Back to Journals » International Medical Case Reports Journal » Volume 14
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) Probably Caused by DSG2 p.Val149Ile Mutation as Genetic Background When Carrying with Heterozygous PRRT2 p.Arg217ProfsTer8 Mutation: A Case Report
Authors Huang R, Luo Y, Zhao J, Su K, Lei Y, Li Y
Received 9 March 2021
Accepted for publication 15 April 2021
Published 12 May 2021 Volume 2021:14 Pages 307—313
DOI https://doi.org/10.2147/IMCRJ.S309668
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Rui Huang,1,* YinHua Luo,2,* Jingbo Zhao,1 Ke Su,1 YuHua Lei,1 Yuanhong Li1
1Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Enshi Clinical College of Wuhan University, Enshi Prefecture, 445000, Hubei Province, People’s Republic of China; 2Department of Central Hospital of Tujia and Miao Autonomous Prefecture, Hubei University of Medicine, Shiyan, 442000, Hubei Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuanhong Li
Cardiovascular Disease Center, Central Hospital of Tujia and Miao Autonomous Prefecture, Enshi Clinical College of Wuhan University, Enshi Prefecture, No. 158 Wuyang Avenue, Enshi City, Hubei Province, People’s Republic of China
Email [email protected]
Background: ARVC is a rare genetic-related disease characterized by fibrous fat replacement in the ventricular myocardium, caused by mutations in genes encoding for the desmosomal proteins, such as the desmoglein-2 gene (DSG2). It is reported in the literature that other genetic factors may play a role in disease penetrance. Herein, we report a Chinese proband with ARVC, which was probably caused by DSG2 p.Val149Ile mutation as genetic background when carrying heterozygous PRRT2 p.Arg217ProfsTer8 mutation.
Case Presentation: A 17-year-old male with a history of paroxysmal kinesigenic dyskinesia (PKD) presented to the hospital for syncope induced by ventricular tachycardia. According to relevant clinical data and the diagnostic criteria of ARVC, a precise positive diagnosis of ARVC was finally made. Gene testing revealed that the patient carried a DSG2 heterozygous missense mutation (NM_001943: exon5: c.445G>A, p.Val149Ile) as well as frameshift mutation of PRRT2 (NM_001256442: exon2: p. Arg217Profs Ter8).
Conclusion: This is the first time to report a Chinese proband with ARVC and a history of PKD carrying both DSG2 p. val149ile mutation and PRRT2 p. Arg217ProfsTer8 mutation, which can provide a new direction for gene screening of patients with ARVC and further supplements for its diagnostic criteria.
Keywords: arrhythmogenic right ventricular cardiomyopathy, ARVC, desmosomal proteins, desmoglein-2 gene, DSG2
Introduction
ARVC is a reasonably rare genetic cardiomyopathy characterized by the progressive fibro-fatty replacement of ventricular myocardial tissue. Syncope caused by ventricular tachycardia (VT) or ventricular fibrillation (VF) is the primary clinical manifestation of ARVC. However, it can also be the initial expression of the disease. The pathogenesis of ARVC is related to the mutation of the genes encoding for the desmosomal proteins in myocytes, such as DSG2, Plakoglobin (JUP), Desmocollin 2 (DSC2), Plakophilin 2 (PKP2), and Desmoplakin (DSP), and causing dysfunctional cell binding, myocytes death, and subsequent fibrofatty replacement of the affected right ventricle (RV). Nevertheless, there are sparse reports about ARVC and even fewer ARVC cases caused by DSG2 mutations in the literature. Currently, approximately 76 pathogenic mutation sites on DSG2 are related to ARVC,1 including the furin-cleavage site of the DSG2 protein, nt 134, nt 143, exon 9, exon 14, intron 12. Besides, it is reported in relevant research that other genetic factors may also play a role in disease penetrance of ARVC.2 PKD is now recognized as the most common type of paroxysmal movement disorder, with the clinical feature of recurrent and transient dyskinesia. PRRT2 has been proved to be the primary causative gene of PKD, transmitted in an autosomal manner.
Herein, we report a Chinese proband with ARVC and a history of PKD for the first time. Gene testing revealed that the patient carried a DSG2 heterozygous missense mutation (NM_001943: exon5: c.445G>A, p. Val149Ile) frameshift mutation of PRRT2 (NM_001256442:exon2:p. Arg217ProfsTer8), which is considered to be correlated to PKD.
Case Presentation
A case of a 17-year-old male with a history of PKD was presented to the hospital due to syncope during exercise in a gymnasium. After emergency personnel performed cardiopulmonary resuscitation (CPR) on the patient at the scene, the patient was rushed to the emergency center. An electrocardiogram (ECG) indicated ventricular tachycardia, with a left bundle branch block-like pattern indicating right ventricular outflow tract (RVOT) origin and a heart rate of 250 beats/min (Figure 1A). Moreover, the patient was immediately given 200 joules of synchronized electrical resuscitation to restore sinus rhythm and then transferred to our hospital’s heart center for further evaluation and treatment. As for his family, no similar history and situation were discovered.
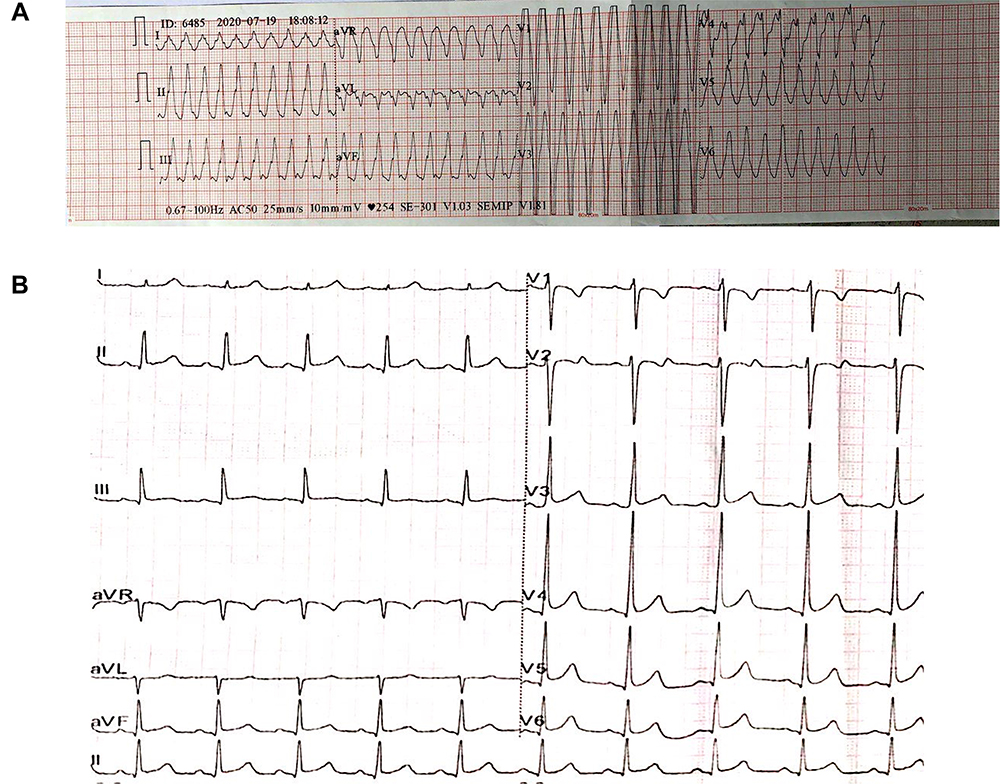 |
Figure 1 (A) Pre-hospital ECG showed ventricular tachycardia with a heart rate of 250 beats/min. (B) Admission ECG. |
In terms of physical examination, the blood pressure was 110/80mmHg, the heart rate was 88 beats/min, and the cardiac examination showed no positive signs. The ECG on admission was normal (Figure 1B). Transthoracic echocardiography revealed that the left ventricle’s (LV) end-diastolic diameter and RV were 36mm, 27mm, respectively, and the ejection fraction was 58%. Furthermore, subtle regional wall motion and echo abnormalities were found in the apex of the RV’s anterior wall (Figure 2). Subsequently, coronary angiography was performed, indicating that the blood flow in the coronary arteries was normal. Intracardiac electrophysiological studies of RV stimulation easily induced rapid ventricular tachycardia with a heart rate of 250–280 beats/min originating from the RV with various forms (Figure 3). The patient developed chest pain and amaurosis and undergone electrical cardioversion again during the operation.
 |
Figure 2 (A) The left ventricular long-axis four-chamber view showed localized wall thinning of the RV apex (bold arrow) and abnormal echo in the posterior basal segment (thin arrow). (B) The short-axis two-chamber view of the left ventricle indicated abnormal wall motion and localized dilation during diastole in RV, similar to a mini ventricular aneurysm (black arrow). |
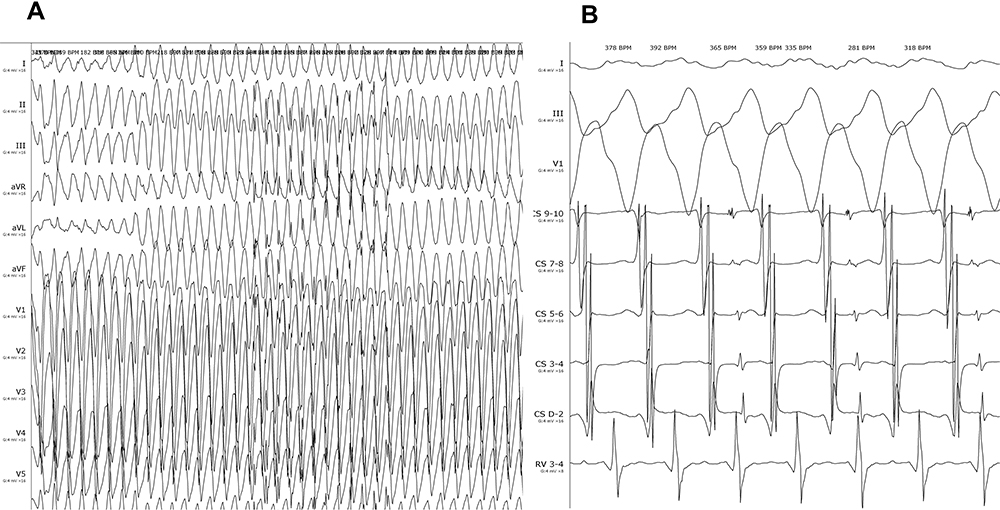 |
Figure 3 Intracardiac electrophysiological studies of right ventricular stimulation easily induced rapid ventricular tachycardia with a heart rate of 250–280 beats/min originating from the RV with various forms. (A) Body surface electrocardiogram, paper speed 25mm/s. (B) Intracardiac electrogram, paper speed 100mm/s. |
MRI of the long-axis and short-axis of the heart confirmed the typical RV segmental wall motion abnormalities, that is, an akinetic zone presented inferiorly in the RV’s apex. The anterior wall was slightly thinned, and there was a signal change, suggesting myocardial fat infiltration (Figure 4).
 |
Figure 4 Myocardial MRI showed that the segmental RV wall was locally thinned, and the abnormal signal indicated fat infiltration (bold arrow). (A) Left ventricular long-axis four-chamber view, bright blood FIESTA sequence. (B) Short axis section of the left ventricle, black blood Double IR sequence. |
According to these results and the diagnostic criteria of ARVC3,4 (the patient met two significant criteria: regional RV akinesia or dyskinesia; LBBB-type ventricular tachycardia), a precise positive diagnosis of ARVC was finally made.
After obtaining the informed consent, we conducted whole-exome sequencing (WES) on the patient and his family to confirm our diagnosis. Gene testing revealed that the patient carried a DSG2 heterozygous missense mutation (NM_001943: exon5: c.445G>A, p. Val149Ile) as well as a frameshift mutation of PRRT2 (NM_001256442:exon2:p. Arg217Profs Ter8) (Figures 5 and 6). Moreover, the pedigree analysis showed that the patient’s mother and sister carried the same DSG2 mutation, while his father did not (Figures 5 and 7). On the contrary, the patient’s father carried the same mutation of PRRT2, while his mother and sister did not (Figures 6 and 7). What is more, not his mother and sister but his father were absolutely asymptomatic.
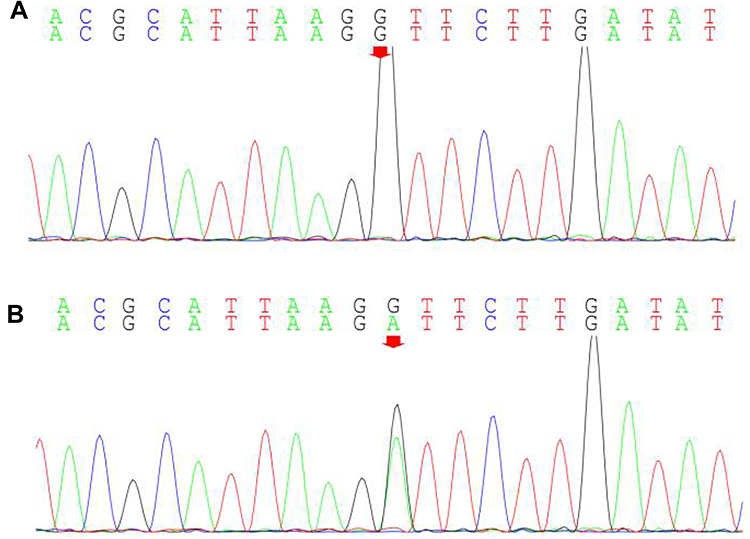 |
Figure 5 Sequencing results of the DSG2 mutation. (A) The father of the proband did not carry the pathogenic gene. (B) The proband, his mother and sister carried a mutation in the DSG2 gene. Sequence chromatogram indicated a G to A transition of nucleotide 445. |
 |
Figure 6 Sequencing results of the PRRT2 mutation. (A) The proband and his father carried a mutation in the PRRT2 gene. (B) The mother and sister of the proband did not carry the pathogenic gene. Sequence chromatogram indicated a C loss of nucleotide 641. |
 |
Figure 7 The pedigrees of the proband’s family affected by PKD and ARVC in our study. Symbols with a slash through them indicate deceased individuals. |
Because of the high risk of sudden cardiac death, we recommend implanting a cardioverter defibrillator (ICD) for the patient. Due to economic reasons, the patient refused, and we gave the patient metoprolol orally. No serious arrhythmias and complications occurred during the 6-month follow-up.
Discussion
ARVC is a rare cardiac disorder, which is inherited in an autosomal dominant trait, with an estimated incidence of 1:1000–1:5000.3 The severity and incidence of male patients are significantly higher than that of female patients.
It is accepted that genetic testing must be performed on individuals who were suspected of having a genetic disorder. Thus, we sequenced the proband and his families’ WES, and the results showed that the patient carried both DSG2 and PRRT2 gene mutations. Based on the corresponding results from the OMIM database, functional damage of desmosome protein was predicted by SIFT, POLYPHEN2, and Mutation Taster software, respectively. The results of the software all showed that the mutation caused by DSG2 was a pathogenic variation, as well as the mutation caused by PRRT2.5
As reported, multiple mutation sites in DSG2 can lead to ARVC, and its possible pathogenic mechanisms are as follows. As the only subtype expressed in cardiomyocytes, desmoglein 2 is a crucial component of the desmosomes’ transmembrane structure. The mutation of DSG2 can disrupt the “connexome”, which is the protein coordinated network at the intercalated disks composed of desmosomes, sodium channels, and gap-junction proteins, and cause DSG2, DSC2-mediated cell adhesion to decrease. While the former can lead to the disruption of intercellular connections, and the latter can result in myocyte detachment and cell death.6 Furthermore, there are a total of 76 DSG2 mutation sites as reported, and we discovered a new missense mutation site in the DSG2 (exon5: c.445G > A, p.Val149Ile), which was not absent in the general population but had never been associated with ARVC in the relevant literature.
PRRT2 is the major causative gene of PKD, which is most commonly transmitted in an autosomal dominant manner. The patient we reported had an unexplained PKD history and had been treated with carbamazepine for a long time. Strangely, the proband’s father with the same PRRT2 mutation gene was normal. Regrettably, there were limited clinical data, including electroencephalogram (EEG), for detailed information about his symptoms, which prevented us from exploring the relationship between PRRT2 penetration and PKD.
Many ARVC studies in the last two decades have examined how other genes affect ARVC’s penetrance and heterogeneity. Based on the hypothesis of SamiQadri,7 the co-mutation of the DSG2 gene and other genes may increase the penetrance of ARVC, but there have not been large-scale population genetic surveys and many relevant data to bear out the abovementioned hypothesis. This hypothesis may explain why the proband’s mother and sister were completely asymptomatic. It turned out that the proband’s mother and sister carried the same DSG2 mutation as the proband but not the PRRT2 mutation.
Hence, it was suspected that PRRT2 might play a role in ARVC’s penetrance. Nonetheless, there were still some flaws in our hypothesis. Firstly, there was a possibility that the sister and mother of the proband may be in the early stage of the disease. Secondly, we did not further discuss this hypothesis’s molecular mechanism and verify it by animal experiments.
Gene testing counts for ARVC. The importance of gene detection for ARVC lies in exploring its pathogenic mechanism and its diagnosis and treatment. Although the latest diagnostic criteria have been published,3 the diagnosis of ARVC is challenging due to the multiple phenotypes, non-characteristic imaging examinations and clinical manifestations, and limitations in studying the molecular mechanisms of ARVC. Epsilon wave is an ECG phenomenon with relatively high specificity in the diagnosis of ARVC, but the incidence is only about 30%. Endomyocardial biopsy is regarded as the gold standard for the diagnosis of ARVC but is very rarely used for ARVC diagnosis because of poor sensitivity and the availability of advanced imaging and genetic testing. Gene screening is relatively specific and non-invasive, which can significantly improve the early diagnosis rate of ARVC. In our case, a new mutation site on DSG2 was discovered through WES, which can provide a brand-new direction for gene screening of suspected and further complements its diagnostic criteria.
As for the treatment of ARVC, clinical management mainly includes life management, drug therapy, catheter ablation, ICD implantation, and heart transplantation.8 Implanting ICD or restricting strenuous exercise can effectively reduce the incidence of adverse events, such as sudden cardiac death.
This paper’s ultimate goal is to illustrate more evidence for gene screening by providing new gene mutation sites to improve the detection rate of healthy gene carriers or patients in the early incubation stage, thereby reducing the incidence of adverse outcomes. A hypothetical example was proposed: heterozygous DSG2 (NM_001943: exon5: c.445G>A, p.Val149Ile) as a genetic background may be susceptible to ARVC when coexisting with heterozygous PRRT2 (NM_001256442: exon2: p. Arg217ProfsTer8). However, there are still more clinical studies to bear out the hypothesis.
Abbreviations
ARVC, arrhythmogenic right ventricular cardiomyopathy; DSG2, desmoglein-2 gene; PKD, paroxysmal kinesigenic dyskinesia; VT, ventricular tachycardia; VF, ventricular fibrillation; JUP, plakoglobin; DSC2, desmocollin 2; PKP2, plakophilin 2; DSP, desmoplakin; RV, right ventricle; LV, left ventricle; CRP, cardiopulmonary resuscitation; ECG, electrocardiogram; RVOT, right ventricular outflow tract; WES, whole-exome sequencing; EEG, electroencephalogram.
Data Sharing Statement
All relevant data supporting the conclusions of this article are included within the article.
Ethical Approval
The need for institutional ethics approval for this case report was waived.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and accompanying images.
Acknowledgment
Rui Huang and Yinhua Luo should be regarded as co-first authors.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agreed to be accountable for all aspects of the work.
Funding
This work was supported by Enshi Tujia and Miao Autonomous Prefecture Bureau of Science and Technology (Grant Number E20180002) and the Graduate Education Innovation Project of Hubei Minzu University (MYG2020010)(Yuanhong Li). The funding bodies played no role in designing the study, collection, analysis, and interpretation of the data in writing the manuscript.
Disclosure
The authors of this article declare that they have no conflicts of interest.
References
1. Hamilton-Craig C, McGavigan A, Semsarian C, et al. The cardiac society of Australia and New Zealand position statement on the diagnosis and management of arrhythmogenic right ventricular cardiomyopathy (2019 Update). Heart Lung Circ. 2020;29(1):40–48. doi:10.1016/j.hlc.2019.02.194
2. Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–1179. doi:10.1161/CIRCULATIONAHA.105.583674
3. Castaños Gutiérrez SL, Kamel IR, Zimmerman SL. Current concepts on diagnosis and prognosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Thorac Imaging. 2016;31(6):324–335. doi:10.1097/RTI.0000000000000171
4. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806–814. doi:10.1093/eurheartj/ehq025
5. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi:10.1038/gim.2015.30
6. Hariharan V, Asimaki A, Michaelson JE, et al. Arrhythmogenic right ventricular cardiomyopathy mutations alter shear response without changes in cell-cell adhesion. Cardiovasc Res. 2014;104(2):280–289. doi:10.1093/cvr/cvu212
7. Awad MM, Dalal D, Cho E, et al. DSG2 mutations contribute to arrhythmogenic right ventricular dysplasia/cardiomyopathy. Am J Hum Genet. 2006;79(1):136–142. doi:10.1086/504393
8. Corrado D, Wichter T, Link MS, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Eur Heart J. 2015;36(46):3227–3237. doi:10.1093/eurheartj/ehv162
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.