Back to Journals » OncoTargets and Therapy » Volume 10
ARHGAP1 overexpression inhibits proliferation, migration and invasion of C-33A and SiHa cell lines
Received 6 May 2016
Accepted for publication 10 August 2016
Published 7 February 2017 Volume 2017:10 Pages 691—701
DOI https://doi.org/10.2147/OTT.S112223
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr William C. Cho
Jun-ping Li,1 Yang Liu,2 Yi-hua Yin3
1Department of Gynecology and Obstetrics, Huashan Hospital North, 2Institute of Antibiotics, Huashan Hospital, Fudan University, 3Department of Gynecology, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai, People’s Republic of China
Abstract: ARHGAP1, also known as RhoGAP, RhoGAP1, CDC42GAP and p50rhoGAP, is officially named Ras homology (Rho) GTPase-activating protein 1, which is one of the key members of RhoGAPs. Growing evidences demonstrate that several RhoGAPs are suppressed or downregulated in cancers. Thus, the aim of this study was to explore the effects of ARHGAP1 on cervical carcinoma cells. The human cervical carcinoma cells C-33A and SiHa were transduced with lentivirus targeting ARHGAP1 (lenti-ARHGAP1). Cellular proliferation, migration and invasion assays, as well as quantitative real-time polymerase chain reaction and Western blot assays, were performed in the control, negative control (infected with lentivirus) and ARHGAP1+-infected groups. Results showed that overexpression of ARHGAP1 markedly inhibited the proliferation of both C-33A and SiHa cells at 24 h, 48 h and 72 h in a time-dependent manner (n=3, P<0.01). Migration and invasion of C-33A and SiHa cells were suppressed after the transduction with lenti-ARHGAP1 compared with the controls (n=3, P<0.01). In addition, several tumor cellular process-related proteins, such as matrix metallopeptidase 2, zinc finger E-box binding homeobox 1, Cyclin B1, twist family bHLH transcription factor 1 and proliferating cell nuclear antigen, were all downregulated in ARHGAP1-overexpressed C-33A and SiHa cells and proved to be targets of ARHGAP1. This study indicated that ARHGAP1 may have a positive function on antitumor activity in the treatment of cervical cancer.
Keywords: cervical carcinoma, ARHGAP1, tumor cellular process-related protein
Introduction
Cervical carcinoma is a cancer arising from the cervix, and it is the fourth most common cause of death in women.1,2 Despite the fact that cervical cancer is preventable, this tumor currently afflicts ~5 million women per year, with more than half of them experiencing death, ~70% of which occur in developing countries.2 Ras homology (Rho) GTPase-activating proteins (RhoGAPs) are emerging as a new class of biomarkers for various cancers.3 To expand the understanding of the pathogenesis of cervical cancer, we embarked a research on the founding member of the RhoGAP family, ARHGAP1, which encodes Rho GTPase-activating proteins and is also known as RhoGAP, RhoGAP1, CDC42GAP and p50rhoGAP. Growing reports have proved that Rho GTPases are involved in various cellular functions and as negative regulators of the Rho proteins; RhoGAPs play critical roles in gene expression, cell control, migration, invasion and normal development.4,5 In addition, many RhoGAPs, including deleted in liver cancer (DLC) and ARHGAP8, are found to be downregulated or deleted in different types of cancers.6 It has been proved that by negatively regulating the activity of Rho proteins, DLC-1, also known as ARHGAP7, suppresses the growth of cancer cells.7 Further studies have indicated that downexpression of DLC-1 or DLC-2 is associated with various cancer types, such as DLC-1 in ovary, colon, kidney, prostate, breast and gastric cancers, and DLC-2 in hepatocellular carcinomas.8,9 Structurally, DLC contains a conserved GTPase-activating protein (GAP) for RhoGAP domain,8 and the tumor-suppressive function of DLC relies on its RhoGAP activity.9 Given that the Rho GTPases are well-known regulators of cell migration and may influence cell invasion even in metastasis, it is generally supposed that the relationship between RhoGAPs and cancer is regulated by the Rho pathway,10 while it is unknown whether it can be regulated by Rho-independent pathways. To date, although specific functions for ARHGAP1 in disease relevance are poorly understood, it has been previously proved that ARHGAP1 is necessary for transforming growth factor-β-induced invasion in cancer cells,11 and it has biochemical GAP activity toward Rho and Cdc42.12–14 Moreover, it is generally assumed that ARHGAP1 is a potential target of both oncogenic and antitumor microRNAs.11,15
In the current study, we provide insights into one of the key RhoGAP members, ARHGAP1, and aim to explore the association between ARHGAP1 and cervical cancer. C-33A and SiHa cell lines were determined as our study target and transduced with reconstructed lentivirus targeting ARHGAP1 (lenti-ARHGAP1). After treatment with the overexpression of ARHGAP1, cell proliferation was identified by Cell Counting Kit-8 (CCK-8), and Transwell assay was performed to detect the migration and invasion of cells. Besides, the expression levels of several tumor-related genes were determined by quantitative real-time polymerase chain reaction (qRT-PCR) and Western blot. Through the abovementioned work, we basically confirmed the function of ARHGAP1 on cervical cancer.
Patients and methods
Cervical cancer patients and tissues
In this study, 35 patients with cervical cancer admitted to the Shanghai First Maternity and Infant Hospital were enrolled, with all patients having complete pathological and clinical follow-up data. The expression levels of ARHGAP1 in cervical cancer tissues and peritumoral tissues were detected by qRT-PCR. Tissues were resected when the patients were undergoing definitive surgery, with peritumoral tissues being within ~5 cm of the tumor margin. Informed written consent was obtained from all the patients or from their advisers according to the ethics committee guidelines. Ethical approval for the study was provided by the independent ethics committee, Shanghai First Maternity and Infant Hospital.
Cervical cancer cell culture
Human cervical cancer cells HeLa, CaSKi, C-33A, SiHa and C4-1 were obtained from Shanghai Institute of Cellular Biology (Shanghai, People’s Republic of China). All the cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) with 100 U/mL penicillin, 100 μg/mL streptomycin and 10% heat-inactivated fetal bovine serum (FBS; Gibco BRL, Rockville, MD, USA). In addition, the incubator (Thermo Fisher Scientific Inc., Waltham, MA, USA) was set to 37°C, 100% humidity and 5% CO2.
Construction of lentivirus vector and overexpression of ARHGAP1
The recombinant lentivirus expression vector was generated by transient cotransduction. The core plasmid pLVX-AcGFP-C1 (Clontech) was used to construct the recombinant plasmids, pLVX-AcGFP-C1-ARHGAP1 and pLVX-AcGFP-C1-shRNA-ARHGAP1, by double enzyme restriction and ligation. The packaging plasmids, psPAX2 and pMD2G, purchased from Addgene Company, together with the recombinant plasmids pLVX-AcGFP-C1-ARHGAP1, pLVX-AcGFP-C1-shRNA-ARHGAP1 or noncarrier plasmid pLVX-AcGFP-C1, were transiently transduced into 293T cells (ATCC, Manassas, VA, USA). Thus, the lenti-ARHGAP1 (ARHGAP1+), lenti-shRNA-ARHGAP1 (ARHGAP1−), lentivirus (negative control, NC) and lenti-shRNA-NC were obtained.
Lentivirus transduction
C-33A and SiHa cells in logarithmic growth phase were collected and seeded in 96-well culture plates in triplicate at a density of 3×105 cells/well and incubated in 100 μL medium undisturbed at 37°C and 5% CO2. After culturing for 1 day, for each kind of cells, the original medium in triplicate was pipetted and replaced with 100 μL DMEM (Hyclone, SH30024.01B; control), 100 μL DMEM containing 0.1 μL lentivirus (NC) or 100 μL DMEM containing 0.1 μL lenti-ARHGAP1. Each of the time above was regarded as one study group, with cells without transduction as the control group. All the transfected cells in different groups were collected and processed for the subsequent cell proliferation, Transwell, qRT-PCR and Western blot assays.
Cell proliferation assay
CCK-8 (SAB Company, USA) was used to assess the effects of ARHGAP1 on the viability of cervical cancer cell. Briefly, after transfecting the cells for 0 h, 12 h, 48 h and 72 h, CCK-8 reagent was added to each group of wells with 1:10 (v/v) per 100 μL medium. All the cells were further cultured in an incubator (37°C, 100% humidity and 5% CO2; Thermo Fisher Scientific Inc.) for 1 h. Then, the end point of incubation and the cell viability were evaluated. The optical density at 450 nm was determined for the supernatant of each well by a microplate reader. All the experiments were performed at least three times.
Transwell assay
For cell migration assay, C-33A and SiHa cells were transduced with lentivirus or lenti-ARHGAP1 in triplicate, respectively. Different transduction groups were starved with serum-free DMEM for 24 h. Cells were digested for 5 min by 0.25% trypsin (Gibco, Shanghai, People’s Republic of China) and resuspended in DMEM containing 1% FBS. After counting, cells were diluted to 1×105 cells/mL, seeded in a 24-plate Transwell and then continued to incubate for 48 h. Then, the cells were fixed for 10 min using 1 mL/well of 4% paraformaldehyde (JRDUN biotech, Shanghai, People’s Republic of China), stained with Giemsa (JRDUN) for 30 min and washed three times with 1× phosphate-buffered saline (PBS). Finally, the cells were removed from Transwell inserts without migrating cells using a cotton swab and placed under 200× microscope (Olympus, Shenzhen, People’s Republic of China) for counting the number of cells.
For cell invasion, Transwell room with 24-well plates and 8 μm pore size (Sigma, San Francisco, CA, USA) was washed with 1× PBS for 5 min before the assay was performed. The inserts were coated with 80 μL matrigel (dilution at 1:2; BD Bioscience). Cells were transferred to the upper matrigel chamber with a density of 1×105 cells/mL in 0.5 mL serum-free medium; meanwhile, the lower chamber was added with 0.75 mL complete medium containing 10% FBS as chemoattractants. Cells were incubated at 37°C for 48 h. After that, the cells that were able to pass through the filter were fixed and stained by 1 mL 0.5% crystal violet for 30 min. The numbers of invaded cells in five randomly selected high-power fields were counted under the microscope.
qRT-PCR assay
After the transduction with lentivirus (model control) and lenti-ARHGAP1, the mRNA levels of ARHGAP1 in C-33A and SiHa cells were quantified by qRT-PCR, with no transduction cells as a control. Total RNA was isolated from cells using Trizol Reagent (Invitrogen, Tokyo, Japan) and detected by agarose gel electrophoresis. As the template for PCR reactions, cDNA was synthesized from 5 μg of RNA using avian myeloblastosis virus reverse transcriptase (Fermentas, Waltham, MA, USA). qRT-PCR reactions were performed using SYBR® Green 10× Supermix (Takara, Kyoto, Japan) in a 25 μL total volume and on Roche Light Cycler® 480II System (Roche Diagnostics Ltd., Basel, Switzerland). Primer pairs (Table 1) for human genes were designed using the Primer Express Software (Applied Biosystems, Shanghai, People’s Republic of China). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the internal control. The PCR procedure was as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 45 s; one cycle of 95°C for 15 s, 60°C for 1 min; one cycle of 95°C for 15 s, 60°C for 15 s. All PCR reactions were performed in triplicate, and the relative expression levels of different groups were calculated by normalizing to the mRNA expression level of GAPDH using ΔΔCT method.
 | Table 1 Primers used in qRT-PCR analysis |
Western blot analysis
Cultured or transfected cells were harvested and washed twice with PBS. Then, the cells were lysed in ice-cold radio immunoprecipitation assay buffer (Beyotime, Shanghai, People’s Republic of China) containing 0.01% protease and phosphatase inhibitor (Sigma, Shanghai, People’s Republic of China) and incubated on ice for 30 min. Cell lysis was centrifuged at 12,000× g for 10 min at 4°C, and the proteins in supernatant were obtained (20-30 μg) and separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, then transferred electrophoretically to a polyvinylidene fluoride membrane (Millipore, Shanghai, People’s Republic of China). The membrane was blocked with 5% bovine serum albumin in tris-buffered saline (TBS) and Polysorbate 20 (also known as Tween 20) and incubated with primary antibodies against ARHGAP1 (Ab194425, 1:1,000 dilution; Abcam), matrix metallopeptidase 2 (MMP2; Ab92536, 1:1,000 dilution; Abcam), zinc finger E-box binding homeobox 1 (ZEB1; Ab124512, 1:1,000 dilution; Abcam), Cyclin B1 (CCNB1; #12231, 1:1,000 dilution; Cell Signaling Technology, Inc.), twist family bHLH transcription factor 1 (Twist; Ab175430, 1:500 dilution; Abcam), proliferating cell nuclear antigen (PCNA; Ab92552, 1:5,000 dilution; Abcam) and GAPDH (#5174, 1:1,500 dilution; CST). Blots were then incubated for 1 h at 37°C with goat anti-mouse or anti-rabbit secondary antibody (Beyotime), and the intensities were measured using enhanced chemiluminescence (Thermo Fisher Scientific Inc.).
Statistical analysis
All the experiments were performed with values expressed as mean ± standard deviation. Data were analyzed by the median and the Kruskal–Wallis test. In all statistical comparisons, P<0.05 was considered statistically significant.
Results
Lower expression of ARHGAP1 in tumor tissue than normal tissue
The expression data of ARHGAP1 gene were obtained at The Cancer Genome Atlas for the projects of cervical carcinoma. As shown in Figure 1A (P<0.001), the mRNA expression of ARHGAP1 was much lower in tumor tissues than in normal tissues. To verify the biological function of ARHGAP1 further, qRT-PCR was used for detecting the expression levels of ARHGAP1 in both cervical carcinoma and paracancerous tissues from 35 patients. As shown in Figure 1B (P<0.001), an obvious increase was observed in ARHGAP1 expression of paracancerous tissues compared with cervical carcinoma tissues. The results indicated that ARHGAP1 possibly play a positive role in inhibiting cervical carcinoma.
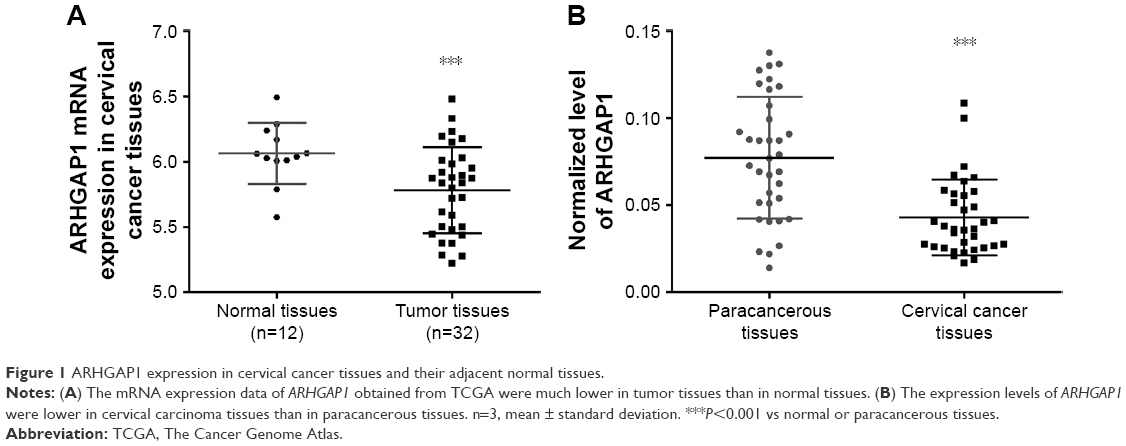 | Figure 1 ARHGAP1 expression in cervical cancer tissues and their adjacent normal tissues. |
Next, we explored the association between ARHGAP1 expression and clinical characteristics. Patients with cervical carcinoma were divided into two groups according to the median level of ARHGAP1. Evaluation of ARHGAP1 expression in 35 patients with cervical carcinoma (Shanghai First Maternity and Infant Hospital) with different clinicopathological features revealed that the lower ARHGAP1 expression was correlated with the larger tumor size and lymphatic invasion (Table 2). However, we did not find any association between ARHGAP1 expression levels and other clinical pathological features including patients’ age, pathologic stages, distant metastasis and depth of tumor invasion (Table 2).
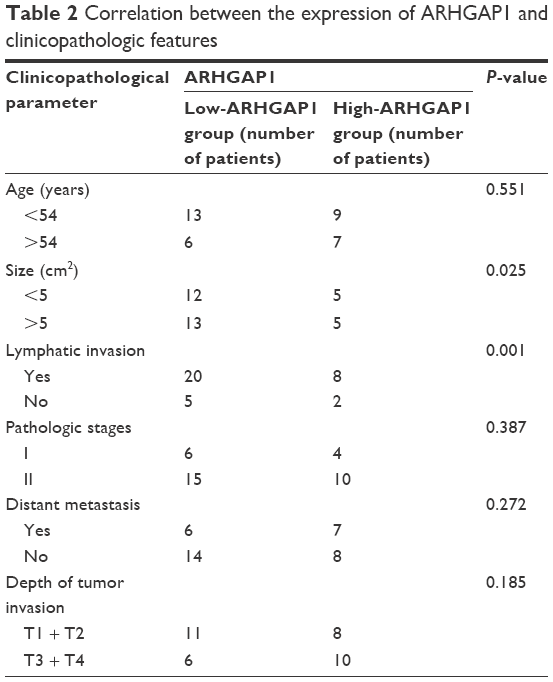 | Table 2 Correlation between the expression of ARHGAP1 and clinicopathologic features |
ARHGAP1 expression in cervical carcinoma cell lines
As the first step to explore the effects of ARHGAP1 on the biology behavior of cervical carcinoma cells, the mRNA expression and protein levels of ARHGAP1 in various cervical carcinoma cell lines including HeLa, CaSKi, C-33A, SiHa and C4-1 were detected by RT-PCR and Western blot, respectively. Results showed that ARHGAP1 mRNA expression in C-33A and SiHa cell lines was markedly lower than the others and was highest in HeLa cells (P<0.001). Unsurprisingly, the protein levels of ARHGAP1 in C-33A and SiHa cells were also much lower than the others, with the C-33A cell presenting the lowest protein level (Figure 2A and B).
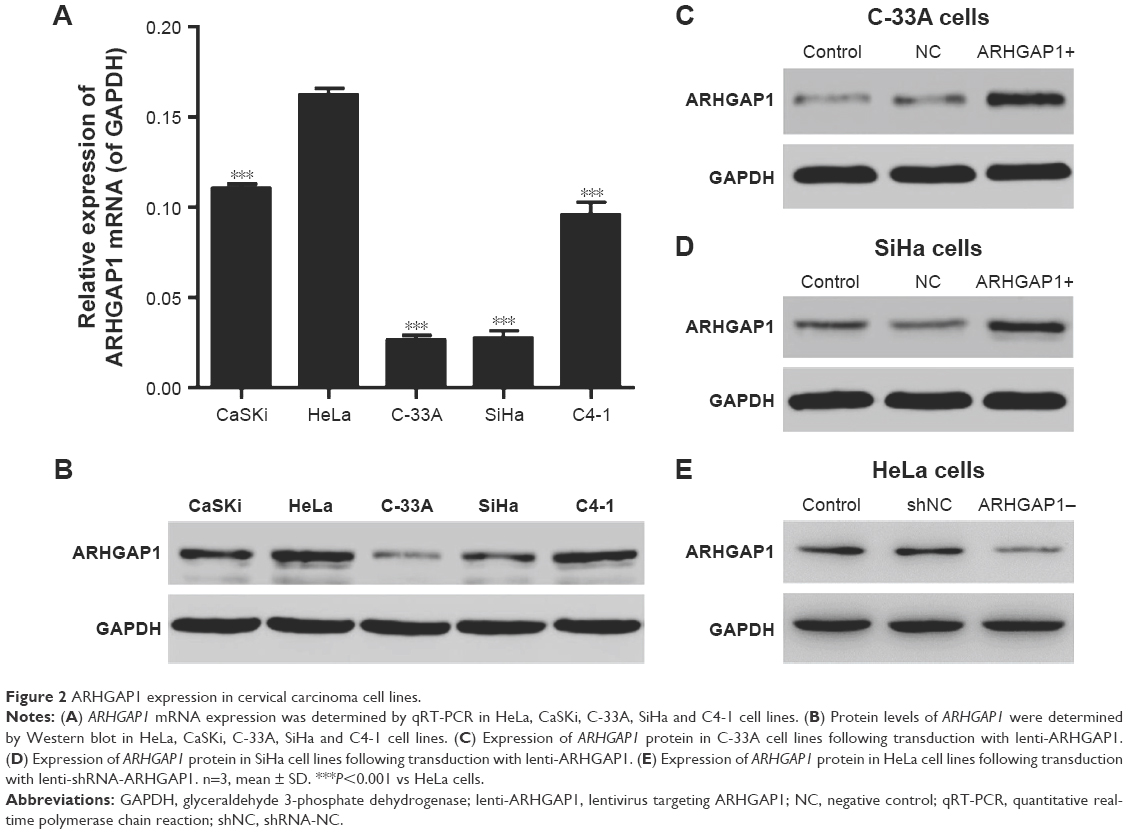 | Figure 2 ARHGAP1 expression in cervical carcinoma cell lines. |
Expression of ARHGAP1 in cervical carcinoma cells after transduction
To detect whether the recombinant lenti-ARHGAP1 was constructed successfully, Western blot assay was performed in C-33A and SiHa cells. The ARHGAP1 levels of the control and lentivirus-transfected cells were substantially identical, both of which were declined notably in comparison with the lenti-ARHGAP1-transfected cells. In addition, this result occurred in both C-33A (Figure 2C) and SiHa cells (Figure 2D), while lenti-shRNA-ARHGAP1-transfected HeLa cells showed significantly decreased expression of ARHGAP1 (Figure 2E), suggesting that ARHGAP1 overexpression and knockdown system were achieved and could be used for the subsequent studies.
ARHGAP1 overexpression inhibited cell viability of C-33A and SiHa cells
The effects of ARHGAP1 overexpression on the cell growth and proliferation of C-33A and SiHa cell lines were measured by CCK-8 assay. The results showed that after transduction for 24 h, 48 h and 72 h, the cell viability of both C-33A and SiHa cells was weakened markedly in the lenti-ARHGAP1 group in a time-dependent manner (n=3, P<0.001), compared with that of unprocessed control cells and lentivirus-transduced cells (Figure 3A and C). The mean cell proliferation rates of C-33A and SiHa cells that were transfected with lenti-ARHGAP1 for 72 h were significantly obstructed compared with that of the controls (n=3, P<0.001; Figure 3B and D). However, the mean cell proliferation rates of HeLa cells that were transfected with lenti-shRNA-ARHGAP1 for 72 h were significantly increased compared with that of the controls (n=3, P<0.001; Figure 3E and F).
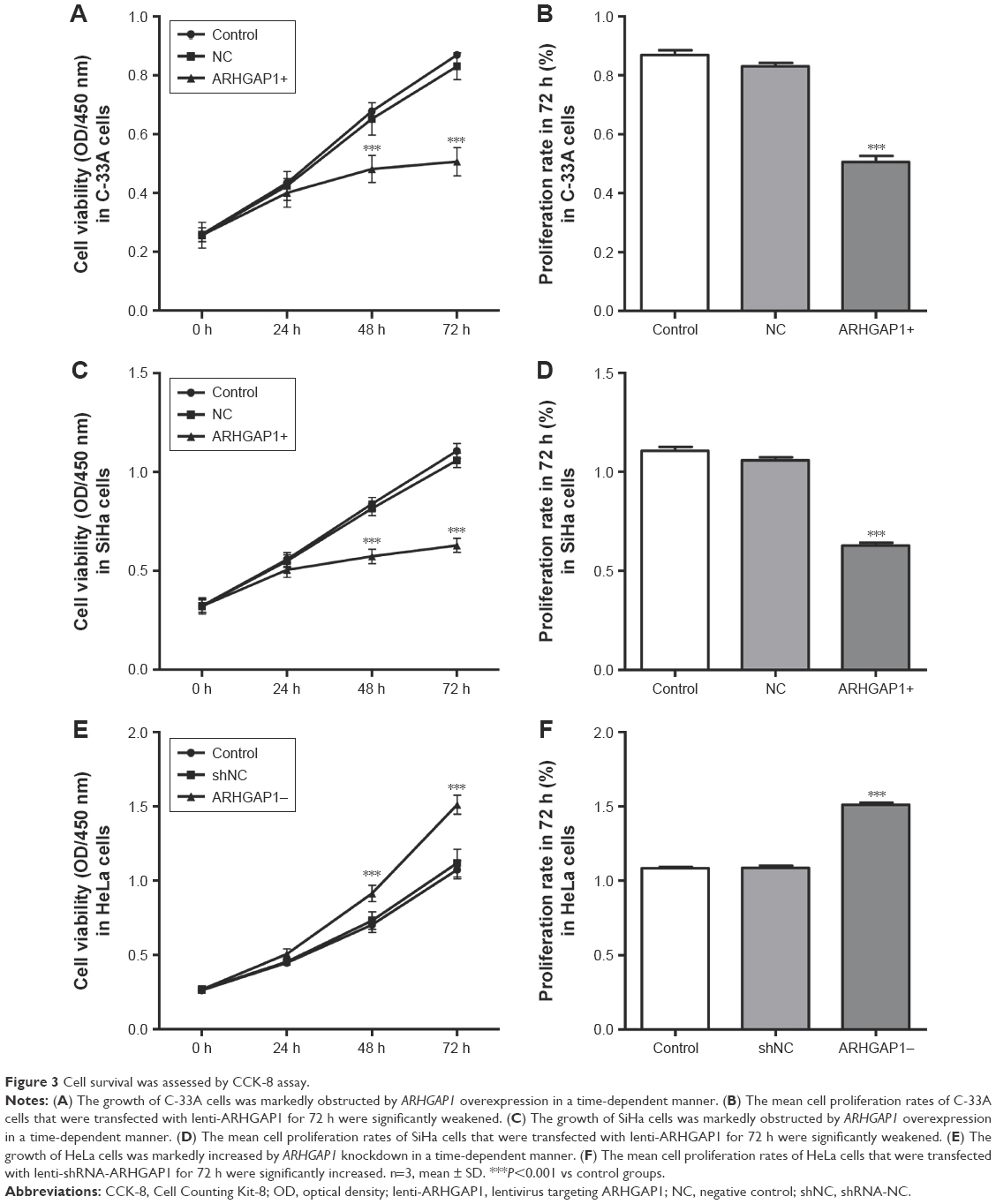 | Figure 3 Cell survival was assessed by CCK-8 assay. |
ARHGAP1 overexpression inhibited cell migration and invasion of C-33A and SiHa cells
To figure out the effects of ARHGAP1 on cell invasion and migration of C-33A and SiHa cells, the Transwell assays were performed, and the results are shown in Figure 4. Compared with the control and NC groups, treatment with ARHGAP1 obviously suppressed the migration ability of C-33A cells (Figure 4A), and the number of migrating C-33A cells in the abovementioned three groups was 125±8, 116±7 and 73±7, respectively (Figure 4B). Not surprisingly, the migration ability of SiHa cells was inhibited, too (Figure 4C), with the number of migrating SiHa cells in the control, NC and ARHGAP1+ groups being 116±6, 107±6 and 58±6, respectively (Figure 4D). However, the migration ability of HeLa cells was increased, with the number of migrating HeLa cells in the control, NC and ARHGAP1− groups being 106±8, 105±10 and 235±9, respectively (Figure 4E and F).
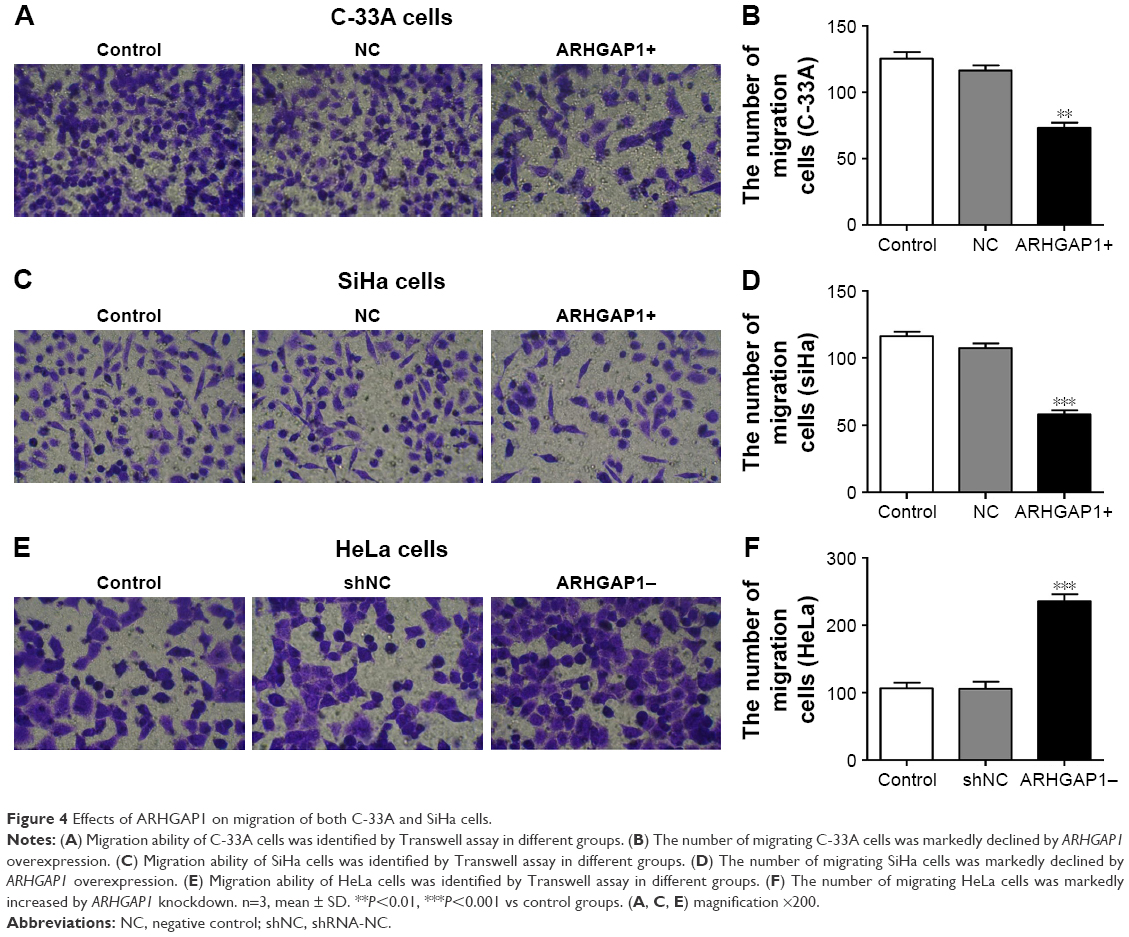 | Figure 4 Effects of ARHGAP1 on migration of both C-33A and SiHa cells. |
Similar results were observed in Transwell invasion assays. After transduction with lenti-ARHGAP1, the invasion ability of both C-33A and SiHa cells was suppressed (Figure 5A and C), with the invasion number of lenti-ARHGAP1-transfected cells was 48±4 and 29±3, respectively, much less than the controls (Figure 5B and D). After transduction with lenti-shRNA-ARHGAP1, the invasion ability of HeLa cells was increased (Figure 5E), with the invasion number of lenti-shRNA-ARHGAP1 transfected cells was 108±3, much higher than the controls (60±4; Figure 5F). All the results suggested that the overexpression of ARHGAP1 played a negative role in cell migration and invasion of C-33A and SiHa cells.
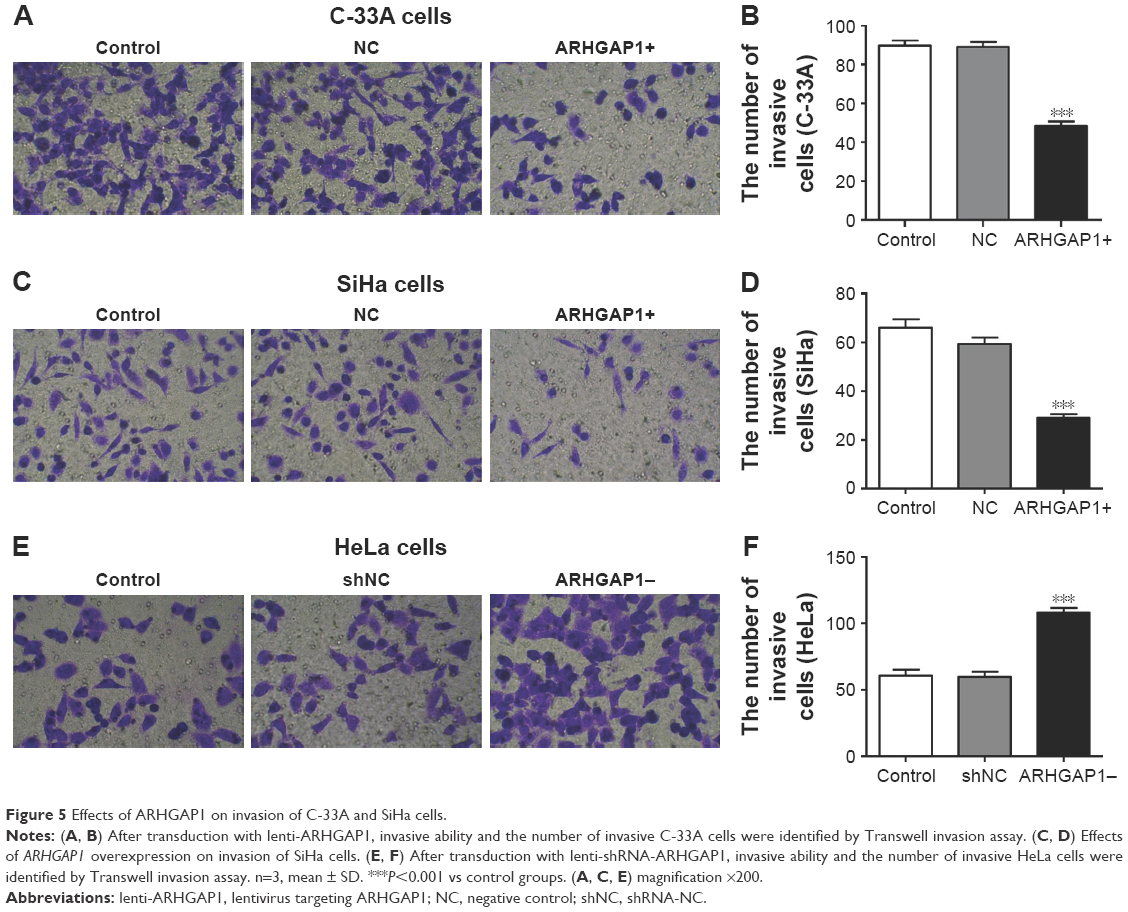 | Figure 5 Effects of ARHGAP1 on invasion of C-33A and SiHa cells. |
ARHGAP1 overexpression caused downregulation of several tumor-related genes in C-33A and SiHa cells
To clarify the mechanism of biological behavior of C-33A and SiHa cells induced by ARHGAP1, the mRNA expression and protein levels of several tumor-related genes in C-33A and SiHa cells were detected by qRT-PCR and Western blot, respectively. As revealed in Figure 6A and B, ARHGAP1 overexpression evidently decreased both the mRNA expression and protein levels of all the proteins, including MMP2, ZEB1, CCNB1, Twist and PCNA in C-33A cells. Likewise, the same effects of ARHGAP1 on all the proteins were occurred in SiHa cells (Figure 6C and D). However, ARHGAP1 knockdown evidently increased both the mRNA expression and protein levels of all the proteins, including MMP2, ZEB1, CCNB1, Twist and PCNA in HeLa cells (Figure 6E and F). In addition, the results indicated that the way ARHGAP1 impaired the cell proliferation, invasion and migration probably may be via MMP2, ZEB1, CCNB1, Twist or PCNA.
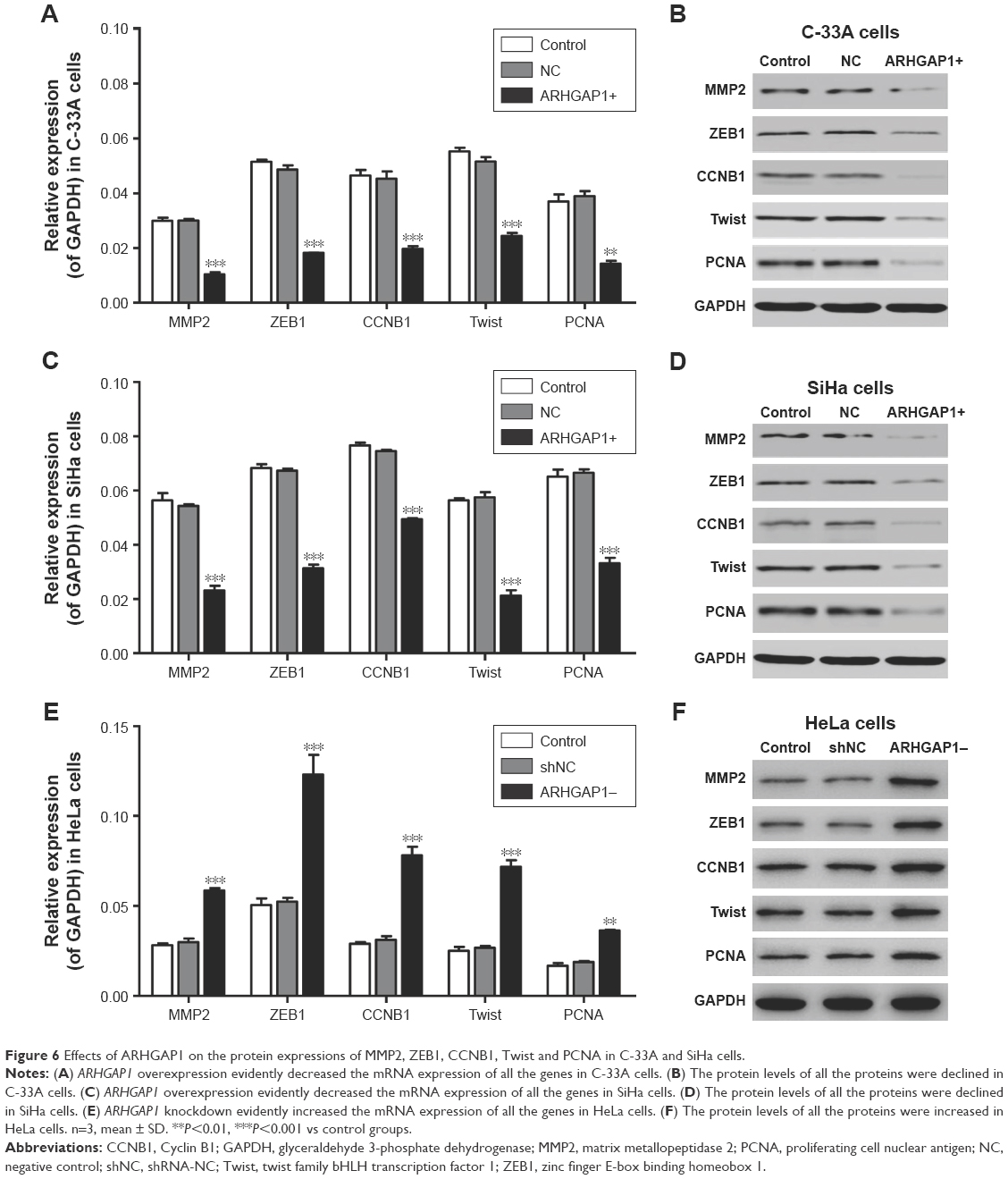 | Figure 6 Effects of ARHGAP1 on the protein expressions of MMP2, ZEB1, CCNB1, Twist and PCNA in C-33A and SiHa cells. |
Discussion
ARHGAP1 was first isolated from human spleen tissue. As the founding member of RhoGAP family, ARHGAP1 is officially named Rho GTPase-activating protein 1.12,16,17 With the involvement of RhoGAPs in cancer phenotypes that are becoming increasingly apparent, most studies have been exploring the link of Rho GTPases in all stages during cancer progression.18,19 However, currently, most relationships between RhoGAP proteins and various kinds of cancers have not been characterized, and thus, we need a thorough scan to determine the functions of RhoGAPs in different types of cancers. In addition, as the founding member of RhoGAPs, ARHGAP1 may act as a study example.
In the current study, we found that ARHGAP1 of patients with cervical cancer showed higher expression in normal tissues than in tumor tissues, and decreased ARHGAP1 expression was correlated with the larger tumor size and lymphatic invasion, which was consistent with our bioinformatics analysis results and the existing reports. Overexpression of ARHGAP1 markedly inhibited the cell viability and suppressed the cell capability of migration as well as invasion in the two cell lines. Our results mentioned earlier reveal a basis for the functions of ARHGAP1 on cervical cancer. Currently, there are no reports about the function of ARHGAP1 in cervical cancer, while several reports are available on the role of ARHGAP1 and its family members in breast cancer, liver cancer and several other cancers. Therefore, transduction of ARHGAP1 into MCF7 cells (human breast adenocarcinoma cell line) can significantly inhibit cell growth,20 which means that ARHGAP1 may play a negative role in cancer cell viability. Besides, there were reports that DLC-1 can suppress cancer cell growth,7 and the downexpression of DLC-1 or DLC-2 is associated with various cancer types, such as DLC-1 in ovary, colon, kidney, prostate, breast and gastric cancers, and DLC-2 in hepatocellular carcinomas.8,9 In addition, Holeiter et al21 demonstrated that depletion of DLC3 (the Rho-specific protein) leads to mislocalization of E-cadherin and catenins, which is associated with impaired cell aggregation and increased migration. Liu et al22 proved that leukocyte migration is regulated by the motorized RhoGAP myosin IXB (Myo9b) through controlling RhoA signaling. Overall, these findings identify ARHGAP1 as a novel component of the antitumor machinery without tumor specificity. However, Xu et al reported that ArhGAP1 can promote the transcription function of p53, which indicates that ARHGAP1 genes likely function as tumor suppressors.6
To explore the mechanism at the molecular level, the levels of MMP2, ZEB1, CCNB1, Twist, and PCNA genes in C-33A + ARHGAP1 and SiHa + ARHGAP1 cells were detected by qRT-PCR and Western blot and resulted to be decreased. Ran et al23 found that the downregulation of ZEB1 expression may reduce the proliferation and motility of cervical cancer cells, suggesting that ZEB1 might be a potential therapeutic target for cervical cancer. Besides, CCNB1 depletion or stable gene silencing of CCNB1 inhibits proliferation and induces apoptosis in human tumor cells.24,25 Twist gene in the cervical cancer cell, CaSKi, is highly expressed and the silent Twist gene could inhibit the cell proliferation of CaSKi and promote its apoptosis.26 All the abovementioned reports showed a common point that the downregulation of ZBE1, CCNB1 and Twist inhibits the cancer cell proliferation, indicating that the inhibition of cervical cancer cell viability by ARHGAP1 overexpression may be via ZBE1, CCNB1 and Twist. In addition, it is well known that matrix metalloproteinases (MMPs) play a critical role in the development and invasion of tumor. As a member of MMPs, MMP2 is a secreted or membrane-associated protein, capable in digesting extracellular matrix components, and may have a role in the migration of tumor cells during invasion.27,28 In addition, Moroz et al29 proved that it is through the downregulation of MMP2 and MMP9 that human prostate cancer cell invasion is inhibited by finasteride. Neoplastic cells and stromal fibroblastic cells undergoing activation of ZEB1 and Twist due to growth factors produced by the tumor are transformed after treatment with epithelial–mesenchymal transition (EMT).30 However, epithelial expression of twist is associated with a poor prognosis, hinting at its importance in the spread of breast carcinoma.23,30 Carcinomas metastatic to the lung showed a significantly higher expression of involvement of ZEB1 and twist in EMT in primary lung tumors than primary lung tumors, indicating the probable importance of ZEB1 and Twist in the metastatic process.31 Positive Twist expression seems to be a useful marker in patients with cervical cancer likely to have an unfavorable clinical outcome.32 Upregulation of PCNA is closely associated with high-risk human papillomavirus and progression of cervical intraepithelial neoplasia, but does not predict disease outcome in cervical cancer.33 PCNA protein expression increases during cervical tumorigenesis.34 Overall, the way ARHGAP1 impaired the cell invasion and migration probably may be via MMP2, Twist or PCNA.
Conclusion
Our findings suggest that the overexpression of ARHGAP1 markedly inhibited the proliferation of cervical cancer cells in a time-dependent manner and suppressed the migration and invasion of cancer cells indicating that ARHGAP1 was a tumor suppressor for cervical cancer. Furthermore, several tumor cellular process-related proteins were downregulated in ARHGAP1-overexpressed C-33A and SiHa cells, which proved to be targets of ARHGAP1. This study implied that ARHGAP1 was a novel factor of antitumor activity and had a positive function on antitumor activity in the treatment of cervical cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Brooks SE, Chen TT, Ghosh A, Mullins CD, Gardner JF, Baquet CR. Cervical cancer outcomes analysis: impact of age, race, and comorbid illness on hospitalizations for invasive carcinoma of the cervix. Gynecol Oncol. 2000;79(1):107–115. | ||
Newmann SJ, Garner EO. Social inequities along the cervical cancer continuum: a structured review. Cancer Causes Control. 2005;16(1):63–70. | ||
Kandpal RP. Rho GTPase activating proteins in cancer phenotypes. Curr Protein Pept Sci. 2006;7(4):355–365. | ||
Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–635. | ||
Johnstone CN, Castellví-Bel S, Chang LM, et al. ARHGAP8 is a novel member of the RHOGAP family related to ARHGAP1/CDC42GAP/p50RHOGAP: mutation and expression analyses in colorectal and breast cancers. Gene. 2004;336(1):59–71. | ||
Xu J, Zhou X, Wang J, et al. RhoGAPs attenuate cell proliferation by direct interaction with p53 tetramerization domain. Cell Rep. 2013;3(5):1526–1538. | ||
Wong CM, Yam JWP, Ching YP, et al. Rho GTPase-activating protein deleted in liver cancer suppresses cell proliferation and invasion in hepatocellular carcinoma. Cancer Res. 2005;65(19):8861–8868. | ||
Ching YP, Wong CM, Chan SF, et al. Deleted in liver cancer (DLC) 2 encodes a RhoGAP protein with growth suppressor function and is underexpressed in hepatocellular carcinoma. J Biol Chem. 2003;278(12):10824–10830. | ||
Liao YC, Lo SH. Deleted in liver cancer-1 (DLC-1): a tumor suppressor not just for liver. Int J Biochem Cell Biol. 2008;40(5):843–847. | ||
Rosenthal DT, Brenner JC, Merajver SD. Rho proteins in cancer. In: van Golen KL, editor. Rho GTPases Cancer. New York, NY: Springer New York; 2010:29–42. | ||
Ahn Y-H, Gibbons DL, Chakravarti D, et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest. 2012;122(9):3170–3183. | ||
Barfod E, Zheng Y, Kuang W, et al. Cloning and expression of a human CDC42 GTPase-activating protein reveals a functional SH3-binding domain. J Biol Chem. 1993;268(35):26059–26062. | ||
Ridley AJ, Self AJ, Kasmi F, et al. rho family GTPase activating proteins p190, bcr and rhoGAP show distinct specificities in vitro and in vivo. EMBO J. 1993;12(13):5151. | ||
Zhang B, Zheng Y. Regulation of RhoA GTP hydrolysis by the GTPase-activating proteins p190, p50RhoGAP, Bcr, and 3BP-1. Biochemistry. 1998;37(15):5249–5257. | ||
Ouchida M, Kanzaki H, Ito S, et al. Novel direct targets of miR-19a identified in breast cancer cells by a quantitative proteomic approach. PLoS One. 2012;7(8):e44095. | ||
Garrett MD, Self AJ, van Oers C, Hall A. Identification of distinct cytoplasmic targets for ras/R-ras and rho regulatory proteins. J Biol Chem. 1989;264(1):10–13. | ||
Lancaster CA, Taylor-Harris PM, Self AJ, Brill S, van Erp HE, Hall A. Characterization of rhoGAP. A GTPase-activating protein for rho-related small GTPases. J Biol Chem. 1994;269(2):1137–1142. | ||
Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093–2101. | ||
Nagaraja GM, Kandpal RP. Chromosome 13q12 encoded Rho GTPase activating protein suppresses growth of breast carcinoma cells, and yeast two-hybrid screen shows its interaction with several proteins. Biochem Biophys Res Commun. 2004;313(3):654–665. | ||
Peck J, Douglas G 4th, Wu CH, Burbelo PD. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528(1–3):27–34. | ||
Holeiter G, Bischoff A, Braun A, et al. The RhoGAP protein deleted in liver cancer 3 (DLC3) is essential for adherens junctions integrity. Oncogenesis. 2012;1:e13. | ||
Liu Z, Xu Y, Zhang X, Song J, Sorokin L, Bähler M. The motorized RhoGAP myosin IXb (Myo9b) in leukocytes regulates experimental autoimmune encephalomyelitis induction and recovery. J Neuroimmunol. 2015;282:25–32. | ||
Ran J, Lin DL, Wu RF, et al. ZEB1 promotes epithelial-mesenchymal transition in cervical cancer metastasis. Fertil Steril. 2015;103(6):1606–1614. | ||
Yuan J, Yan R, Krämer A, et al. Cyclin B1 depletion inhibits proliferation and induces apoptosis in human tumor cells. Oncogene. 2004;23(34):5843–5852. | ||
Yuan J, Krämer A, Matthess Y, et al. Stable gene silencing of cyclin B1 in tumor cells increases susceptibility to taxol and leads to growth arrest in vivo. Oncogene. 2006;25(12):1753–1762. | ||
Sun Z, Zhang D, Cui Y, Cheng L, Cao J, Wu X. Research on chemotherapy efficacy of twist gene on cervical cancer cells to paclitaxel. Pak J Pharm Sci. 2014;27(5 suppl):1713–1718. | ||
Mitra A, Chakrabarti J, Chattopadhyay N, Chatterjee A. Membrane-associated MMP-2 in human cervical cancer. J Environ Pathol Toxicol Oncol. 2003;22(2):93–100. | ||
Mitra A, Chakrabarti J, Banerji A, Das S, Chatterjee A. Culture of human cervical cancer cells, SiHa, in the presence of fibronectin activates MMP-2. J Cancer Res Clin Oncol. 2006;132(8):505–513. | ||
Moroz A, Delella FK, Almeida R, et al. Finasteride inhibits human prostate cancer cell invasion through MMP2 and MMP9 downregulation. PLoS One. 2013;8(12):e84757. | ||
Soini Y, Tuhkanen H, Sironen R, et al. Transcription factors zeb1, twist and snai1 in breast carcinoma. BMC Cancer. 2011;11:1. | ||
Merikallio H, Kaarteenaho R, Pääkkö P, et al. Zeb1 and twist are more commonly expressed in metastatic than primary lung tumours and show inverse associations with claudins. J Clin Pathol. 2011;64(2):136–140. | ||
Shibata K, Kajiyama H, Ino K, et al. Twist expression in patients with cervical cancer is associated with poor disease outcome. Ann Oncol. 2008;19(1):81–85. | ||
Branca M, Ciotti M, Giorgi C, et al; HPV-PathogenISS Study Group. Up-regulation of proliferating cell nuclear antigen (PCNA) is closely associated with high-risk human papillomavirus (HPV) and progression of cervical intraepithelial neoplasia (CIN), but does not predict disease outcome in cervical cancer. Eur J Obstet Gynecol Reprod Biol. 2007;130(2):223–231. | ||
Astudillo H, Perez M, Silva J, et al. P53, Bcl-2, PCNA expression and apoptotic rates during cervical tumorigenesis. Ann N Y Acad Sci. 2003;1010:771–774. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.