lower extremities. Upon assuming a recumbent position at night, this fluid redistributes from the legs to the neck region, resulting in upper airway narrowing that may precipitate or exacerbate OSAS. Reducing daytime leg fluid accumulation may consequently ameliorate OSAS severity.9,10
The Respiratory Arousal Threshold (AT), representing the physiological measure of respiratory effort required to initiate sleep arousal, represents another relevant factor.11 Hyperventilation following arousal events can induce central sleep apnea, affecting vagus and hypoglossal nerve output, decreasing airway sphincter activity, and increasing OSAS susceptibility.12 This mechanism affects approximately half of patients with moderate to severe OSAS, contributing to daytime sleepiness and attentional deficits.13
Loop gain, quantifying respiratory stability and central apnea susceptibility, constitutes an additional important factor.14 Elevated loop gain in OSAS may reflect heightened sensitivity to CO2 fluctuations, potentially decreasing upper airway dilator activity and promoting recurrent respiratory events.15
Emerging research suggests associations between gut microbiota alterations and OSAS, with microbiota fluctuations linked to intermittent hypoxia and sleep fragmentation.16 In summary, OSAS represents a complex interplay between anatomical and functional factors, with sleep fragmentation and upper respiratory failure serving as key drivers. Future investigations should integrate multi-omics approaches to further elucidate molecular networks and identify novel targets for precision interventions.
Pathogenesis of Ischemic Stroke
Ischemic stroke constitutes an acute, focal neurological disorder resulting from inadequate cerebral blood flow. This may occur when blood clots obstruct circulation, either through local thrombus formation (thrombotic stroke) or embolic migration from distant sites (embolic stroke). Additionally, IS can arise from structural or functional abnormalities in cerebral vasculature that impair adequate blood supply.17
The pathophysiology of IS involves complex cascade reactions including energy metabolism impairment and excitotoxicity, oxidative stress and free radical injury, and inflammation with immune activation.
Energy Metabolism Impairment and Excitotoxicity
Cerebral ischemia reduces neuronal NAD+, an essential cofactor in mitochondrial respiratory chain function and DNA repair enzymes (including PARP1 and Sirtuins), leading to energy depletion and mitochondrial respiratory chain blockade. During ischemia, neurons shift from the pentose phosphate pathway (PPP) to glycolysis, reducing NADPH and increasing oxidative stress. In excitotoxicity, cerebral ischemia triggers glutamate release from neurons and astrocytes, with subsequent glutamate receptor overactivation causing excessive calcium influx that initiates downstream toxic signaling pathways.18
Oxidative Stress and Free Radical Damage
Both ROS and RNS contribute to oxidative stress injury in IS, with ROS serving as the primary mediators. Mitochondrial dysfunction following ischemia and reperfusion increases ROS production. Pathological ROS effects manifest through three major categories: lipid peroxidation, protein oxidation, and DNA damage. ROS also induce neuronal apoptosis via mitochondrial and death receptor pathways (such as Fas/FasL). Under inflammatory conditions with intact blood-brain barrier, ROS activate specific signaling pathways that promote protease and inflammatory mediator release, ultimately leading to blood-brain barrier disruption.17
Inflammation and Immune Activation
Local cerebral blood flow disruption following IS can induce hypoxic neuronal death and activate both microglia and peripheral immunity. Microglia release proinflammatory factors through activation of related signaling pathways and recruit neutrophils and monocytes for infiltration. Neutrophils produce matrix metalloproteases (MMP-9) that damage the blood-brain barrier and generate neutrophil extracellular traps (NETs) that exacerbate ischemia; Th1/Th17 cells produce IFN-γ and IL-17, activating astrocytes and amplifying inflammation. Concurrently, regulatory T cells (Treg) inhibit excessive immune responses and promote repair through IL-10 and PD-1 mechanisms.19
The pathological cascade centers on disturbances in energy metabolism, oxidative stress, and inflammation. Targeting these components (for instance, through ROS production inhibition or microglial polarization modulation) may represent crucial strategies for minimizing neuronal injury.
Mechanisms of OSAS-Induced and Exacerbated Ischemic Stroke
Although the association between OSAS and IS has been extensively investigated, the precise causal relationships remain incompletely elucidated.20,21 Nevertheless, substantial evidence confirms that OSAS significantly contributes to IS development and progression, with varying OSAS severity conferring differential IS risk. Patients with moderate or severe OSAS face elevated IS risk compared to those with mild or no OSAS.22
OSAS is known to promote IS through cerebral hemodynamic alterations and atherosclerosis, with established evidence demonstrating that OSAS dramatically increases stroke risk independent of other risk factors.23 Research indicates that OSAS induces oxidative stress and inflammation via intermittent hypoxia and sleep fragmentation, characterized by reactive oxygen species activation resulting from oxidative stress and inflammation triggered by activated hypoxia-inducible factors (HIFs).24,25
OSAS-Induced IS
Inflammation links both cerebral hemodynamics and atherosclerosis in OSAS-related IS pathogenesis. Regarding cerebral blood flow, severe OSAS affects cerebral circulation through cyclic vasodilation and constriction, sympathetic nervous system activation, and impacts on cerebral autoregulatory mechanisms. Inflammation itself may alter cerebral hemodynamics. OSAS induces transient cerebral blood flow changes that can result in cerebral tissue ischemia.25
Concerning atherosclerosis, OSAS can induce vascular endothelial disorders that exacerbate atherosclerosis in major arteries, potentially leading to cerebral blood flow disruptions and subsequent IS.26,27 Importantly, OSAS-induced IS does not operate through isolated mechanisms but rather involves intermittent hypoxia and inflammatory responses that both directly impair cerebrovascular function and activate the NLRP3 inflammasome, which associates with atherosclerotic pathological processes. Inflammasome activation in atherosclerotic plaques may promote plaque instability, thereby increasing cardiovascular event risk. According to the “inflammatory aging” theory linking aging with inflammation, inflammatory markers may predict atherosclerotic stroke risk.28
It should be emphasized that OSAS-induced IS involves multiple interconnected pathways, with intermittent hypoxia and inflammatory responses triggered by OSAS directly impairing cerebrovascular function while indirectly facilitating atherosclerotic plaque development and rupture through activation of molecular pathways such as the NLRP3 inflammasome. Furthermore, research indicates that activation of these inflammatory pathways contributes not only to stroke onset but also plays crucial roles in post-stroke injury amplification. For example, intermittent hypoxia exacerbates neuronal death through mitochondrial dysfunction and oxidative stress, while persistent inflammasome activation aggravates blood-brain barrier disruption and neuroinflammatory infiltration.
OSAS Exacerbates IS Injury
Based on the aforementioned mechanisms, the aggravating effects of OSAS on IS injury primarily manifest in three domains: energy metabolism disturbance, oxidative stress, and immune activation.
Regarding energy metabolism and excitotoxicity, OSAS worsens post-ischemic injury; intermittent hypoxia in IS parallels post-ischemic conditions, leading to mitochondrial dysfunction. Intermittent hypoxic preconditioning has been found to exacerbate mitochondrial permeability transition pore opening, accelerating neuronal dysfunction.29
Concerning oxidative stress and free radical damage, intermittent hypoxia promotes excessive ROS and RNS release, exacerbating ischemic perfusion injury after IS and leading to neuronal death and infarct expansion. Animal studies demonstrate that severe intermittent hypoxia (6% O2) significantly increases brain tissue levels of malondialdehyde and 8-hydroxydeoxyguanosine.30 Concurrently, antioxidant enzyme activities are suppressed, suggesting that OSAS contributes to stroke injury amplification.
Regarding inflammation and immune activation, intermittent hypoxia upregulates pro-inflammatory factors and promotes neutrophil infiltration by activating specific inflammatory signaling pathways. Clinical studies reveal direct correlations between inflammatory markers in OSAS patients and stroke severity, with certain inflammatory responses reversible through treatment, as indicated in reference31 suggesting that inflammation represents a key pathway through which OSAS exacerbates brain injury in IS.
Influence of Age and Sex on Inflammatory Mechanisms
The inflammatory landscape linking OSAS to ischemic injury is fundamentally altered by biological variables, particularly sex and age. While OSAS is traditionally characterized by male predominance, the risk profile shifts dramatically in females following menopause. This surge suggests that the withdrawal of estrogen compromises its established anti-inflammatory and neuroprotective barriers, thereby increasing susceptibility to stroke.32,33 Furthermore, the aging process introduces “inflammaging”—a baseline state of chronic, low-grade inflammation. This pre-existing condition likely lowers the threshold for neuroinflammatory cascades triggered by intermittent hypoxia.34 This mechanistic vulnerability is substantiated by clinical evidence confirming that severe OSAS remains a potent independent predictor of stroke in the elderly, countering earlier assumptions that this risk attenuates with age.35 Consequently, distinguishing the specific roles of pathways such as TLR4 and NLRP3 requires rigorous stratification by demographic factors to avoid generalized conclusions that mask subgroup-specific mechanisms.36
Effects of Sleep Apnea on Ischemic Brain Injury in Animal Models
Intermittent hypoxia represents the most prominent sleep apnea feature, and researchers have frequently utilized this characteristic to model sleep apnea, more often analyzing respiratory control instability in OSAS patients and intermittent hypoxia effects on systemic physiology than simulating upper airway collapse through surgical means.37
Two similar experiments investigated intermittent hypoxia in ischemic brain injury. The first found that OSAS significantly increased MCAO-induced brain damage after ischemia-reperfusion in MCAO model mice (subjected to obstructive sleep apnea with AHI ≥15 for 3 days and 3 hours), suggesting that OSAS adversely affects brain damage recovery following IS episodes.38 The second experiment revealed significantly increased MCAO-induced brain damage after ischemia-reperfusion in male rats treated with intermittent hypoxia compared to non-hypoxic controls, with the intermittent hypoxia group exacerbating overall neurological injury from cerebral ischemia-reperfusion in rats.39 Both experiments concluded that intermittent hypoxia contributes to ischemic brain injury, with the first experiment additionally employing a 3D model to develop a novel experimental OSAS model using an OSAS device.
While intermittent hypoxia (IH) models effectively mimic oxygen desaturation, they fundamentally fail to reproduce the mechanical airway obstruction defining human OSAS. Consequently, these models lack critical pathophysiological features such as profound negative intrathoracic pressure swings and the specific autonomic fragmentation seen in patients.40 This discrepancy likely simplifies the inflammatory landscape, potentially explaining why neuroprotective agents that show promise in rodent IH paradigms often fail to translate into clinical efficacy for stroke patients. Future translational success depends on developing models that incorporate actual airway occlusion to simulate the full hemodynamic and inflammatory syndrome.20
Regulatory Mechanisms of Signaling Pathways in OSAS-Induced and Exacerbated IS
In this review, we specifically prioritize the PI3K/Akt, Toll-like receptor (TLR), and NLRP3 inflammasome pathways due to their extensively validated roles at the intersection of intermittent hypoxia and cerebral ischemic injury. Current evidence suggests these three pathways represent the primary signaling hubs linking OSAS-induced oxidative stress to the neuroinflammatory cascade in ischemic stroke. While other signaling mechanisms, such as the JAK/STAT and MAPK pathways, are also implicated in inflammatory regulation, the direct crosstalk between intermittent hypoxia and the specific triad of PI3K/Akt, TLR, and NLRP3 offers the most robust translational evidence to date.
PI3K/Akt Signaling Pathway
The PI3K/Akt signaling pathway comprises Akt and serine/threonine kinase PI3K. Human cells express three PI3K classes, with Class I PI3Ks particularly relevant to IS. In this pathway, PI3K activation occurs through receptor-coupled tyrosine kinases and heterotrimeric G proteins. Akt activation typically occurs downstream of PI3K, with Akt phosphorylation serving as a PI3K activation marker.
PI3K activation promotes conversion of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) to phosphatidylinositol-3,4,5-trisphosphate (PI(3,4,5)P3), also termed PIP3. PIP3 functions as a second messenger mediating Akt and other effector protein activation. Akt activation proves crucial for various physiological processes including cell growth, survival, metabolism, and migration.41
The PI3K/Akt signaling axis can activate immune cells such as macrophages to regulate inflammation. In macrophages, this pathway transduces signals from cytokines and Toll-like receptors.42 Beyond macrophages, the PI3K/Akt pathway plays important roles in microglial transformation, promoting polarization from pro-inflammatory M1 to anti-inflammatory M2 phenotypes. Inhibition of this pathway may impede this transition, resulting in increased inflammation.43
The PI3K/Akt pathway has been implicated in various diseases including obesity, type 2 diabetes, spinal cord injury, idiopathic pulmonary fibrosis, cancer, and neurodegenerative disorders, where its dysregulation may either contribute to disease pathogenesis or offer therapeutic potential.44–48
OSAS Influences the PI3K/Akt Signaling Pathway in IS
The interaction between ROS and the PI3K/Akt pathway is complex and context-dependent. Emerging evidence suggests a biphasic response: while physiological or transient levels of ROS may function as signaling molecules to activate Akt and promote cell survival during early hypoxic stress, the pathological scenario differs significantly. In the context of severe OSAS, the chronic and excessive accumulation of ROS overwhelms antioxidant defenses, leading to the oxidative modification of key signaling proteins. This sustained oxidative stress predominantly inhibits the PI3K/Akt axis—often through the inactivation of PTEN or direct oxidation of Akt kinases—thereby abrogating its neuroprotective capacity and shifting the cellular fate toward apoptosis and inflammation.49,50 PI3K/Akt pathway activation induces NF-κB pathway expression, leading to TNF-α, IL-8, IL-6 and CRP production.51 Simultaneously, reduced GSK3β expression decreases anti-inflammatory factor expression (such as CD206, TGFβ),52 as illustrated in Figure 1, promoting inflammation.
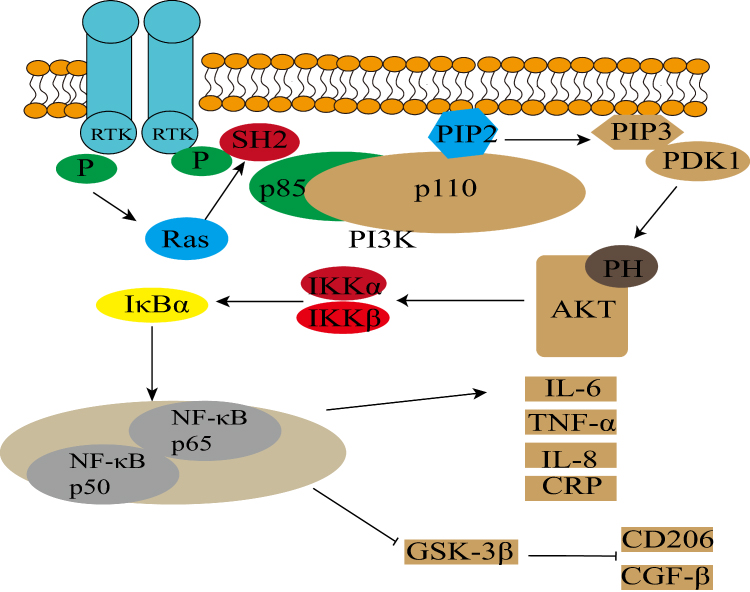 |
Figure 1 The figure illustrates that chronic intermittent hypoxia (CIH) activates the PI3K/Akt signaling pathway, which stimulates the release of pro-inflammatory factors (e.g., TNF-α, IL-6, IL-8) through the NF-κB signaling pathway. Additionally, it suppresses GSK-3β expression, thereby intensifying neuroinflammation. In the figure, t-arrows denote inhibition, while solid arrows denote activation. |
Conversely, the PI3K/Akt signaling pathway inhibits M1 to M2 microglial conversion, with M1 microglia and macrophage activation resulting in neuroinflammation. Neuroinflammation associates with acute stroke, blood-brain barrier disruption, neuronal damage, and other critical neurological outcomes. Blood-brain barrier damage following IS intensifies inflammation, permitting peripheral immune cell migration (eg., macrophages) into the brain, thereby worsening neuroinflammation.53–55
Toll-Like Receptor Signaling Pathway
Toll-like receptors (TLRs) constitute type I transmembrane proteins consisting of multiple extracellular leucine-rich repeats and a Toll/IL-1 receptor structure.56 The TLR family represents a crucial component of innate immunity by recognizing molecular markers of microbial pathogens. TLR activation can induce diverse inflammatory gene expression through signaling pathway activation, with cellular responses influenced by pathogens and intracellular adapter proteins.57
Based on findings from Daniel Fische and colleagues, the myddosome (a signaling complex containing MyD88) operates throughout the TLR pathway and regulates its activity, representing a critical TLR signaling component. Myddosome disruption or inhibition may diminish TLR signaling pathway contributions to inflammation and autoimmune diseases.58 Similarly, targeting the myddosome or suppressing its expression could reduce Toll-like receptor pathway impacts in these contexts.
The TLR4 pathway, similar to the PI3K/Akt pathway, demonstrates dual functionality: it can both induce inflammation leading to cell death and regulate inflammation.59 TLRs associate with diverse diseases including infection, autoimmunity, chronic inflammation, cancer, diabetes, neuroimmunity, and lifestyle disorders.60 Reviewing TLR signaling pathways may prove beneficial for treating or alleviating these.
OSAS Affects the Signaling Pathway of Toll-Like Receptor in IS
OSAS can damage central nervous system cells (such as neurons), leading to damage-associated molecular pattern (DAMP) release including Heat Shock Protein (HSP60) and High Mobility Group Box 1 (HMGB1). These DAMPs activate through Toll-like receptor interactions on microglial cells, inducing inflammation.61
Research by Javier R. Caso’s team demonstrated that TLR4-deficient mice exhibited reduced infarcts and less severe inflammation following ischemia, suggesting TLR4 involvement in ischemia-induced brain damage and inflammation.62 Similar to the PI3K/Akt signaling pathway, TLR2/MyD88 or TLR4/NF-κB signaling pathway activation promotes associated inflammatory mediator production. TLR2/MyD88 activation results in neuroinflammation.63,64
Comparable to PI3K/Akt signaling, the TLR4 signaling pathway demonstrates dual roles, either promoting inflammatory responses causing cell death or regulating inflammation.59 TLR4 plays significant roles in atherosclerosis, with TLR4 promoting adhesion molecule expression and inflammatory cell invasion that accelerate plaque progression. Low-dose TLR ligands induce ischemic tolerance and reduce infarct volume.65 Notably, TLR2 effects on IS primarily drive inflammatory responses, though under certain circumstances may provide neuroprotection through neurosteroid modulation.66
NLRP3 Inflammasome
Inflammasomes represent intracellular multimeric protein complexes that activate inflammatory caspase-1, with inflammasome activation constituting a primary inflammatory pathway.67 Inflammasomes serve as crucial innate immunity components, regulating inflammatory responses through inflammatory cysteine protease activation.68 Among these complexes, the NLRP3 inflammasome has been most extensively studied.
The NLRP3 inflammasome comprises three proteins: NLRP3, ASC (apoptosis-associated speck-like protein containing a CARD domain), and the effector protein procaspase-1.69 NLRP3 activation mechanisms include potassium, calcium, and chloride efflux; reactive oxygen species generation; mitochondrial dysfunction; large nonspecific pore formation; and lysosomal rupture.70 Activation pathways divide into classical and non-classical routes that promote pro-inflammatory cytokine maturation and release (such as IL-1β and IL-18).71
Hypoxia induces HIF-1α production, which can co-localize with NLRP3,72 potentially enhancing NLRP3 inflammasome activation through this interaction.73 Classical pathway activation requires two distinct signals: a “priming” signal triggered by pattern recognition receptors such as TLR4, and an activation signal facilitating NLRP3, ASC, and pro-caspase-1 assembly.74 The non-classical pathway detects cytoplasmic lipopolysaccharide and bacterial mRNA, directly activating the NLRP3 inflammasome without NLRP3 involvement.
The NLRP3 inflammasome has been implicated in various disorders including Ehrlichiosis, sepsis, metabolic diseases, neurodegenerative conditions, autoinflammatory syndromes, cancer, and atherosclerosis.75,76
OSAS Regulates NLRP3 in IS
OSAS-induced intermittent hypoxia causes mitochondrial dysfunction and excessive mtROS production. Excessive ROS can damage mitochondria and induce mitochondrial membrane potential loss and cellular component release, such as mtDNA. This mtDNA can activate the NLRP3 inflammasome, thereby promoting inflammasome assembly and inflammatory factor production,77 as shown in Figure 2.
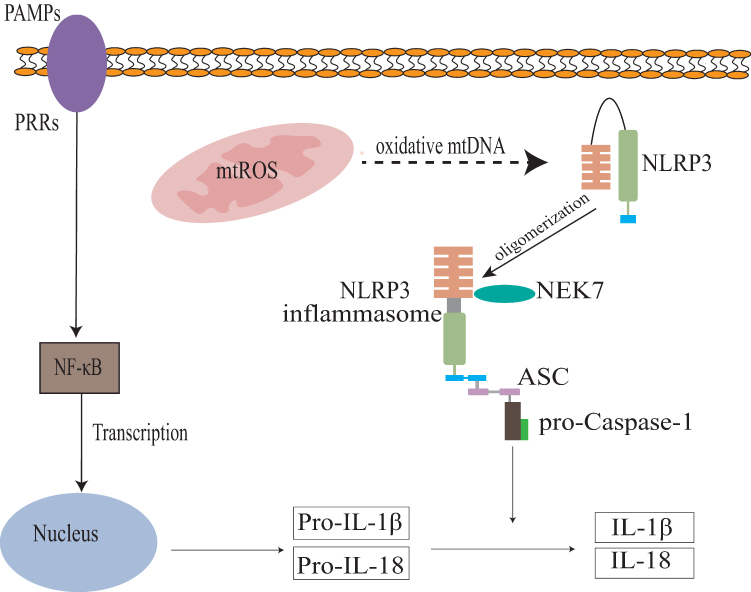 |
Figure 2 Intermittent hypoxia triggers mitochondrial ROS (mtROS) leakage, which activates the NLRP3 inflammasome. This activation promotes the caspase-1-mediated maturation and release of IL-1β and IL-18, ultimately inducing cellular pyroptosis. |
The NLRP3 inflammasome also activates through OSAS’s Toll-like receptor signaling pathway.77 Chronic intermittent hypoxia in the NLRP3 inflammasome elevates NLRP3, cleaved caspase-1, and caspase-1 levels, leading to cellular pyroptosis.78
The NLRP3 inflammasome plays pivotal roles in IS pathogenesis, with its activation closely linked to inflammation exacerbation. The NLRP3 inflammasome constitutes a multiprotein complex that monitors cellular damage and regulates various signaling pathways including AMPK/Nrf2/NLRP3, TAK1/JNK/NLRP3, DRP1/NLRP3, ROS/TXNIP/NLRP3, TLR4/NF-κB/NLRP3, and others. These pathways enhance pro-inflammatory cytokine release such as IL-1β and IL-18, thereby driving inflammatory responses following ischemic injury.79,80
Therapeutic Strategies Targeting Signaling Pathways in IS
As summarized in Table 1, all experimental interventions targeting inflammatory signaling pathways in middle cerebral artery occlusion rats ultimately yielded favorable outcomes. Beneficial effects in IS treatment were achieved through modulation of these signaling pathways.
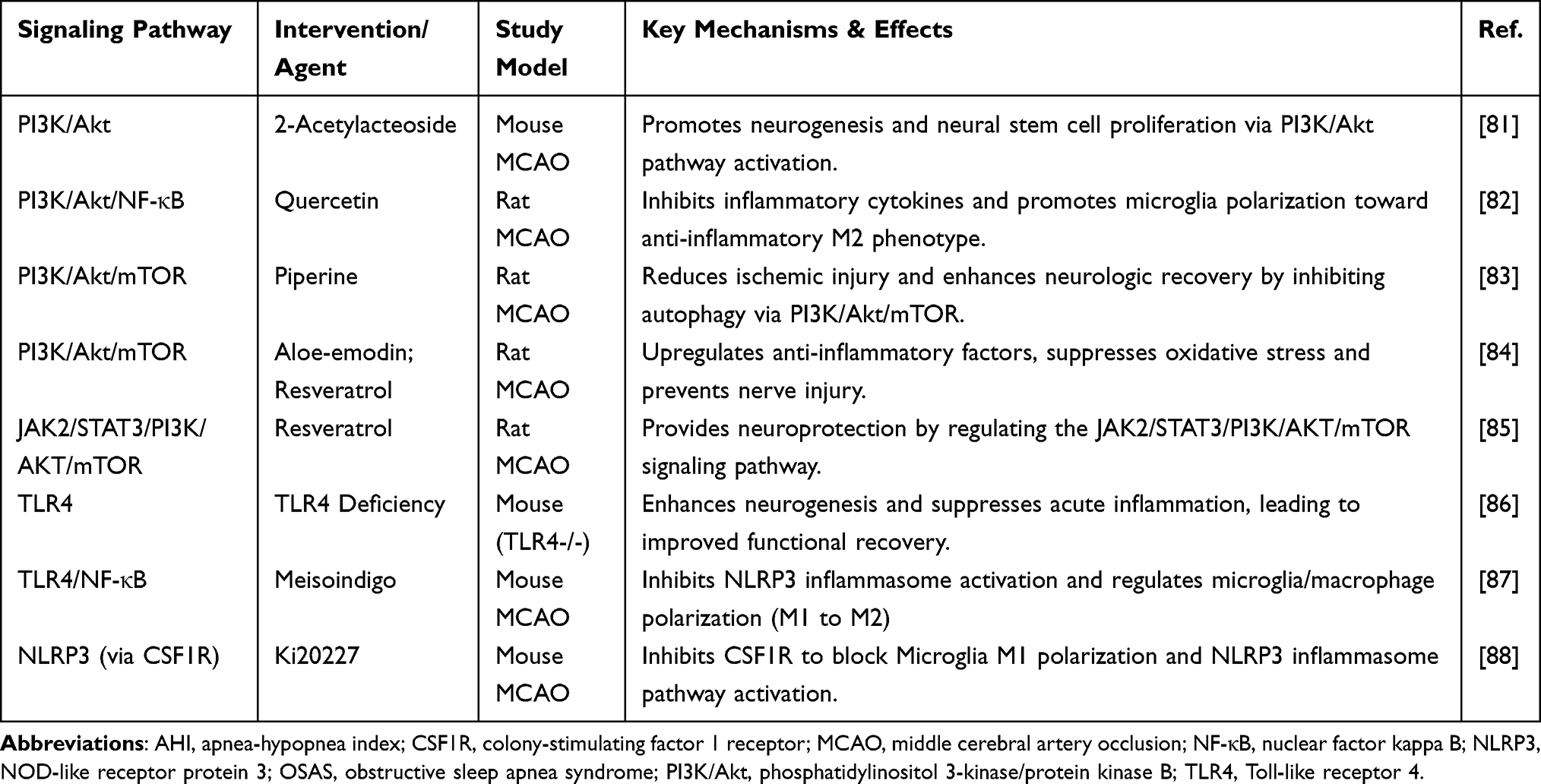 |
Table 1 Summary of Potential Therapeutic Agents Targeting Inflammatory Signaling Pathways in OSAS-Related Ischemic Stroke |
In the PI3K/Akt signaling pathway, pathway activation promotes neural stem cell proliferation and neurogenesis while reducing M1 microglia numbers to minimize neuroinflammation. By interfering with PI3K/Akt/mTOR and PI3K/Akt/NF-κB signaling, inflammation prevention and IS-related neuroinflammation reduction become possible. Tanshinone modulation may exert neuroprotective effects.89
Novel approaches include Qilong Capsule, a Chinese herbal compound that inhibited the TLR4/NF-κB signaling pathway, alleviating brain ischemia/reperfusion inflammation and promoting nerve recovery.90 Therapeutic targeting of Toll-like receptors may effectively reduce IS-associated inflammation and neurological damage.
Regarding therapeutic strategies, NLRP3 inflammasome inhibition has been proposed as a potential treatment approach. Numerous inhibitors have demonstrated efficacy in preclinical models, including small molecule compounds, type I interferons, microRNAs, nitric oxide, and nuclear factor erythroid 2 (Nrf2).91 For instance, MCC950, a small molecule inhibiting NLRP3 inflammasome assembly by blocking caspase-1 activation, reduces IL-1β maturation and release, thereby mitigating ischemic brain injury in animal models.92
Furthermore, various natural products and herbs including artemisinin, curcumin, ginsenosides Rd, paeoniflorin, and metformin demonstrate NLRP3 inflammasome modulatory effects and have shown efficacy in reducing IS damage.93,94 Modulation of IS-related signaling pathways may positively influence OSAS outcomes, suggesting that OSAS therapy could prove beneficial in IS management.
Clinical Research Perspectives
Impact of OSAS in Recovery from IS
Timely and consistent implementation of effective secondary prevention strategies in patients experiencing first stroke or transient ischemic attack could reduce stroke burden by up to 25%.95
Consequently, intervention in post-stroke patients proves significant.OSAS occurs in 67.5% of IS patients,96 indicating broad OSAS effects in IS populations. OSAS significantly negatively impacts IS recovery time. OSAS patients experience fragmented sleep architecture and recurrent nocturnal apnea, resulting in impaired sleep quality during post-stroke recovery. This may influence nerve function recovery and cognitive improvement. OSAS has been found to worsen cognitive impairment, delay recovery, and affect patients’ functional independence and physical recovery.97
Additionally, OSAS negatively affects cardiovascular systems in stroke patients. Intermittent hypoxemia and sleep fragmentation due to OSAS may cause blood pressure variability and increased cardiovascular workload.98 In conclusion, OSAS exerts comprehensive and profound impacts on stroke recovery. Therefore, OSAS recognition and treatment in stroke patients proves essential for optimizing recovery outcomes and reducing long-term stroke burden.
Prognostic Results of Treatment of OSAS for IS
Clinical OSAS treatment with continuous positive airway pressure ventilation is associated with reduced recurrence rates in IS patients with OSAS demonstrating greater impact than in cardiovascular disease. This effect relates to OSAS treatment duration, with earlier CPAP administration yielding better outcomes.99 CPAP may similarly reduce the incidence of new vascular events and for patients unable to undergo CPAP treatment, CPAP therapy absence can increase new cerebrovascular event probability five-fold.100
For IS patients with OSAS, CPAP therapy has been linked to reduced mortality in observational cohorts.101 In older adults, CPAP therapy adherence reduces stroke recurrence rates, with adherence associated with recurrence frequency. CPAP treatment efficacy was mitigated by poor treatment adherence, with many patients responding poorly to CPAP. OSAS intervention in IS patients requires alternative therapies, potentially achieved through bilevel positive airway pressure ventilation, automatic adjustable continuous positive airway pressure ventilation, and oral appliances.99
Multiple research groups have examined CPAP therapy effects on OSAS in stroke patients, as well as differences between CPAP and other therapies. Stroke patient studies, summarized in Table 2, demonstrate various treatment outcomes.
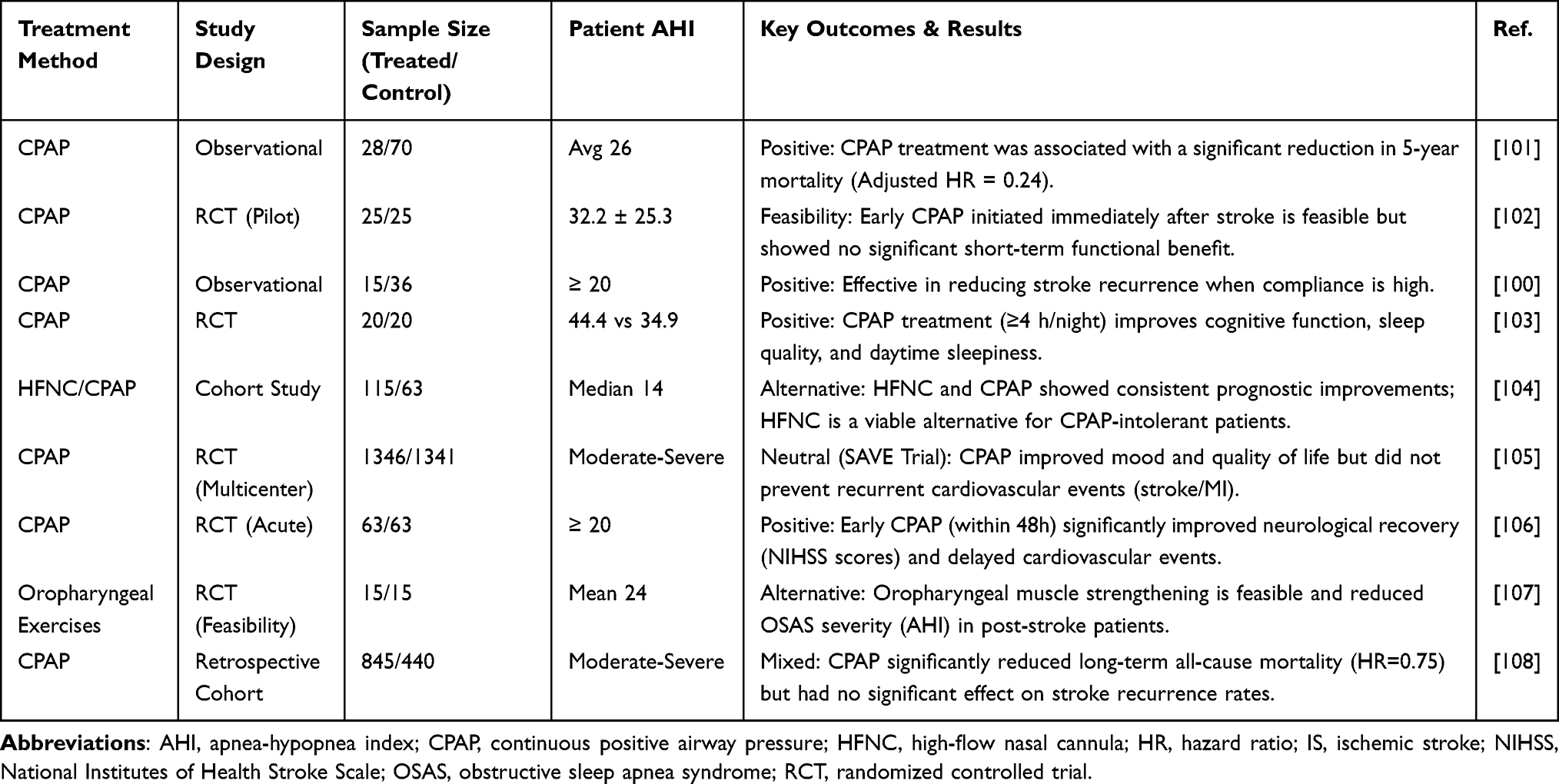 |
Table 2 Clinical Evaluation of OSAS Interventions in Patients with Ischemic Stroke |
As evident from Table 2, CPAP remains the current OSAS treatment mainstay. However, other modalities can treat OSAS with comparable efficacy in cases of poor patient compliance, highlighting the importance of demonstrating high-quality experiment feasibility regarding CPAP effects on stroke in OSAS.
Alternative Therapeutic Strategies:CPAP into lerance is frequent among stroke survivors secondary to facial paresis, aphasia, or cognitive deficits, necessitating alternative interventions. Mandibular advancement devices (MADs) offer a viable strategy for mild-to-moderate OSAS by mechanically enlarging the upper airway, subject to adequate dentition and motor function.109 Positional therapy serves as a cost-effective, non-invasive adjunct specifically for position-dependent OSAS phenotypes.110 Additionally, High-Flow Nasal Cannula (HFNC) therapy delivers positive airway pressure with superior comfort profiles compared to traditional masks.104 Despite their physiological benefits, the specific efficacy of these non-CPAP modalities in reducing post-stroke inflammatory markers remains to be validated in rigorous randomized trials.
Impact of Comorbidities on Inflammatory
Risk Obesity, type 2 diabetes, and hypertension fundamentally alter the pathogenic landscape of OSAS-related stroke. In obesity, adipose tissue functions as an endocrine organ, releasing basal IL-6 and TNF-α to “prime” the immune system.111 This chronic inflammatory state synergizes with intermittent hypoxia, amplifying endothelial injury beyond the effects of either condition alone. Concurrently, hyperglycemic conditions in diabetes trigger NLRP3 inflammasome activation.112 On this primed background, OSAS acts as a critical “second hit,” where intermittent hypoxia accelerates neurovascular damage beyond what either condition causes alone.113
Conclusion and Future Perspectives
Current evidence suggests that OSAS may exacerbate ischemic stroke outcomes via the PI3K/Akt, TLR, and NLRP3 inflammatory pathways. Clinically, variable CPAP adherence underscores the potential value of adjunctive anti-inflammatory strategies and early OSAS screening. Future research should prioritize developing specific NLRP3/TLR4 inhibitors, identifying prognostic biomarkers, and conducting large-scale longitudinal cohorts to further investigate the association between OSAS endotypes and stroke severity.Observational studies have consistently linked CPAP therapy to improved neurological recovery and reduced recurrence of vascular events in stroke patients with OSAS. Conversely, the large-scale multicenter SAVE trial found that CPAP did not significantly reduce the composite endpoint of cardiovascular events, despite improvements in mood and quality of life. This discrepancy is widely attributed to suboptimal treatment adherence in the trial cohort (mean usage < 3.3 hours/night), suggesting that higher compliance is essential for achieving prognostic cardiovascular benefits.Additionally, common stroke medications such as statins and ARBs may also mitigate inflammation in OSAS patients, warranting further investigation.
Conclusion
This review examines OSAS and IS mechanisms and their interactions, primarily through inflammatory pathways. Animal experiments demonstrate that early OSAS identification and management prove important for reducing IS risk. However, limited experimental data necessitate further investigation into specific OSAS damage mechanisms and pathways in IS. In OSAS and IS contexts, OSAS treatment may reduce relapse and death risks, though more treatment options remain needed. Although current research provides important insights into OSAS-IS relationships, further exploration is required to elucidate their specific molecular mechanisms and other interaction pathways.
Abbreviations
AHI, Apnea-Hypopnea Index; IS, Ischemic Stroke; BBB, Blood-Brain Barrier; CPAP, Continuous Positive Airway Pressure; DAMPs, Damage-Associated Molecular Patterns; HIF-1α, Hypoxia-Inducible Factor 1 Alpha; HMGB1, High Mobility Group Box 1; IL-1β, Interleukin-1 Beta; IL-6, Interleukin-6; IL-8, Interleukin-8; IL-10, Interleukin-10; IL-17, Interleukin-17; IL-18, Interleukin-18; MMP-9, Matrix Metalloproteinase-9; mtROS, Mitochondrial Reactive Oxygen Species; NF-κB, Nuclear Factor Kappa B; NLRP3, NOD-like Receptor Protein 3; OSAS, Obstructive Sleep Apnea Syndrome; PI3K/Akt, Phosphatidylinositol 3-Kinase/Protein Kinase B; ROS, Reactive Oxygen Species; RNS, Reactive Nitrogen Species; TLR, Toll-like Receptor; TNF-α, Tumor Necrosis Factor Alpha; Treg, Regulatory T Cells.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We sincerely thank the Jiujiang Precision Clinical Medicine Research Center staff and the students Weixin Zhou and Zhiyuan Xie.
Author Contributions
Zihao Zhang: Formal Analysis, Project Administration, Validation, Writing – Original Draft.
Ziqing Zhu: Formal Analysis, Project Administration, Validation, Writing – Original Draft.
Zhiying Chen: Data Curation, Formal Analysis, Validation, Writing – Original Draft, Writing – Review & Editing.
Xiaoqun Liu: Conceptualization, Data Curation, Resources, Validation, Writing – Original Draft.
Wenmin Yu: Validation, Writing – Original Draft.
Toheed Aqdas: Validation, Writing – Original Draft, Writing – Review & Editing.
Rui Yuan: Conceptualization, Data Curation, Resources, Supervision, Validation, Writing – Original Draft.
All gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was sponsored by Jiangxi Provincial Health Commission Science and Technology Plan project (202311506 to ZYC), Jiangxi Provincial Administration of Traditional Chinese Medicine science and technology plan project (2022A322 to ZYC), Youth Foundation of Natural Science Foundation of Jiangxi Province (20224BAB216045 to ZYC), Research and Reform Project on Education and Teaching in Ordinary Colleges and Universities of Jiangxi Province (JXJG-24-17-20 to ZYC), Jiangxi Provincial Department of Education Science and Technology Research Project (GJJ2501801 to LXQ).
Disclosure
The authors report no conflict of interest.
References
1. Gottlieb DJ, Punjabi NM. Diagnosis and management of obstructive sleep apnea: a review. JAMA. 2020;323:1389. doi:10.1001/jama.2020.3514
2. Benjafield AV, Ayas NT, Eastwood PR, et al. Estimation of the global prevalence and burden of obstructive sleep apnoea: a literature-based analysis. Lancet Respir Med. 2019;7(8):687–15. doi:10.1016/S2213-2600(19)30198-5
3. Pu L, Wang L, Zhang R, et al. Projected global trends in ischemic stroke incidence, deaths and disability-adjusted life years from 2020 to 2030. Stroke. 2023;54:1330–1339. doi:10.1161/STROKEAHA.122.040073
4. Lv R, Liu X, Zhang Y. Pathophysiological mechanisms and therapeutic approaches in obstructive sleep apnea syndrome. Signal Transduct Target Ther. 2023;8. doi:10.1038/s41392-023-01496-3
5. Mayer P, Pepin JL, Bettega G, et al. Relationship between body mass index, age and upper airway measurements in snorers and sleep apnoea patients. Eur Respir J. 1996;9(9):1801–1809. doi:10.1183/09031936.96.09091801
6. Bonsignore MR, Saaresranta T, Riha RL. Sex differences in obstructive sleep apnoea. Eur Respir Rev. 2019;28:190030. doi:10.1183/16000617.0030-2019
7. Neelapu BC, Kharbanda OP, Sardana HK, et al. Craniofacial and upper airway morphology in adult obstructive sleep apnea patients: a systematic review and meta-analysis of cephalometric studies. Sleep Med Rev. 2017;31:79–90. doi:10.1016/j.smrv.2016.01.007
8. Brennan L, Kirkham FJ, Gavlak JC. Sleep-disordered breathing and comorbidities: role of the upper airway and craniofacial skeleton. NSS. 2020;12:907–936. doi:10.2147/NSS.S146608
9. Brown DL, Yadollahi A, He K, et al. Overnight rostral fluid shifts exacerbate obstructive sleep apnea after stroke. Stroke. 2021;52:3176–3183. doi:10.1161/STROKEAHA.120.032688
10. White LH, Bradley TD. Role of nocturnal rostral fluid shift in the pathogenesis of obstructive and central sleep apnoea. J Physiol. 2013;591:1179–1193. doi:10.1113/jphysiol.2012.245159
11. Yanagimori M, Fernandes MD, Garcia ML, et al. Respiratory arousal threshold among patients with isolated sleep apnea and with comorbid insomnia (COMISA). Sci Rep. 2023;13:7638. doi:10.1038/s41598-023-34002-4
12. Lee JJ, Sundar KM. Evaluation and management of adults with obstructive sleep apnea syndrome. Lung. 2021;199:87–101. doi:10.1007/s00408-021-00426-w
13. El-Solh AA, Lawson Y, Wilding GE. Impact of low arousal threshold on treatment of obstructive sleep apnea in patients with post-traumatic stress disorder. Sleep Breath. 2021;25:597–604. doi:10.1007/s11325-020-02106-0
14. Pal A, Martinez F, Akey MA, et al. Breathing rate variability in obstructive sleep apnea during wakefulness. J Clin Sleep Med. 2022;18:825–833. doi:10.5664/jcsm.9728
15. Schmickl CN, Orr JE, Sands SA, et al. Loop gain as a predictor of blood pressure response in patients treated for obstructive sleep apnea: secondary analysis of a clinical trial. Ann ATS. 2024;21:296–307. doi:10.1513/AnnalsATS.202305-437OC
16. Neroni B, Evangelisti M, Radocchia G, et al. Relationship between sleep disorders and gut dysbiosis: what affects what? Sleep Med. 2021;87:1–7. doi:10.1016/j.sleep.2021.08.003
17. Orellana-Urzúa S, Rojas I, Líbano L, Rodrigo R. Pathophysiology of ischemic stroke: role of oxidative stress. CPD. 2020;26:4246–4260. doi:10.2174/1381612826666200708133912
18. Neves D, Salazar IL, Almeida RD, Silva RM. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023;328:121814. doi:10.1016/j.lfs.2023.121814
19. DeLong JH, Ohashi SN, O’Connor KC, Sansing LH. Inflammatory responses after ischemic stroke. Semin Immunopathol. 2022;44:625–648. doi:10.1007/s00281-022-00943-7
20. Biose IJ, Bakare AB, Wang H, Gressett TE, Bix GJ. Sleep apnea and ischemic stroke— a perspective for translational preclinical modelling. Sleep Med Rev. 2024;75:101929. doi:10.1016/j.smrv.2024.101929
21. Alexiev F, Brill A-K, Ott SR, et al. Sleep-disordered breathing and stroke: chicken or egg? J Thorac Dis. 2018;10:S4244–S4252. doi:10.21037/jtd.2018.12.66
22. Nacafaliyev V, Ortan P, Sayin SS. Relationship between obstructive sleep apnoea syndrome and silent brain infarction. Postgrad Med J. 2023;99:731–735. doi:10.1136/pmj-2022-141911
23. Yaggi HK, Kernan WN, Brass LM, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. New Engl J Med. 2005;353:2034–2041. doi:10.1056/NEJMoa043104
24. Lavalle S, Masiello E, Iannella G, et al. Unraveling the complexities of oxidative stress and inflammation biomarkers in obstructive sleep apnea syndrome: a comprehensive review. Life. 2024;14(4):425. doi:10.3390/life14040425
25. Gregori-Pla C, Zirak P, Cotta G, et al. How does obstructive sleep apnea alter cerebral hemodynamics? SLEEP. 2023;46:zsad122. doi:10.1093/sleep/zsad122
26. Maida CD, Norrito RL, Daidone M, Tuttolomondo A, Pinto A. Neuroinflammatory mechanisms in ischemic stroke: focus on cardioembolic stroke, background, and therapeutic approaches. IJMS. 2020;21:6454. doi:10.3390/ijms21186454
27. Redline S, Azarbarzin A, Peker Y. Obstructive sleep apnoea heterogeneity and cardiovascular disease. Nat Rev Cardiol. 2023;20:560–573. doi:10.1038/s41569-023-00846-6
28. Koutsaliaris IK, Moschonas IC, Pechlivani LM, Tsouka AN, Tselepis AD. Inflammation, oxidative stress, vascular aging and atheroscleroticischemic stroke. CMC. 2022;29:5496–5509. doi:10.2174/0929867328666210921161711
29. Jackman KA, Zhou P, Faraco G, et al. Dichotomous effects of chronic intermittent hypoxia on focal cerebral ischemic injury. Stroke. 2014;45:1460–1467. doi:10.1161/STROKEAHA.114.004816
30. Kaczmarek E, Bakker JP, Clarke DN, et al. Molecular biomarkers of vascular dysfunction in obstructive sleep apnea. PLoS One. 2013;8:e70559. doi:10.1371/journal.pone.0070559
31. Jelic S, Padeletti M, Kawut SM, et al. Inflammation, oxidative stress, and repair capacity of the vascular endothelium in obstructive sleep apnea. Circulation. 2008;117:2270–2278. doi:10.1161/CIRCULATIONAHA.107.741512
32. Tariq MB, Lee J, McCullough LD. Sex differences in the inflammatory response to stroke. Semin Immunopathol. 2023;45:295–313. doi:10.1007/s00281-022-00969-x
33. Martins FO, Conde SV. Gender differences in the context of obstructive sleep apnea and metabolic diseases. Front Physiol. 2021;12:792633. doi:10.3389/fphys.2021.792633
34. Antonaglia C, Fabozzi A, Steffanina A, et al. Walking the fine line between OSA and aging. Sleep Breath. 2025;29:195. doi:10.1007/s11325-025-03343-x
35. Munoz R, Duran-Cantolla J, Martínez-Vila E, et al. Severe sleep apnea and risk of ischemic stroke in the elderly. Stroke. 2006;37:2317–2321. doi:10.1161/01.STR.0000236560.15735.0f
36. Niu P, Li L, Zhang Y, et al. Immune regulation based on sex differences in ischemic stroke pathology. Front Immunol. 2023;14:1087815. doi:10.3389/fimmu.2023.1087815
37. Kim LJ, Freire C, Curado TF, Jun JC, Polotsky VY. The role of animal models in developing pharmacotherapy for obstructive sleep apnea. J Clin Med. 2019;8(12):2049. doi:10.3390/jcm8122049
38. Cananzi SG, White LA, Barzegar M, et al. Obstructive sleep apnea intensifies stroke severity following middle cerebral artery occlusion. Sleep Med. 2020;67:278–285. doi:10.1016/j.sleep.2020.01.014
39. Zhao Y-N, Guo X-F, Li J-M, et al. mTOR/autophagy pathway in the hippocampus of rats suffering intermittent hypoxia preconditioning and global cerebral ischemia-reperfusion. Oncotarget. 2017;8:23353–23359. doi:10.18632/oncotarget.15058
40. Ryan S. Mechanisms of cardiovascular disease in obstructive sleep apnoea. J Thorac Dis. 2018;10:S4201–S4211. doi:10.21037/jtd.2018.08.56
41. Bilanges B, Posor Y, Vanhaesebroeck B. PI 3‐kinase isoforms at the crossroads of signalling and vesicular traffic. Nat Rev Mol Cell Biol. 2019;20(9):515–534. doi:10.1038/s41580-019-0129-z
42. Acosta-Martinez M, Cabail MZ. The PI3K/Akt pathway in meta-inflammation. IJMS. 2022;23:15330. doi:10.3390/ijms232315330
43. Tian Y, Liu B, Li Y. Activation of RARα receptor attenuates neuroinflammation after SAH via promoting M1-to-M2 phenotypic polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-κB pathway. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.839796
44. Rai SN, Dilnashin H, Birla H, et al. The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res. 2019;35:775–795. doi:10.1007/s12640-019-0003-y
45. Wang J, Hu K, Cai X, et al. Targeting PI3K/AKT signaling for treatment of idiopathic pulmonary fibrosis. Acta Pharmaceutica Sinica B. 2022;12:18–32. doi:10.1016/j.apsb.2021.07.023
46. Li Q, Li Z, Luo T, Shi H. Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy. Mol Biomed. 2022;3:47. doi:10.1186/s43556-022-00110-2
47. Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–1496. doi:10.7150/ijbs.27173
48. Xiao C-L, Yin W-C, Zhong Y-C. The role of PI3K/Akt signalling pathway in spinal cord injury. Biomed Pharmacother. 2022;156:113881. doi:10.1016/j.biopha.2022.113881
49. Liu Z, Huang M, Hong Y, et al. Isovalerylspiramycin I suppresses non-small cell lung carcinoma growth through ROS-mediated inhibition of PI3K/AKT signaling pathway. Int J Biol Sci. 2022;18:3714–3730. doi:10.7150/ijbs.69989
50. Chen K, Li Y, Zhang X, et al. The role of the PI3K/AKT signalling pathway in the corneal epithelium: recent updates. Cell Death Dis. 2022;13:513. doi:10.1038/s41419-022-04963-x
51. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Sig Transduct Target Ther. 2020;5:209.
52. Xue R, Wan Y, Sun X, et al. Nicotinic mitigation of neuroinflammation and oxidative stress after chronic sleep deprivation. Front Immunol. 2019;10:2546. doi:10.3389/fimmu.2019.02546
53. Wang H, Zhang S, Xie L, Zhong Z, Yan F. Neuroinflammation and peripheral immunity: focus on ischemic stroke. Int Immunopharmacol. 2023;120:110332. doi:10.1016/j.intimp.2023.110332
54. Candelario-Jalil E, Dijkhuizen RM, Magnus T. Neuroinflammation, stroke, blood-brain barrier dysfunction, and imaging modalities. Stroke. 2022;53:1473–1486. doi:10.1161/STROKEAHA.122.036946
55. Woodburn SC, Bollinger JL, Wohleb ES. The semantics of microglia activation: neuroinflammation, homeostasis, and stress. J Neuroinflammation. 2021;18(1):258. doi:10.1186/s12974-021-02309-6
56. Moresco EMY, LaVine D, Beutler B. Toll-like receptors. Curr Biol. 2011;21:R488–R493. doi:10.1016/j.cub.2011.05.039
57. Kawai T, Akira S. TLR signaling. Semin Immunopathol. 2007;19:24–32. doi:10.1016/j.smim.2006.12.004
58. Fisch D, Zhang T, Sun H, et al. Molecular definition of the endogenous Toll-like receptor signalling pathways. Nature. 2024;631:635–644. doi:10.1038/s41586-024-07614-7
59. Kumari S, Dhapola R, Sharma P, et al. The impact of cytokines in neuroinflammation-mediated stroke. Cytokine Growth Factor Rev. 2024;78:105–119. doi:10.1016/j.cytogfr.2024.06.002
60. Chen Y-H, Wu K-H, Wu H-P. Unraveling the complexities of toll-like receptors: from molecular mechanisms to clinical applications. IJMS. 2024;25:5037. doi:10.3390/ijms25095037
61. Kiernan EA, Smith SMC, Mitchell GS, Watters JJ. Mechanisms of microglial activation in models of inflammation and hypoxia: implications for chronic intermittent hypoxia. J Physiol. 2016;594:1563–1577. doi:10.1113/JP271502
62. Caso JR, Pradillo JM, Hurtado O, et al. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation. 2007;115:1599–1608. doi:10.1161/CIRCULATIONAHA.106.603431
63. Li W, Yu Y, Li D, et al. TLR2 deficiency attenuated chronic intermittent hypoxia-induced neurocognitive deficits. Int Immunopharmacol. 2020;81:106284. doi:10.1016/j.intimp.2020.106284
64. Lin Z, Lin H, Yu X, Zheng Y, Cheng S. TLR4 mediates inflammation and hepatic fibrosis induced by chronic intermittent hypoxia in rats. Mol Med Rep. 2020;22:651–660. doi:10.3892/mmr.2020.11134
65. Ashayeri Ahmadabad R, Khaleghi Ghadiri M, Gorji A. The role of Toll-like receptor signaling pathways in cerebrovascular disorders: the impact of spreading depolarization. J Neuroinflammation. 2020;17. doi:10.1186/s12974-020-01785-6
66. Tajalli-Nezhad S, Karimian M, Beyer C, Atlasi MA, Azami Tameh A. The regulatory role of Toll-like receptors after ischemic stroke: neurosteroids as TLR modulators with the focus on TLR2/4. Cell Mol Life Sci. 2019;76(3):523–537. doi:10.1007/s00018-018-2953-2
67. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. IJMS. 2019;20:3328. doi:10.3390/ijms20133328
68. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Molecular Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
69. Fu J, Wu H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Ann Rev Immunol. 2023;41:301–316. doi:10.1146/annurev-immunol-081022-021207
70. Xu Y, Yang Y, Chen X, et al. NLRP3 inflammasome in cognitive impairment and pharmacological properties of its inhibitors. Transl Neurodegener. 2023;12:49. doi:10.1186/s40035-023-00381-x
71. Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R. Focus on the role of NLRP3 inflammasome in diseases. IJMS. 2020;21:4223. doi:10.3390/ijms21124223
72. Ke Q, Costa M. Hypoxia-inducible Factor-1 (HIF-1). Mol Pharmacol. 2006;70:1469–1480. doi:10.1124/mol.106.027029
73. Jiang Q, Ding Y, Li F, et al. Modulation of NLRP3 inflammasome-related-inflammation via RIPK1/RIPK3-DRP1 or HIF-1α signaling by phenothiazine in hypothermic and normothermic neuroprotection after acute ischemic stroke. Redox Biol. 2024;73:103169. doi:10.1016/j.redox.2024.103169
74. Song M, Wang J-X, Sun Y-L, et al. Tetrandrine alleviates silicosis by inhibiting canonical and non-canonical NLRP3 inflammasome activation in lung macrophages. Acta Pharmacol Sin. 2022;43:1274–1284. doi:10.1038/s41401-021-00693-6
75. Sharma AK, Ismail N. Non-canonical inflammasome pathway: the role of cell death and inflammation in ehrlichiosis. Cells. 2023;12(22):2597. doi:10.3390/cells12222597
76. Sharma BR, Kanneganti T-D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat Immunol. 2021;22(5):550–559. doi:10.1038/s41590-021-00886-5
77. Wu X, Gong L, Xie L, et al. NLRP3 deficiency protects against intermittent hypoxia-induced neuroinflammation and mitochondrial ROS by promoting the PINK1-Parkin pathway of mitophagy in a murine model of sleep apnea. Front Immunol. 2021;12:628168. doi:10.3389/fimmu.2021.628168
78. Bai C, Zhu Y, Dong Q, Zhang Y. Chronic intermittent hypoxia induces the pyroptosis of renal tubular epithelial cells by activating the NLRP3 inflammasome. Bioengineered. 2022;13(3):7528–7540. doi:10.1080/21655979.2022.2047394
79. Puleo MG, Miceli S, Di Chiara T, et al. Molecular mechanisms of inflammasome in ischemic stroke pathogenesis. Pharmaceuticals. 2022;15:1168. doi:10.3390/ph15101168
80. Long J-X, Tian M-Z, Chen X-Y, et al. The role of NLRP3 inflammasome-mediated pyroptosis in ischemic stroke and the intervention of traditional Chinese medicine. Front Pharmacol. 2023;14:1151196. doi:10.3389/fphar.2023.1151196
81. Wang M, Xing S, Liu Y, et al. 2-Acetylacteoside improves recovery after ischemic stroke by promoting neurogenesis via the PI3K/Akt pathway. Free Radic Biol Med. 2024;225:415–429. doi:10.1016/j.freeradbiomed.2024.10.268
82. Li L, Jiang W, Yu B, et al. Quercetin improves cerebral ischemia/reperfusion injury by promoting microglia/macrophages M2 polarization via regulating PI3K/Akt/NF-κB signaling pathway. Biomed Pharmacother. 2023;168:115653. doi:10.1016/j.biopha.2023.115653
83. Zhang Y, Yang M, Yuan Q, et al. Piperine ameliorates ischemic stroke-induced brain injury in rats by regulating the PI3K/AKT/mTOR pathway. J Ethnopharmacol. 2022;295:115309. doi:10.1016/j.jep.2022.115309
84. Xian M, Cai J, Zheng K, et al. Aloe-emodin prevents nerve injury and neuroinflammation caused by ischemic stroke via the PI3K/AKT/mTOR and NF-κB pathway. Food Funct. 2021;12:8056–8067. doi:10.1039/D1FO01144H
85. Hou Y, Wang K, Wan W, et al. Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/mTOR pathway after stroke in rats. Genes Dis. 2018;5:245–255. doi:10.1016/j.gendis.2018.06.001
86. Moraga A, Gómez-Vallejo V, Cuartero MI, et al. Imaging the role of toll-like receptor 4 on cell proliferation and inflammation after cerebral ischemia by positron emission tomography. J Cereb Blood Flow Metab. 2016;36:702–708. doi:10.1177/0271678X15627657
87. Gu L, Xiong X. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-κB signaling pathway. Front Cell Neurosci. 2019;13.
88. Du X, Xu Y, Chen S, Fang M. Inhibited CSF1R alleviates ischemia injury via inhibition of Microglia M1 polarization and NLRP3 pathway. Neural Plast. 2020;2020:1–11. doi:10.1155/2020/8825954
89. Ashayeri Ahmadabad R, Mirzaasgari Z, Gorji A, Khaleghi Ghadiri M. Toll-like receptor signaling pathways: novel therapeutic targets for cerebrovascular disorders. IJMS. 2021;22:6153. doi:10.3390/ijms22116153
90. Lyu J, Liu Y, Liu F, et al. Therapeutic effect and mechanisms of traditional Chinese medicine compound (Qilong capsule) in the treatment of ischemic stroke. Phytomedicine. 2024;132:155781. doi:10.1016/j.phymed.2024.155781
91. Alishahi M, Farzaneh M, Ghaedrahmati F, et al. NLRP3 inflammasome in ischemic stroke: as possible therapeutic target. Int J Stroke. 2019;14(6):574–591. doi:10.1177/1747493019841242
92. Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556–559. doi:10.1038/s41589-019-0277-7
93. Yang K, Zeng L, He Q, et al. Advancements in research on the immune-inflammatory mechanisms mediated by NLRP3 inflammasome in ischemic stroke and the regulatory role of natural plant products. Front Pharmacol. 2024;15:1250918. doi:10.3389/fphar.2024.1250918
94. Wang Y, Yuan H, Shen D, et al. Artemisinin attenuated ischemic stroke induced pyroptosis by inhibiting ROS/TXNIP/NLRP3/Caspase-1 signaling pathway. Biomed Pharmacother. 2024;177:116894. doi:10.1016/j.biopha.2024.116894
95. Hankey GJ. Secondary stroke prevention. Lancet Neurol. 2014;13:178–194. doi:10.1016/S1474-4422(13)70255-2
96. Liu X, Lam DC-L, Chan KPF, et al. Prevalence and determinants of sleep apnea in patients with stroke: a meta-analysis. J Stroke Cerebrovascular Dis. 2021;30:106129. doi:10.1016/j.jstrokecerebrovasdis.2021.106129
97. Zhu R, Ouyang C, Ma R, Wang K. Obstructive sleep apnea is associated with cognitive impairment in minor ischemic stroke. Sleep Breath. 2022;26:1907–1914. doi:10.1007/s11325-022-02575-5
98. Kohler M, Stradling JR. Mechanisms of vascular damage in obstructive sleep apnea. Nat Rev Cardiol. 2010;7:677–685. doi:10.1038/nrcardio.2010.145
99. Mohamed B, Yarlagadda K, Self Z, et al. Obstructive sleep apnea and stroke: determining the mechanisms behind their association and treatment options. Transl Stroke Res. 2024;15:239–332.
100. Martínez-García MÁ, Galiano-Blancart R, Román-Sánchez P, et al. Continuous positive airway pressure treatment in sleep apnea prevents new vascular events after ischemic stroke. Chest. 2005;128:2123–2129. doi:10.1378/chest.128.4.2123
101. Martínez-García MÁ, Soler-Cataluña JJ, Ejarque-Martínez L, et al. Continuous positive airway pressure treatment reduces mortality in patients with ischemic stroke and obstructive sleep apnea: a 5-year follow-up study. Am J Respir Crit Care Med. 2009;180:36–41. doi:10.1164/rccm.200808-1341OC
102. Minnerup J, Ritter MA, Wersching H, et al. Continuous positive airway pressure ventilation for acute ischemic stroke: a randomized feasibility study. Stroke. 2012;43:1137–1139. doi:10.1161/STROKEAHA.111.637611
103. Kim H, Im S, Park JI, et al. Improvement of cognitive function after continuous positive airway pressure treatment for subacute stroke patients with obstructive sleep apnea: a randomized controlled trial. Brain Sciences. 2019;9:252. doi:10.3390/brainsci9100252
104. Li Z, Pang M, Zhang J, et al. Effect of ventilation modalities on the early prognosis of patients with poststroke sleep apnea. Ann Clin Transl Neurol. 2024;11:355–367. doi:10.1002/acn3.51956
105. McEvoy RD, Antic NA, Heeley E, et al. CPAP for prevention of cardiovascular events in obstructive sleep apnea. N Engl J Med. 2016;375(10):919–931. doi:10.1056/NEJMoa1606599
106. Parra O, Sanchez-Armengol A, Bonnin M, et al. Early treatment of obstructive apnoea and stroke outcome: a randomised controlled trial. Eur Respir J. 2011;37(5):1128–1136. doi:10.1183/09031936.00034410
107. Marzouqah R, Dharmakulaseelan L, Colelli DR, et al. Strengthening oropharyngeal muscles as an approach to treat post‐stroke obstructive sleep apnea: a feasibility randomised controlled trial. Journal of Sleep Research. 2024;33:e14086. doi:10.1111/jsr.14086
108. Suusgaard J, West AS, Ponsaing LB, et al. Stroke recurrence and all-cause mortality in CPAP-treated sleep-disordered-breathing patients. J Stroke Cerebrovascular Dis. 2025;34:108204. doi:10.1016/j.jstrokecerebrovasdis.2024.108204
109. Uniken Venema JAM, Rosenmöller BRAM, de Vries N, et al. Mandibular advancement device design: a systematic review on outcomes in obstructive sleep apnea treatment. Sleep Med Rev. 2021;60:101557. doi:10.1016/j.smrv.2021.101557
110. Suzuki M, Funayama Y, Homma M, et al. Effect of position therapy and oral devices on sleep parameters in patients with obstructive sleep apnea. Eur Arch Otorhinolaryngol. 2021;278:4545–4550. doi:10.1007/s00405-021-06817-2
111. Tasali E, Pamidi S, Covassin N, Somers VK. Obstructive sleep apnea and cardiometabolic disease: obesity, hypertension, and diabetes. Circ Res. 2025;137:764–787. doi:10.1161/CIRCRESAHA.125.325676
112. Hong P, Gu R-N, Li F-X, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflammation. 2019;16:121. doi:10.1186/s12974-019-1498-0
113. Reutrakul S, Mokhlesi B. Obstructive Sleep Apnea and Diabetes. Chest. 2017;152:1070–1086. doi:10.1016/j.chest.2017.05.009
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.