Back to Archived Journals » Oncolytic Virotherapy » Volume 3
Applications of coxsackievirus A21 in oncology
Authors Bradley S, Jakes A, Harrington K, Pandha H, Melcher A, Errington-Mais F
Received 21 October 2013
Accepted for publication 8 January 2014
Published 10 April 2014 Volume 2014:3 Pages 47—55
DOI https://doi.org/10.2147/OV.S56322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Stephen Bradley,1 Adam D Jakes,1 Kevin Harrington,2 Hardev Pandha,3 Alan Melcher,1 Fiona Errington-Mais1
1Leeds Institute of Cancer and Pathology, Cancer Research UK and Experimental Cancer Medicine Centre, St James' University Hospital, Leeds, UK; 2Division of Cancer Biology, The Institute of Cancer Research, London, UK; 3Oncology Department, Faculty of Health and Medical Sciences, University of Surrey, Guildford, UK
Abstract: The clinical management of cancer continues to be dominated by macroscopic surgical resection, radiotherapy, and cytotoxic drugs. The major challenge facing oncology is to achieve more selective, less toxic and effective methods of targeting disseminated tumors, a challenge oncolytic virotherapy may be well-placed to meet. Characterization of coxsackievirus A21 (CVA21) receptor-based mechanism of virus internalization and lysis in the last decade has suggested promise for CVA21 as a virotherapy against malignancies which overexpress those receptors. Preclinical studies have demonstrated proof of principle, and with the results of early clinical trials awaited, CVA21 may be one of the few viruses to demonstrate benefit for patients. This review outlines the potential of CVA21 as an oncolytic agent, describing the therapeutic development of CVA21 in preclinical studies and early stage clinical trials. Preclinical evidence supports the potential use of CVA21 across a range of malignancies. Malignant melanoma is the most intensively studied cancer, and may represent a “test case” for future development of the virus. Although there are theoretical barriers to the clinical utility of oncolytic viruses like CVA21, whether these will block the efficacy of the virus in clinical practice remains to be established, and is a question which can only be answered by appropriate trials. As these data become available, the rapid journey of CVA21 from animal studies to clinical trials may offer a model for the translation of other oncolytic virotherapies from laboratory to clinic.
Keywords: coxsackievirus A21, CVA21, cavatak, oncolytic virotherapy
Introduction
Cancer continues to be a leading cause of death, accounting for 8.2 million deaths worldwide in 2012. Notwithstanding the combined efforts of public health initiatives; epidemiologists; and clinical, surgical and radiation oncologists, cancer is set to remain a leading cause of mortality with incidence projected to rise from an estimated 12 million cases in 2012 to 22 million within the next twenty years.1 Traditionally, efforts have sought to exploit the rapid turnover of cancer cells using cytotoxic chemotherapy and radiotherapy, and strategies which combine surgical resection with chemotherapy/radiotherapy have been developed to confer the optimal prognosis for patients with solid tumors. Remarkable improvements have been made in some malignancies, such as breast cancer; however, for malignancies such as lung, brain, and pancreatic cancer little improvement in survival has been observed over recent years.2 Treatment of metastatic cancer remains a largely palliative exercise, with no treatments definitively and significantly altering the natural history of the disease. Meanwhile, the cost and toxicity of treatment regimens, which often provide limited benefit, impose difficult decisions on health professionals and patients, particularly in advanced stages of the disease.
The intractability of malignant disease and the heavy toll of treatment on quality of life have prompted innovative approaches. One such strategy is harnessing and adapting viruses, either naturally occurring or genetically modified, as oncolytic viruses (OVs) to lyse malignant cells and stimulate an antitumor immune response. Such an approach allows viruses to exploit the natural characteristics of malignant disease, sparing normal cells and avoiding the adverse effects often associated with chemotherapy. In addition, the immune stimulation associated with virus treatment may help to generate long-term immune control of cancer cell growth. However, to date, no oncolytic virotherapy has become part of standard anticancer therapy.
An emerging oncolytic virus, coxsackievirus A21 (CVA21), may stand on the cusp of such practical clinical application, although researchers are still establishing the capabilities of this virus against malignancy. This review outlines the wider context of oncolytic virotherapy and describes current evidence supporting the use of CVA21 for the treatment of malignant disease. The barriers associated with clinical implementation of CVA21 and its development as a novel anticancer agent are also discussed.
Oncolytic viruses and virotherapy
The concept of utilizing viruses as cancer therapies is not new. The idea originated over a century ago, and virotherapy experimentation later gained popularity in the 1950s, when patients were treated with a variety of virus preparations.3 Viruses are known to have tropisms which predispose them to infect specific cell types, and some naturally occurring viruses preferentially replicate in tumor cells which, due to defective antiviral response mechanisms, are more susceptible than healthy cells. Their main mode of action within a tumor mass is thought to be through infiltration and replication, leading to destruction of tumor tissue/oncolysis.4 As well as direct oncolysis, OVs can also promote host antitumor immunity,5 and it is hoped that viral replication within the tumor, systemic dissemination, and the generation of antitumor immune priming could all be effective against widespread metastatic disease.
The advent of molecular techniques controlling transcription and translation facilitated major advances in oncolytic virotherapy in the 1990s, using genetic modification to engineer and deliver viruses with more specific antitumor effects.4 Subsequently, two distinct strategies utilizing either naturally occurring OVs (eg, Newcastle-disease virus, reovirus, and CVA21), with inherent cytotoxicity against tumor cells, or genetically modified viruses (eg, adenovirus, vaccinia and herpes simplex virus), which are genetically engineered to selectively target tumor cells,6 were developed.
Since research into oncolytic virotherapy began in earnest in the 1990s, interest has been focused around a dozen or so OVs, with herpes simplex, measles, Newcastle-disease virus, reovirus, vaccinia, and adenovirus being the most well characterized.3 The first government approved oncolytic virotherapy was licensed by the People’s Republic of China in 2005 for the treatment of head and neck cancer, namely the genetically modified H101 adenovirus.7
Phase III trials remain to be conducted for almost all candidate OVs, although the results of some Phase I/II trials demonstrate grounds for optimism in this emerging field of oncology. Most significantly, a herpes simplex virus encoding granulocyte macrophage-colony stimulating factor (talimogene laherparepvec or T-VEC) injected intratumorly led to regression of melanoma in eight of 50 patients in a Phase II study,8 and the consequent Phase III trial has recently been reported to have met its primary endpoint of durable response rate; overall survival data are still awaited. Equally encouraging is the observation that OVs so far appear to be safe, with Phase I studies suggesting large doses can be tolerated without dose-limiting toxicities.9
CVA21
CVA21 is an enterovirus and, in common with other members of the Picornaviridae family, is unenveloped and contains a positive-sense, single-stranded RNA genome, enclosed in an icosahedral capsid. The virus has been classified into two groups, A and B, based on their effects in murine models. Group A contains 23 serotypes that impact mainly on skeletal muscle while group B contains only 6 serotypes and affects a wider variety of tissue types. In humans, the virus is clinically significant as one of many organisms responsible for mild upper respiratory tract infections (coryza), spread by aerosol transmission,10,11 and as such, its clinical manifestation is indistinguishable from several other viruses.
Oncolytic CVA21 is manufactured commercially as CAVATAK™ by Viralytics Limited (Sydney, NSW, Australia), and has been developed from the wild type Kuykendall strain. The receptor-based mechanisms of attachment and cell internalization have been extensively modeled and are well understood,12 and there is evidence that other group A serotypes of coxsackie could yield similar oncolytic potential.13 CVA21 infection is characterized by its interaction with the intercellular adhesion molecule-1 (ICAM-1, also termed CD54), which acts as the primary receptor for attachment and internalization, and decay-accelerating factor (DAF, or CD55), a coreceptor acting as a secondary point of virus attachment.14
ICAM-1 is a cell surface glycoprotein that is expressed on various cell types including leukocytes, epithelial cells, and endothelial cells15 and has a number of immunological roles, including binding to leukocyte function-associated antigen 1 during adhesion of leucocytes to endothelial cells. The recruitment of ICAM-1 as a viral receptor is common among many of the Picornaviridae family of viruses. DAF is expressed ubiquitously on almost all cells, including erythrocytes, and its primary role is in regulating complement responses,14 but DAF is also targeted by several other enteroviruses, including serotypes of echovirus and coxsackie B virus. CVA21 attachment to DAF is not sufficient to infect host cells, as internalization is dependent on ICAM-1 expression; DAF is thought to act as a membrane concentration receptor, which accumulates virus at the cell surface, optimizing viral entry via ICAM-1.16 Interestingly, a bioselected coxsackie variant has been produced that is solely dependent on DAF for both invasion and lysis.16 Variants with altered cell surface specificity may be of significance for tumors that overexpress DAF and/or underexpress ICAM-1. Although the expression of ICAM-1 and DAF are proposed to underpin the oncolytic properties of CVA21, it is possible that future investigations will uncover alternative mechanisms required for cancer-specific viral replication.
The production of such variants notwithstanding, the existence of CVA21 as a wild-type virus probably limits the prospects of deliberate manipulation or engineering.4 On the other hand, the virus’s ubiquity and well understood, albeit irksome, role in coryza, may represent significant advantages in terms of safety and achieving regulatory approval.
The promise of exploiting CVA21 preferentially to lyse cancer cells lies in the observation that the ICAM-1 and DAF receptors are overexpressed in various cancer cells, compared to nonmalignant tissue. This has been demonstrated in melanoma, breast, colon, endometrial, head and neck, pancreatic, and lung cancers as well as in multiple myeloma and malignant glioma.17–21 Furthermore, increased expression of ICAM-1 on metastatic disease, and the role for ICAM-1 in metastasis formation,22–24 make CVA21 targeting of metastatic disease a particularly attractive strategy.
It is hoped that CVA21 will be effective as an oncolytic agent, both through direct invasion and lysis, and by stimulation of host antitumor immune mechanisms; however, the immunogenic potential of CVA21 remains unknown, and investigations using relevant model systems are required to clarify the role of the immune system in the efficacy of CVA21 therapy.
Evidence supporting CVA21 as a novel anticancer agent
Research into the potential use of CVA21 for a variety of malignancies is ongoing, spanning preclinical in vitro studies and Phase I/II clinical trials. These studies have investigated the efficacy of CVA21 in melanoma, multiple myeloma, breast, lung, and prostate cancer models, and key findings are summarized in Table 1.
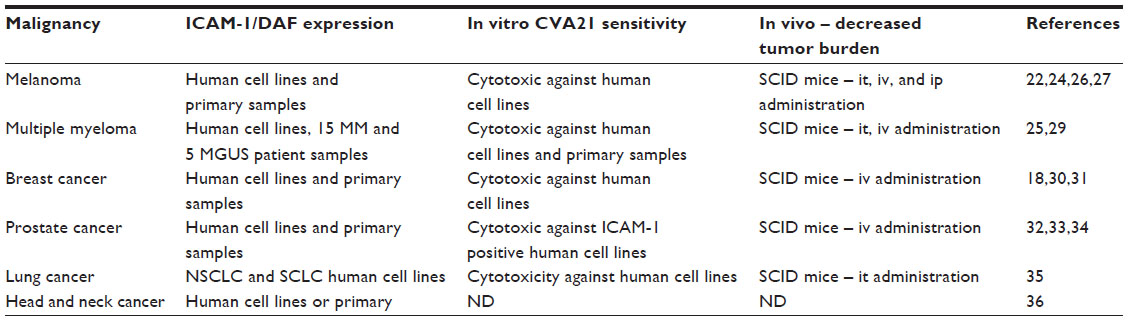 | Table 1 Summary of preclinical studies with CVA21 |
Melanoma
Because melanoma is a malignancy with marked overexpression of ICAM-1 and DAF compared to normal cells,25 with lesions amenable to injection, studies of CVA21 in melanoma are the most advanced, and may come to be regarded as the “test case” proof-of-principle for the efficacy of CVA21. The characteristic upregulation of ICAM-1 in melanoma is well established; indeed, it has been considered a clinically relevant marker of prognosis.22,24 In addition, a role for ICAM-1 in the generation of metastases has been suggested, as it facilitates cell–cell interactions between malignant melanocytes and circulating lymphocytes and spread of the disease.26
Experiments published in 200427 established that upregulation of ICAM-1 in melanoma could be exploited for CVA21 therapy. These data used two human melanoma cell lines, one chemotherapy-resistant and one derived from primary melanoma, and demonstrated in vitro oncolysis. In vivo, the authors proceeded to show that mouse xenograft tumors were reduced when treated with CVA21 by intratumoral, intravenous, and intraperitoneal administration.26 Interestingly, the same group later demonstrated that other group A coxsackie viruses (13, 15, and 18) exhibited similar oncolytic properties and were not susceptible to neutralizing antibodies (NABs) against CVA21. This raises the prospect of a future “multivalent” approach, using different serotypes to overcome potential immunological barriers.13
CVA21 investigations have progressed to clinical trials, with Phase I safety trials being completed in 2009 and a Phase II (6 months) and extension trial (48 weeks) currently recruiting in the US. The Phase II trial (CALM [CAVATAK in Late stage Melanoma])28 is currently underway across nine sites in the US. The trial has a primary endpoint assessing progression-free survival at 6 months as well as further assessments of safety. The trial’s investigators aim to include 54 patients with unresectable stage IIIc-IV M1c melanoma, 38 of whom have so far been recruited. Participants will be treated intratumorally on days 1, 3, 5, 8, and 21, then every 3 weeks for an additional five injections. In addition, Phase I trials are currently in development to examine intravenous administration of CVA21 to solid tumors, including melanoma, as well as lung, prostate, and bladder cancers.
Multiple myeloma
A 2006 preclinical study sought to investigate CVA21-mediated oncolysis, both in vitro and in vivo, in multiple myeloma. Flow cytometry first confirmed ICAM-1 and DAF expression in four multiple myeloma cell lines. In vitro, CVA21 caused oncolysis, and in vivo, tumor regression was observed in mouse xenografts, although toxicity was noted in these immunologically compromised mice, with development of hind limb paralysis after treatment. Pathology demonstrated widespread myositis as well as complete tumor ablation.29
A further ex vivo study tested the virus against bone marrow samples derived from 15 patients (ten with multiple myeloma and five with monoclonal gammopathy of undetermined significance). This study confirmed strong expression of ICAM-1 in the multiple myeloma cells compared to normal peripheral blood mononuclear cells. As for melanoma, preferential selectivity of CVA21 for myeloma, with greater lysis than control peripheral blood mononuclear cells, was found. Myeloma cell lines also supported greater virus replication while normal mononuclear cells were not productively infected.25 Finally, the researchers showed that CVA21 could purge malignant plasma cells from bone marrow samples of patients with malignant myeloma and monoclonal gammopathy of undetermined significance, suggesting that CVA21 could be used as a purging agent prior to autologous stem cell transplantation.25
Breast cancer
In common with melanoma, examination of surface markers on metastatic breast cancer has revealed overexpression of ICAM-1, prompting speculation again that this receptor is itself linked to the development of metastatic disease.18,30 A 2009 study31 used four breast cancer cell lines as well as a single normal breast cell line and demonstrated ICAM-1 and DAF overexpression on malignant cells in comparison to the normal line. Antitumor activity was observed in vitro, and mice injected with tumor cells to induce metastatic disease, treated with CVA21, showed no detectable metastases after 42 days, while untreated controls developed heavy tumor burden. Furthermore, mice with tumor xenografts treated with a single injection of intravenous CVA21 had reduced tumor burden compared to controls.
Prostate cancer
As with the other cancers discussed above, malignant prostate cells displayed elevated levels of ICAM-1 and DAF;32 ICAM-1 was absent from normal prostatic epithelium.33 One study used three prostate cancer cell lines, two of which expressed high levels of ICAM-1, while the third expressed relatively low ICAM-1 but increased DAF.34 Lysis was observed on treatment with CVA21 in vitro, though this was significantly reduced when ICAM-1 expression was low. An adapted variant of CVA21, CVA21-DAFv, which internalizes through the DAF receptor,16 demonstrated more lysis against the low ICAM-1 cell line, providing further evidence that receptor expression and virus internalization are key factors in conferring sensitivity to CVA21. In fact, CVA21-DAFv achieved more lysis across all three lines at lower viral doses than CVA21, perhaps through additive utilization of both DAF and ICAM-1 receptors. In vivo, mouse prostate tumor xenograft models demonstrated that systemic administration of CVA21 and CVA21-DAFv achieved tumor regression. The authors also noted that high viral titers of CVA21 and CVA21-DAFv persisted in mice with no perceivable tumor burden at the time of sacrifice, and postulated that the viruses may be capable of active replication in micrometastases, although this finding could alternatively be related to viral persistence in immunocompromised hosts.
Lung cancer
Lung tumor cells have been shown to strongly express ICAM-1, and initial data35 showed activity of CVA21 in five mouse models of both non-small-cell and small cell lung cancers (NSCLC and SCLC, respectively). Moderate to high levels of ICAM-1 expression were observed, as was cytotoxic CVA21 replication in NSCLC and SCLC cells in vitro. Intratumoral administration of CVA21 in NSCLC xenografts in severe combined immunodeficiency (SCID) mice yielded oncolysis, reduced tumor burden, and increased systemic levels of replicating CVA21 associated with tumor destruction.
Head and neck cancer
A Phase I study to establish preliminary safety and indications of efficacy of CVA21 in ICAM-1- and DAF-expressing squamous cell cancers of head and neck was initiated in 2009, but subsequently discontinued due to a poor recruitment.36 There are no further published data at present detailing the effect of CVA21 in head and neck malignancies; however, these tumors are known to express ICAM-137 and are likely to be regarded as a further potential candidate for CVA21 oncolytic therapy.
Obstacles to achieving virotherapy with CVA21
Virus delivery and the tumor microenvironment
Confirming CVA21 oncolysis in patients remains a priority for clinical trials, and as such, intratumoral delivery strategies continue to predominate trial design. This accounts, in part, for the important role for melanoma in both in vitro and in vivo investigations, as cutaneous lesions of this disease are readily accessible for injected virotherapy. Yet, as melanoma’s propensity to metastasize demonstrates, therapeutic efficacy would require systemic therapy to reach widespread metastatic disease.
Clearly, systemic delivery will present additional hurdles such as, how can we ensure that systemic administration can satisfy the necessary, if as yet notional, “viremic threshold” at the sites of disease? In addition, physiological mechanisms that protect us against infective agents may similarly thwart virus therapy, including extravasation in liver and spleen and the presence of NABs, as discussed below. Such mechanisms may need to be subverted or circumvented in order to optimize results with systemic therapy, and much discussion has already been dedicated to how this might be achieved.4
Meanwhile, the local tumor environment presents another range of challenges. Dense connective tissue within the tumor may impede passage of the virus,38 and additional features of tumors, such as poor lymphatic flow, tend to combine with this dense architecture to elevate interstitial fluid pressures, thus mitigating virus diffusion. Strategies that could help facilitate virus spread through the tissue include disruption of collagen synthesis using the angiotensin II blocking drug Losartan and disruption of the extracellular matrix with injected hyaluronidase. Furthermore, vascular permeability can be enhanced by administration of cytokines such as interleukin 2, tumor necrosis factor-α, and histamine as well as the effects of concurrent chemotherapy.4
Limitations of existing models
Oncolytic virotherapy remains in its infancy, or perhaps more optimistically, in its adolescence.39 Clinical trial data for CVA21, beyond initial safety studies, are not yet available. However, in initial studies of other OVs, it has been noted that the dramatic results heralded by in vitro and murine experimental models have not been achieved in human patients.36 This is not altogether surprising since in vitro cultures do not resemble real pathophysiology, and results achieved in mouse models rarely yield equivalent results in human subjects. Importantly, researchers have relied upon SCID mice xenografts with different tumor models to study CVA21, and it is not clear if the reduced tumor burden achieved in these models would be reflected in mice or humans with an intact immune system. This is particularly important because oncolytic virotherapy is thought to be mediated, at least in part, by the generation of antitumor immunity, which is not possible in immunocompromised mouse models. Unfortunately, immunocompetent animal models are not available to adequately mimic human infection with these viruses,36 although in vitro human models of oncolytic virus-induced immunity may be informative.40,41 Moreover, the xenografting of tumor tissue into mice does not accurately mimic the natural pathology of oncogenesis. The desire to develop more plausible models, including translational clinical trials incorporating biological endpoints, has been expressed and may be crucial to elucidating a more representative picture of the interactions between the host immune system, tumor, and CVA21.6
Immunity: challenges and possible solutions
The way in which the human immune system interacts with CVA21 will be a crucial determinant of its success or failure. Host NABs pose a problem that may undermine the efficacy of CVA21 and other viruses in clinical practice. CVA21 is a wild-type virus to which significant numbers of the population have already developed NABs following prior exposure, and in virus-naive individuals, NABs will inevitably be rapidly produced by a competent immune system upon initial CVA21 treatment. This could particularly limit the efficacy of systemic delivery and the ability of CVA21 to reach disseminated metastatic disease, since circulating virus will be vulnerable to a plethora of antiviral immune effector mechanisms including lymphocytes, macrophages, complement, and virus sequestration within the liver.6,42 Nevertheless, the extent to which the therapeutic effect of CVA21 may be disrupted by NABs in patients is as yet unclear. The experience of preclinical and human trials with other OVs suggest that the role of NABs may vary greatly, depending on the virus and the model used.6 Reovirus, for example, has been shown to persist within resected colorectal cancer liver metastases following systemic delivery to patients, despite the presence of NABs in the circulation prior to treatment.43
The extent to which host immunity may reduce or enhance the efficacy of CVA21 remains to be seen. If antiviral immunity does undermine the effectiveness of CVA21, two strategies have been proposed as potential solutions: suppressing the host immune response or enabling the virus to evade immune recognition.36 The first option is to modulate the immune response prior to administration; this has been achieved in preclinical studies by combination with cyclophosphamide or other chemotherapeutics, which as immunosuppressants can inhibit the production of NABs.44–47 Conversely, immunosuppression may not be helpful if virus-activated innate or adaptive immune effector cells contribute significantly to overall therapy, as is the case for other OVs but remains unknown for CVA21.6
An alternative strategy is to extend the therapeutic window before NABs prevent oncolysis by using a combined approach, deploying different coxsackie virus serotypes sequentially. Such viruses would share the ability of CVA21 to interact with the receptors ICAM-1 and DAF but remain active until the generation of specific NABs. This “multivalent” approach has been demonstrated in principle with group A coxsackie viruses 13, 15, and 18, showing they share the oncolytic properties of CVA21 without being susceptible to CVA21-specific NABs.28 This method may extend the therapeutic window, but such gains remain limited as NABs will inevitably develop. Interestingly, one study in which virus capsids were switched suggested that efforts to preclude NAB propagation did not enhance efficacy48 and may be of limited benefit.
Another approach is to conceal the virus within carrier cells (such as different immune effectors, irradiated tumor cells, or stem cells), which have sometimes been called “Trojan horses.” For example, vesicular stomatitis virus can reach disseminated disease after systemic delivery on irradiated vesicular stomatitis virus-infected murine colon carcinoma cells49 and dendritic cells can “hide” reovirus from the neutralizing effects of NABs.39 However, such ex vivo cell-based delivery strategies have the disadvantage of being clinically complicated and expensive. An alternative approach relies upon synthetic coating of the OV with a chemical compound, concealing the surface of the virus to avoid destruction by NABs and activation of the complement system. For example, adenovirus coated with multivalent copolymers of poly N-(2-hydroxypropyl) methacrylamide evaded destruction by NABs50 and caused higher systemic virus levels by avoiding viral uptake in the liver.51 The efficacy of such concealment strategies for CVA21 has not yet been addressed.
A further strategy developed to evade immunological destruction involves abandoning the full virus altogether. Infectious ribonucleic acid (INA) encoding CVA21 RNA offers an alternative vector. INA can achieve regression of myeloma xenografts in mice comparable to that achieved by the virus itself, although toxicity in the form of myositis was seen. CVA21 may be particularly suitable for INA encoding, as it is a small picornavirus, while other OVs are relatively large by comparison.42 A potential advantage of using smaller molecules, such as picornavirus INA, is that it may extravasate relatively easily from circulation into tumor42 compared to larger viral vectors,52,53 although this barrier may be mitigated by the tendency of tumors to be more permeable (or “leaky”) than healthy tissue. Techniques have been explored to enhance extravasation of OV from the circulation to the tumor; these include preadministration of interleukin 2, tumor necrosis factor-α, and histamine and bradykinin analogues to increase the tumor permeability.4 Again, the relevance to CVA21 of such findings remains to be determined.
Safety, tolerability, and improving specificity
In general, oncolytic virotherapies are well tolerated, even at high doses,9 and few serious toxicities have been observed during Phase I dosing and safety trials.36 The early concerns from some preclinical models, which showed significant myositis29 following CVA21 treatment in SCID mice,29 have not translated into immunocompetent humans, at least upon multiple intratumoural injections and single-dose intravenous delivery.54 Potential respiratory or myositic involvement may be of more significance in multi-dose intravenous delivery of CVA21, highlighting the importance of conducting appropriate early stage Phase I signal-seeking and tolerability studies, which began enrollment in January 2014.
The experience of intravenous adenovirus, reovirus, and vaccinia administration suggests that where flu-like symptoms develop, using acetaminophen (paracetamol) or nonsteroidal anti-inflammatory drugs, together with the desensitization that occurs with gradually increasing doses, increases the tolerability of these agents.55 As the oncolytic virotherapy field progresses, other techniques to improve tumor selectivity will hopefully reduce generalized side effects of viremia. For example, one such approach has engineered CVA21 to express muscle-specific microRNAs, thereby reducing viral replication in muscle tissue and enhancing tumor specificity. Destabilizing the viral genome by microRNA target insertion provides a novel strategy for improved target specificity, reducing nonspecific side effects.57 Importantly, this paper utilized SCID mice, which are severely immunecompromised, hence the relevance of muscle paralysis and nonspecific viral replication warrants further investigations in immunocompetent preclinical models and early stage clinical trials.
Discussion: what lies ahead for CVA21?
As OVs including CVA21 continue their journey from laboratory modeling to clinical trials, a picture of how such agents may be applied in oncology practice is slowly emerging. Studies showing additive or even synergistic effects with conventional anticancer agents (chemotherapy and radiotherapy)57,58 are guiding the most practical path to integration with current clinical practice. However, these combination approaches may not provide optimal conditions for the success of OVs, including CVA21, which may rely on the development of both innate and/or adaptive antitumor immunity, which are undoubtedly suppressed by many chemotherapeutic agents. Combination strategies, however, do align with the pragmatic recognition that OVs are likely to confer, at best, only marginal benefit alone, while offering significant improvements together with other treatment modalities.
CVA21 is at an early stage of preclinical and clinical development, and many questions remain about the mechanisms of CVA21 efficacy and toxicity and whether CVA21 can target chemotherapy/radiotherapy resistant cells. Importantly, the answers to all of these questions could facilitate the development of this agent and help optimize therapy for different types/stages of malignant disease. The importance of host antitumor immunity for the therapeutic efficacy of OV is increasingly recognized, and very little is known about the interaction of CVA21 with different immune effectors. To exploit fully the oncolytic and immune potential of CVA21, a full understanding of both antiviral and antitumor responses is essential. Novel immune modulators are currently an exciting area in cancer research, particularly with the success of ipilimumab for the treatment of melanoma.59 The next generation of blocking antibodies (anti-programmed cell death 1 [PD-1]/anti-PD-1ligand [L]) are currently in early phase clinical testing. The potential of OVs in combination with such immunomodulatory therapeutics is widely anticipated and may provide the path to success for oncolytic virotherapy as viral replication/spread and direct oncolysis may be limited in immunocompetent hosts.
As our knowledge of the tumor microenvironment and its role in tumor growth is expanding, it is important to consider the effect of OVs on different immune effector cells and the supporting tumor infrastructure. It is possible that OVs, as potent immune “danger signals,” could either reverse the immunosuppressive tumor microenvironment or even promote tumor growth by the secretion of inflammatory cytokines. In addition, evaluation of viral receptor expression on primary fresh tumor is essential, as cell lines provide a limited understanding of the biology of the disease. More innovative research using three-dimensional culture systems and primary tumor/stromal cells to better mimic the tumor environment in patients is required to unravel the true potential of oncolytic virotherapy.
The antiviral immune response could limit the efficacy of oncolytic virotherapy, with NABs restricting tumor access, particularly after systemic delivery. Delivery strategies have been investigated to optimize viral delivery and evade immune recognition; however, recent studies suggest that viruses may be carried and protected from neutralization by blood cells after systemic delivery.42 Furthermore, preliminary data from our and other laboratories suggest that in contrast to the accepted dogma that NABs will inhibit antitumor responses, NABs may actually improve therapy in some contexts. This hypothesis requires further investigation but demonstrates how limited our knowledge and understanding is regarding how OVs may interact with the host immune system. It is important to continue the parallel, rather than sequential, development of OVs in the laboratory and clinic, as this will rapidly increase our knowledge and understanding of oncolytic virotherapy, will ensure that any sign of clinical efficacy would be observed sooner rather than later, and will facilitate the development of optimal therapeutic strategies. Thus, early results from the Phase II CALM study indicating a 26.7% overall response rate in injected lesions, as reported at the World Congress of Melanoma in 2013, will arouse interest from clinicians and researchers worldwide.
Whatever the eventual role of CVA21, as for any new agent, the results of Phase I, II, and III clinical trials will sultimately determine its application in routine clinical practice. A Phase II trial of intratumoral injection of CVA21 in melanoma is underway while a Phase I trial of intravenous administration is imminent. Potentially, these trials will reveal a role for the virus in all ICAM-1- and DAF-expressing malignancies. The current journey of CVA21, from eliciting its receptor-based mechanisms of action through to application in cancer therapy, is proving an informative example of the current translation of oncolytic virotherapy from laboratory to clinic. The speed with which CVA21 is moving into clinical testing, without the need for exhaustive preclinical testing in models that are at best a poor mimic of cancer in patients, will help the visibility of OVs within the wider, nonresearch-based clinical oncology community. Future studies with this agent are required to support the development of CVA21 as a novel anticancer agent and to exploit tumor and immune characteristics for optimal therapeutic gain in the treatment of malignant disease.
Disclosure
KH was supported by The Institute of Cancer Research/Royal Marsden Hospital NIHR Biomedical Research Centre. HP has received research unrestricted grants form Viralytics Ltd. SB, ADJ, AM, FEM report no conflicts of interest in this work.
References
http://www.who.int/en/[homepage on the Internet]. Fact Sheet no 297, Cancer. World Health Organization; 2013. Available from: http://www.who.int/mediacentre/factsheets/fs297/en/. Accessed February 4, 2014. | |
Macmillan Cancer Support. Living After Diagnosis: Median Cancer Survival Times. London: Macmillan Cancer Support; 2011. Available from: http://www.macmillan.org.uk/Documents/AboutUs/Newsroom/LivingAfterCancerMedianCancerSurvivalTimes.pdf. Accessed February 4, 2014. | |
Eager RM, Nemunaitis J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011;18(5):305–317. | |
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30(7):658–670. | |
Prestwich RJ, Harrington KJ, Pandha HS, Vile RG, Melcher AA, Errington F. Oncolytic viruses: a novel form of immunotherapy. Expert Rev Anticancer Ther. 2008;8(10):1581–1588. | |
Parato KA, Senger D, Forsyth PA, Bell JC. Recent progress in the battle between oncolytic viruses and tumours. Nat Rev Cancer. 2005;5(12):965–976. | |
Huang PI, Chang JF, Kirn DH, Liu TC. Targeted genetic and viral therapy for advanced head and neck cancers. Drug Discov Today. 2009;14(11–12):570–578. | |
Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27(34):5763–5771. | |
Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–117. | |
Xiao C, Bator-Kelly CM, Rieder E, et al. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure. 2005;13(7):1019–1033. | |
Hyypiä T, Kallajoki M, Maaronen M, et al. Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res. 1993;27(1):71–78. | |
Newcombe NG, Beagley LG, Christiansen D, et al. Novel role for decay-accelerating factor in coxsackievirus A21-mediated cell infectivity. J Virol. 2004;78(22):12677–12682. | |
Au GG, Beagley LG, Haley ES, Barry RD, Shafren DR. Oncolysis of malignant human melanoma tumors by Coxsackieviruses A13, A15 and A18. Virol J. 2011;8:22. | |
Shafren DR, Dorahy DJ, Ingham RA, Burns GF, Barry RD. Coxsackievirus A21 binds to decay-accelerating factor but requires intercellular adhesion molecule 1 for cell entry. J Virol. 1997;71(6):4736–4743. | |
Hopkins AM, Baird AW, Nusrat A. ICAM-1: targeted docking for exogenous as well as endogenous ligands. Adv Drug Deliv Rev. 2004;56(6):763–778. | |
Johansson ES, Xing L, Cheng RH, Shafren DR. Enhanced cellular receptor usage by a bioselected variant of coxsackievirus a21. J Virol. 2004;78(22):12603–12612. | |
Bjørge L, Jensen TS, Matre R. Characterisation of the complement-regulatory proteins decay-accelerating factor (DAF, CD55) and membrane cofactor protein (MCP, CD46) on a human colonic adenocarcinoma cell line. Cancer Immunol Immunother. 1996;42(3):185–192. | |
Regidor PA, Callies R, Regidor M, Schindler AE. Expression of the cell adhesion molecules ICAM-1 and VCAM-1 in the cytosol of breast cancer tissue, benign breast tissue and corresponding sera. Eur J Gynaecol Oncol. 1998;19(4):377–383. | |
Kageshita T, Yoshii A, Kimura T, et al. Clinical relevance of ICAM-1 expression in primary lesions and serum of patients with malignant melanoma. Cancer Res. 1993;53(20):4927–4932. | |
Komi J, Lassila O. Toremifene increases the expression of intercellular adhesion molecule-1 (ICAM-1) on MCF-7 breast cancer cells and Jurkat cells. Scand J Immunol. 2000;51(1):73–78. | |
Murray KP, Mathure S, Kaul R, et al. Expression of complement regulatory proteins-CD 35, CD 46, CD 55, and CD 59-in benign and malignant endometrial tissue. Gynecol Oncol. 2000;76(2):176–182. | |
Johnson JP, Stade BG, Holzmann B, Schwäble W, Riethmüller G. De novo expression of intercellular-adhesion molecule 1 in melanoma correlates with increased risk of metastasis. Proc Natl Acad Sci U S A. 1989;86(2):641–644. | |
Miele ME, Bennett CF, Miller BE, Welch DR. Enhanced metastatic ability of TNF-alpha-treated malignant melanoma cells is reduced by intercellular adhesion molecule-1 (ICAM-1, CD54) antisense oligonucleotides. Exp Cell Res. 1994;214(1):231–241. | |
Johnson JP, Stade BG, Hupke U, Holzmann B, Riethmüller G. The melanoma progression-associated antigen P3.58 is identical to the intercellular adhesion molecule, ICAM-1. Immunobiology. 1988;178(3):275–284. | |
Au GG, Lincz LF, Enno A, Shafren DR. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br J Haematol. 2007;137(2):133–141. | |
Shafren DR, Au GG, Nguyen T, et al. Systemic therapy of malignant human melanoma tumors by a common cold-producing enterovirus, coxsackievirus a21. Clin Cancer Res. 2004;10(1 Pt 1):53–60. | |
Au GG, Lindberg AM, Barry RD, Shafren DR. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int J Oncol. 2005;26(6):1471–1476. | |
Viralytics: Investigator information. [webpage on the Internet]. http://www.viralytics.com/clinical-trial-information/investigator-information. Accessed February 13, 2014. | |
Hadac EM, Russell SJ. 436. Coxsackievirus A21 has potent oncolytic activity in multiple myeloma [Abstract]. Mol Ther. 2006; 13(Suppl 1):S168. | |
Rosette C, Roth RB, Oeth P, et al. Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis. 2005;26(5):943–950. | |
Skelding KA, Barry RD, Shafren DR. Systemic targeting of metastatic human breast tumor xenografts by Coxsackievirus A21. Breast Cancer Res Treat. 2009;113(1):21–30. | |
Rokhlin OW, Cohen MB. Expression of cellular adhesion molecules on human prostate tumor cell lines. Prostate. 1995;26(4):205–212. | |
Liu AY, Roudier MP, True LD. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am J Pathol. 2004;165(5):1543–1556. | |
Berry LJ, Au GG, Barry RD, Shafren DR. Potent oncolytic activity of human enteroviruses against human prostate cancer. Prostate. 2008;68(6):577–587. | |
Yee YWV, Chan SH, Quah MY, Shafren D, Au G. Oncolytic activity of coxsackievirus A21 (CAVATAK) in human lung cancer: a novel targeted anti-cancer strategy. Poster presented at: 14th World Conference on Lung Cancer; July 3–7, 2011; Amsterdam, the Netherlands. | |
Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl Res. 2013;161(4):355–364. | |
Scher RL, Koch WM, Richtsmeier WJ. Induction of the intercellular adhesion molecule (ICAM-1) on squamous cell carcinoma by interferon gamma. Arch Otolaryngol Head Neck Surg. 1993;119(4):432–438. | |
Sauthoff H, Hu J, Maca C, et al. Intratumoral spread of wild-type adenovirus is limited after local injection of human xenograft tumors: virus persists and spreads systemically at late time points. Hum Gene Ther. 2003;14(5):425–433. | |
Hammill AM, Conner J, Cripe TP. Oncolytic virotherapy reaches adolescence. Pediatr Blood Cancer. 2010;55(7):1253–1263. | |
Prestwich RJ, Ilett EJ, Errington F, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15(13):4374–4381. | |
Prestwich RJ, Errington F, Steele LP, et al. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol. 2009;183(7):4312–4321. | |
Hadac EM, Kelly EJ, Russell SJ. Myeloma xenograft destruction by a nonviral vector delivering oncolytic infectious nucleic acid. Mol Ther. 2011;19(6):1041–1047. | |
Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4(138):138ra77. | |
Li H, Zeng Z, Fu X, Zhang X. Coadministration of a herpes simplex virus-2 based oncolytic virus and cyclophosphamide produces a synergistic antitumor effect and enhances tumor-specific immune responses. Cancer Res. 2007;67(16):7850–7855. | |
Ungerechts G, Springfeld C, Frenzke ME, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15(11):1991–1997. | |
Qiao J, Wang H, Kottke T, et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Caner Res. 2008;14(1):259–269. | |
Thomas MA, Spencer JF, Toth K, Sagartz JE, Phillips NJ, Wold WS. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther. 2008;16(10):1665–1673. | |
Kanerva A, Nokisalmi P, Diaconu I, et al. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19(10):2734–2744. | |
Power AT, Wang J, Falls TJ, et al. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol Ther. 2007;15(1):123–130. | |
Fisher KD, Stallwood Y, Green NK, Ulbrich K, Mautner V, Seymour LW. Polymer-coated adenovirus permits efficient retargeting and evades neutralising antibodies. Gene Ther. 2001;8(5):341–348. | |
Green NK, Herbert CW, Hale SJ, et al. Extended plasma circulation time and decreased toxicity of polymer-coated adenovirus. Gene Ther. 2004;11(16):1256–1263. | |
Russell SJ, Peng KW. Viruses as anticancer drugs. Trends Pharmacol Sci. 2007;28(7):326–333. | |
Tseng JC, Granot T, DiGiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010;17(4):244–255. | |
Liauw WS, Chern B, Shafren DR. Phase I, Open-Label, Cohort Study of CAVATAK (Coxsackievirus A21), Given Intravenously to Stage IV Patients Bearing ICAM-1 Expressing Solid Tumours. Poster presented at: EORTC-NCI-AACR Symposium on Molecular Targets and Cancer Therapeutics, 6–9 November 2012, Dublin, Ireland. | |
Reid T, Warren R, Kirn D. Intravascular adenoviral agents in cancer patients: lessons from clinical trials. Cancer Gene Ther. 2002;9(12):979–986. | |
Kelly EJ, Hadac EM, Greiner S, Russell SJ. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat Med. 2008;14(11):1278–1283. | |
Skelding KA, Barry RD, Shafren DR. Enhanced oncolysis mediated by Coxsackievirus A21 in combination with doxorubicin hydrochloride. Invest New Drugs. 2012;30(2):568–581. | |
Simpson GR, Ajaz M, Launchbury FA, et al. Major synergy between Coxsackievirus A21 (CAVATAK™) and radiotherapy or chemotherapy in bladder cancer cell lines, due to up-regulation viral receptors ICAM-1 and DAF. Poster presented at: Seventh International Meeting on Replicating Oncolytic Virus Therapeutics; June 15–18, 2013; Quebec, Canada. Available from: http://www.viralytics.com/media/presentations/130618%20ONCO%20Meeting%20CAVATAK%20and%20Bladder%20Cancer%20Pandha.pdf. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.