Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid–paclitaxel (CLA-PTX) on B16-F10 melanoma
Authors Du R, Zhong T, Zhang W, Song P, Song W, Zhao Y, Wang C, Tang Y, Zhang X ![]() , Zhang Q
, Zhang Q
Received 7 April 2014
Accepted for publication 6 May 2014
Published 24 June 2014 Volume 2014:9(1) Pages 3091—3105
DOI https://doi.org/10.2147/IJN.S65664
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Ruo Du,1 Ting Zhong,1 Wei-Qiang Zhang,1 Ping Song,1 Wen-Ding Song,1 Yang Zhao,1 Chao-Wang,1 Yi-Qun Tang,3 Xuan Zhang,1,2 Qiang Zhang1,2
1Department of Pharmaceutics, 2State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, 3Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing, People’s Republic of China
Abstract: In the present study, we prepared a novel delivery system of iRGD (CRGDK/RGPD/EC)-modified sterically stabilized liposomes (SSLs) containing conjugated linoleic acid–paclitaxel (CLA-PTX). The anti-tumor effect of iRGD-SSL-CLA-PTX was investigated on B16-F10 melanoma in vitro and in vivo. The in vitro targeting effect of iRGD-modified SSLs was investigated in a real-time confocal microscopic analysis experiment. An endocytosis-inhibition assay was used to evaluate the endocytosis pathways of the iRGD-modified SSLs. In addition, the in vitro cellular uptake and in vitro cytotoxicity of iRGD-SSL-CLA-PTX were evaluated in B16-F10 melanoma cells. In vivo biodistribution and in vivo antitumor effects of iRGD-SSL-CLA-PTX were investigated in B16-F10 tumor-bearing mice. The induction of apoptosis by iRGD-SSL-CLA-PTX was evaluated in tumor-tissue sections. Real-time confocal microscopic analysis results indicated that the iRGD-modified SSLs internalized into B16-F10 cells faster than SSLs. The identified endocytosis pathway of iRGD-modified SSLs indicated that energy- and lipid raft-mediated endocytosis played a key role in the liposomes’ cellular uptake. The results of the cellular uptake experiment indicated that the increased cellular uptake of CLA-PTX in the iRGD-SSL-CLA-PTX-treated group was 1.9-, 2.4-, or 2.1-fold compared with that in the CLA-PTX group after a 2-, 4-, or 6-hour incubation, respectively. In the biodistribution test, the CLA-PTX level in tumor tissues from iRGD-SSL-CLA-PTX-treated mice at 1 hour (1.84±0.17 µg/g) and 4 hours (1.17±0.28 µg/g) was 2.3- and 2.0-fold higher than that of CLA-PTX solution at 1 hour (0.79±0.06 µg/g) and 4 hours (0.58±0.04 µg/g). The value of the area under the curve for the first 24 hours in the tumors of iRGD-SSL-CLA-PTX-treated mice was significantly higher than that in the SSL-CLA-PTX and CLA-PTX solution-treated groups (P<0.01). The in vivo antitumor results indicated that iRGD-SSL-CLA-PTX significantly inhibited the growth of B16-F10 tumors compared with the SSL-CLA-PTX or CLA-PTX solution-treatment groups (P<0.01). The results of tumor-cell apoptosis showed that tumors from the iRGD-SSL-CLA-PTX-treated group exhibited more advanced cell apoptosis compared with the control, CLA-PTX solution-, and SSL-CLA-PTX-treated groups. In conclusion, the antitumor effect of iRGD-SSL-CLA-PTX was confirmed on B16-F10 melanoma in vitro and in vivo.
Keywords: peptide-modified liposome, iRGD, CLA-PTX, antitumor effect, in vitro, in vivo
Introduction
Paclitaxel (PTX) is a chemotherapeutic antitumor drug widely used in clinical situations for the treatment of many tumors.1 In order to overcome the side effects of Cremophor® EL (CrEL; BASF, Ludwigshafen, Germany), which is used as a solubilizer in the current PTX-injection formulation named Taxol® (Bristol-Myers Squibb, New York, NY, USA), many CrEL-free PTX-delivery systems have been investigated, such as liposomes, nanoparticles, and micelles.2 These PTX-delivery systems express passive targeting effects because of an enhanced-permeability-and-retention effect in tumor tissues.3,4
Conversion of PTX into a water-soluble conjugate is an important alternative strategy.5–8 Examples of this include the poly(L-glutamic acid)–PTX conjugate (coded as CT-2103), polyethylene glycol (PEG)–PTX conjugate, hyperbranched poly(ether-ester)-PTX conjugate, poly(L-γ-glutamyl-glutamine)–PTX conjugate, N-(2-hydroxypropyl)-methacrylamide–PTX conjugate, PEG–polylactic acid–PTX, and hyaluronic acid–PTX conjugate.9–16 These polymer–PTX conjugates possess many evident advantages compared to conventional polymeric carrier drug-delivery systems, including enhanced water-solubility and antitumor activities.
Another strategy for PTX conversion is to improve its lipophilicity.17–19 Modification of PTX with fatty acid derivatives is one potential approach. It has been reported that docosahexaenoic acid–PTX is currently in Phase III clinical trials.20 According to this strategy, we previously reported a new PTX–fatty acid-derivative conjugate, involving the conjugation of linoleic acid with PTX (CLA-PTX).21 The antitumor effect of CLA-PTX in C6 glioma and B16-F10 melanoma cells was confirmed by its in vitro cytotoxicity.21–23 CLA-PTX also exhibits higher cellular uptake in C6 glioma and B16-F10 melanoma cells compared with PTX. The in vivo antitumor activity of CLA-PTX was confirmed in brain tumor-bearing rats. However, the solvent used for the CLA-PTX formulation for intravenous (IV) administration was a mixture of physiological saline, CrEL, and ethanol (80:10:10, volume:volume:volume [v:v:v]), the same as that used for Taxol.21 Therefore, a novel CLA-PTX microemulsion containing less CrEL compared with the CLA-PTX solution was designed and prepared.22 The antitumor activity of the CLA-PTX microemulsion was confirmed in our in vivo C6 glioma tumor-bearing nude mouse model as well as in a rat model in the treatment of brain tumors.22 In addition, the CLA-PTX microemulsion is safer than CLA-PTX solution due to the reduction in CrEL. Therefore, we considered that the CrEL-free as well as the active targeted modified CLA-PTX-delivery system would have none of the toxicity produced by CrEL and would also exhibit increased antitumor activity.
It has been reported that the iRGD peptide (sequence CRGDK/RGPD/EC) could increase vascular and tissue permeability in a tumor-specific and neuropilin-1-dependent manner.24,25 The targeting activity of iRGD-modified sterically stabilized liposomes (SSLs) has been demonstrated in our previous flow-cytometry and confocal analysis studies in B16-F10 cells.26 However, the cellular uptake pathway of iRGD-modified SSLs in B16-F10 cells was unclear. In addition, we found that the antitumor activity of CLA-PTX solution on B16-F10 bearing mice was similar to that of PTX injection.23 Therefore, we suggested that the antitumor effect of the CLA-PTX formulation after being iRGD-modified would be increased in B16-F10 melanoma cells. This assumption needs to be confirmed in detail.
In the present study, iRGD was selected as a targeting ligand, SSLs were used as a delivery system, and CLA-PTX was selected as a model drug to prepare CLA-PTX-loaded iRGD-modified SSLs (iRGD-SSL-CLA-PTX). Because of overexpression of both αv-integrin receptors and neuropilin-1 (NRP-1), B16-F10 melanoma cells were chosen for the tumor-cell model. The antitumor effect of iRGD-SSL-CLA-PTX was investigated on B16-F10 melanoma in vitro and in vivo. The in vitro targeting effect and endocytosis pathway of iRGD-modified SSLs were investigated. In addition, the in vitro cellular uptake and in vitro cytotoxicity of iRGD-SSL-CLA-PTX were also evaluated. The in vivo biodistribution and the in vivo antitumor effect of iRGD-SSL-CLA-PTX were investigated in B16-F10 tumor-bearing mice. The induction of apoptosis by iRGD-SSL-CLA-PTX was evaluated in tumor-tissue sections.
Materials and methods
Materials
PTX was obtained from Yunnan Hande Bio-Tech (Kunming, People’s Republic of China [PRC]). CLA-PTX was synthesized from CLA and PTX according to our previous report.21 The structure of CLA-PTX is shown in Figure 1. The iRGD peptide (CRGDK/RGPD/EC) was supplied by GL Bio-Chem (Shanghai, PRC). Cholesterol, sulforhodamine B, chlorpromazine, amiloride, filipin, methyl-β-cyclodextrin (M-β-CD) and Tris base were purchased from Sigma-Aldrich (St Louis, MO, USA). Coumarin-6 and Hoechst 33258 were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Egg phosphatidylcholine was purchased from Lipoid (Ludwigshafen, Germany). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG) was obtained from Avanti Polar Lipids (Alabaster, AL, USA). iRGD-PEG-DSPE, as shown in Figure 1, was synthesized according to our previously reported method.26
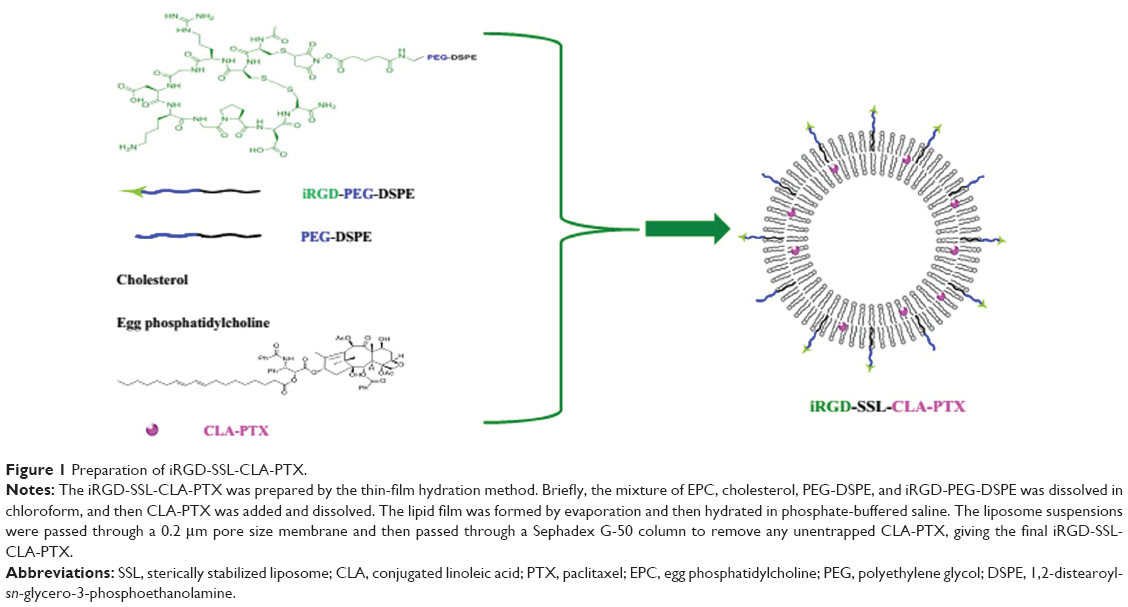 | Figure 1 Preparation of iRGD-SSL-CLA-PTX. |
Cell-culture media, Dulbecco’s Modified Eagle’s Medium DMEM (high glucose), penicillin–streptomycin, fetal bovine serum, and L-glutamine were purchased from Thermo Fisher Scientific. All solvents and other reagents were of analytical grade.
Cells
Murine B16-F10 cells (Chinese Academy of Sciences Cell Bank, Shanghai, PRC) were grown in Dulbecco’s Modified Eagle’s Medium (high glucose) supplemented with 10% fetal bovine serum and 1% glutamine penicillin–streptomycin at 37°C and 5% CO2.
Animals
Female C57BL6/N mice weighing 20–25 g (5–6 weeks old) were obtained from the Experimental Animal Center of Peking University Health Science Center (Beijing, PRC). All care and handling of the animals were performed according to the requirements of the Institutional Authority for Laboratory Animal Care of Peking University. For preparation of the tumor-bearing C57BL6/N mice model, C57BL6/N mice received a subcutaneous inoculation of 100 μL B16-F10 cell suspension (1×106) in the right armpit.
Preparation of iRGD-SSL-CLA-PTX
The iRGD-modified SSLs containing CLA-PTX were prepared as described previously.27 As shown in Figure 1, a mixture of egg phosphatidylcholine, cholesterol, PEG-DSPE, and iRGD-PEG-DSPE (65:30:4:1 molar ratio) was dissolved in chloroform, then CLA-PTX (CLA-PTX:lipid [weight:weight] ratio of 1:20) was added, and the resulting solution was then dried to obtain the lipid film. The resulting thin film was hydrated in phosphate buffered saline (PBS; pH 7.4) and sonicated to produce a liposome suspension. After that, the liposome suspension was passed through a 0.2 μm pore-size membrane, then through a Sephadex G-50 column to remove any free CLA-PTX, obtaining the final iRGD-SSL-CLA-PTX.
To prepare the SSL-CLA-PTX, an identical procedure was performed, except that the equivalent molar iRGD-PEG-DSPE was replaced by PEG-DSPE. The SSL-CLA-PTX was used as nontargeting liposome. Coumarin-6-loaded liposomes (iRGD-SSL-coumarin-6 and SSL-coumarin-6) were prepared exactly the same as for CLA-PTX-loaded liposomes, except that CLA-PTX was replaced by coumarin-6. To prepare the CLA-PTX solution, CLA-PTX was directly dissolved in a mixture of physiological saline, CrEL, and ethanol (80:10:10, v:v:v).
Characterization of iRGD-SSL-CLA-PTX
The particle size and zeta potential of iRGD-SSL-CLA-PTX (0.3 mg/mL) were measured by a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) at 25°C. For measurement of the encapsulation efficiency of iRGD-SSL-CLA-PTX, the final liposomes were passed through the Sephadex G-50 column to remove free CLA-PTX, followed by disruption with 10% Triton X-100 (v:v), then the CLA-PTX in the liposomes was measured by high-performance liquid chromatography (HPLC). Meanwhile, the same amount of iRGD-SSL-CLA-PTX was treated as previously described, except that it was not passed through a Sephadex G-50 column to obtain the total concentration of CLA-PTX. The encapsulation efficiency was calculated using the formula below:
|

In vitro release of CLA-PTX from iRGD-SSL-CLA-PTX
The CLA-PTX released from iRGD-SSL-CLA-PTX was investigated by dialysis under sink conditions. Briefly, a volume of 1 mL iRGD-SSL-CLA-PTX (0.15 mg/mL) was put in a dialysis bag (molecular weight cutoff 8,000–14,000). The dialysis bag was immersed in 40 mL release medium (distilled water containing 0.1% [v:v] Tween 80) and incubated in an orbital shaker at 37°C with mild oscillating at 100 rpm for 48 hours. Each sample (0.5 mL) was taken from the release medium at a predetermined time and replaced with an equal volume of fresh release medium. After the last sample was removed at 48 hours, the solution in the dialysis bag was mixed with the release medium. Then, 10 μL of 10% (v:v) Triton X-100 was added to the release medium, and this mixture was used as a positive control. The concentration of CLA-PTX was determined by HPLC, as described in a later section.
Cellular uptake pathway of iRGD-modified liposomes
To evaluate the effect of inhibitors on the cellular uptake of iRGD-modified SSLs, B16-F10 cells were preincubated with different inhibitors for 30 minutes at 37°C. Briefly, the cells were incubated for 24 hours, then chlorpromazine (10 μg/mL), amiloride (20 μM), filipin (5 μg/mL), or M-β-CD (2.5 mM) was added, followed by incubation for 30 minutes at 37°C. After that, the inhibitor-containing culture medium was replaced with iRGD-SSL-coumarin-6 (the final concentration of coumarin-6 was 150 ng/mL) and incubated for 2 hours at 37°C. Then, the cells were washed three times with PBS solution, harvested by trypsinization, and centrifuged at 1,000 rpm for 5 minutes. After being resuspended in 500 μL PBS medium, the cells were tested using a FACScan™ (BD Biosciences, San Jose, CA, USA). The coumarin-6 in the cells was excited with an argon laser (467 nm), and fluorescence was detected at 502 nm. To investigate the effect of temperature on cellular uptake, the cells without inhibitor treatment were incubated at both 37°C and 4°C, and treated as described earlier.
Real-time confocal microscopic analysis
To study the real-time cellular uptake of iRGD-SSL-coumarin-6, B16-F10 cells (3×105 per dish) were seeded on glass-bottom dishes and allowed to grow and differentiate for 4 days prior to being studied. After that, the medium was removed, and each dish was placed on a microscope stage. Each solution containing SSL-coumarin-6, iRGD-SSL-coumarin-6, or coumarin-6 solution (final drug concentration of coumarin-6, 150 ng/mL) was added to each dish, respectively. The series of images were recorded continuously using a TCS SP2 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany). For quantification of the uptake rate, regions of interest (ROIs) were randomly selected from the cell layer, and their fluorescence intensity was determined using the SP2 confocal software. Each spot on the curve was calculated using the fluorescence intensity of 20 ROIs.
In vitro cellular uptake of iRGD-SSL-CLA-PTX
B16-F10 cells (1×106 cells/well) were seeded in six-well flat-bottom tissue-culture plates for a 24-hour incubation. After that, the medium was replaced by iRGD-SSL-CLA-PTX, SSL-CLA-PTX, or CLA-PTX solution (10 μM), and incubated for 2, 4, or 6 hours at 37°C. After incubation, cells were washed twice with cold PBS and collected using 0.25% trypsin. The cells were centrifuged at 6,000 g for 5 minutes, and the supernatant removed carefully. The cell pellets were added to 0.1 mL 10% sodium dodecyl sulfate to destroy the cells. A volume of 0.1 mL acetonitrile was added to each cell solution to precipitate the protein. After centrifugation at 6,000 g for 5 minutes, a volume of 50 μL supernatant was used for HPLC analysis. Three wells were measured for each sample. Cellular uptake efficiency was calculated using the formula below:
|
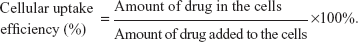
In vitro cytotoxicity
B16-F10 cells (5×103 cells/well) were seeded in 96-well transparent plates and incubated for 24 hours. The medium was removed, and increasing concentrations of iRGD-SSL-CLA-PTX was added. At 48 hours’ incubation, cell viability was determined by sulforhodamine B assay. Absorbance was measured at 540 nm using a 96-well plate reader (model 680; Bio-Rad Laboratories, Hercules, CA, USA). The survival percentages were calculated using the formula: survival % = (A540 nm for the treated cells/A540 nm for the control cells) ×100%, where A540 nm is the absorbance value. Each assay was carried out in triplicate. The half-maximal inhibitory concentration (IC50) values were calculated according to the dose–effect curves.
Biodistribution studies
When the tumor volume reached about 300 mm3, the tumor-bearing mice were randomly assigned to one of three groups: group 1 was given IV CLA-PTX solution, group 2 was given IV SSL-CLA-PTX, and group 3 was given IV iRGD-SSL-CLA-PTX. All the CLA-PTX preparations were injected through the tail veins at a dose of 2 mg CLA-PTX/kg. After drug administration, the mice were killed at 1, 4, 8, 12, and 24 hours. The tumors or excised organs (heart, liver, spleen, lung, kidney, and brain) were collected, blotted with a paper towel, rinsed in saline, blotted to remove excess fluid, weighed, and homogenized. The homogenized tissue samples were extracted using our previously reported method.21 The content of CLA-PTX in each tissue was measured by HPLC under the chromatographic conditions described in the “HPLC analysis of CLA-PTX” section.
In vivo antitumor activity of iRGD-SSL-CLA-PTX
When the tumor volume reached about 100–150 mm3, the tumor-bearing mice were randomly assigned to one of four groups: group 1 was given IV physiological saline as a control, group 2 was given IV CLA-PTX solution, group 3 was given IV SSL-CLA-PTX, and group 4 was given IV iRGD-SSL-CLA-PTX. Each group contained six animals. All the CLA-PTX preparations were injected through the tail veins at a dose of 2 mg CLA-PTX/kg on days 7, 9, 11, 13, and 15. Throughout the study, animals were weighed and tumors measured with calipers twice a week. Tumor volumes were calculated according to the formula: V = length (cm) × width2 (cm2) ×0.5236. On day 19 of tumor inoculation, one or two mice in each group were killed and the tumors collected for the preparation of sections. Terminal deoxynucleotidyl transferase deoxyuridine triphosphate nick-end labeling (TUNEL) staining of the paraffin-embedded tumors was performed according to the standard protocols provided by the manufacturers. The survival time was calculated from the day that the B16-F10 cells were inoculated (day 0) to the day the animal died. Kaplan–Meier survival curves were drawn for each group.
HPLC analysis of CLA-PTX
The content of CLA-PTX was measured by a Waters HPLC system consisting of a 1525 pump, and a 2487 ultraviolet detector (Waters, Milford, MA, USA). The wavelength was set at 227 nm. The mobile phase was started with solvent A (acetonitrile:water 60:40 v:v) for 10 minutes, and then a linear gradient was used to change to solvent B (acetonitrile) at 12 minutes, remaining at solvent B for 20 minutes. The flow was set at 1 mL/min. An ODS 3 C-18 analytical column (5 μm, 250×4.6 mm; Phenomenex, Torrance, CA, USA) was used for chromatographic separation. The retention time of CLA-PTX was approximately 15 minutes.
Statistical analysis
All data are shown as means ± standard deviation. One-way analysis of variance was used to determine the significance of differences among groups, after which post hoc tests with the Bonferroni correction were used for comparisons among the individual groups. Statistical significance was set at P<0.05.
Results
Characterization of iRGD-SSL-CLA-PTX
The average particle size of iRGD-SSL-CLA-PTX was about 93±0.2 nm, with a polydispersity index of 0.19±0.01, as shown in Table 1. The zeta potential of iRGD-SSL-CLA-PTX was slightly negative (−23.97±0.35 mV). The entrapment efficiency of iRGD-SSL-CLA-PTX was 83.22%, which indicated that CLA-PTX was almost completely entrapped within the liposomes. The typical particle size and distribution of iRGD-SSL-CLA-PTX is shown in Figure S1.
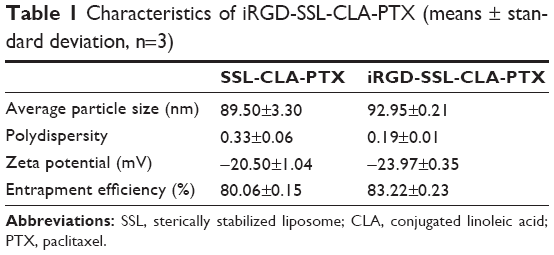 | Table 1 Characteristics of iRGD-SSL-CLA-PTX (means ± standard deviation, n=3) |
In vitro release of CLA-PTX from iRGD-SSL-CLA-PTX
The in vitro released CLA-PTX from iRGD-SSL-CLA-PTX is shown in Figure 2. The CLA-PTX released from the iRGD-SSL-CLA-PTX groups at the 48-hour time point was only 40%. Similar CLA-PTX-release behavior was observed in the SSL-CLA-PTX groups. However, CLA-PTX was rapidly released from the CLA-PTX solution, and this was almost complete within 48 hours.
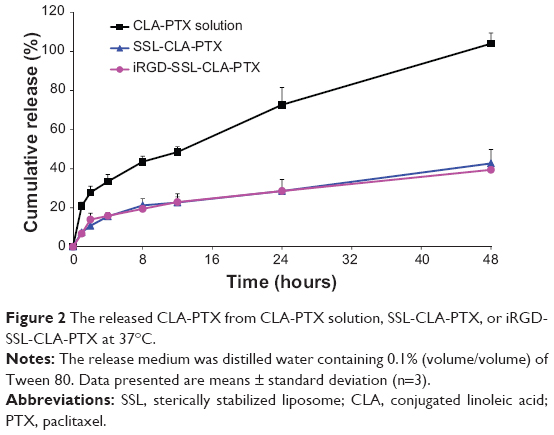 | Figure 2 The released CLA-PTX from CLA-PTX solution, SSL-CLA-PTX, or iRGD-SSL-CLA-PTX at 37°C. |
Cellular uptake pathways
In order to investigate the endocytosis pathway for iRGD-modified SSLs, an endocytosis-inhibition assay was performed. As shown in Figure 3, the rate of uptake of B16-F10 cells was inhibited by 51% at 4°C compared with that at 37°C (P<0.01), and was also inhibited by M-β-CD (down to 76%) (P<0.01), showing energy- and lipid raft-mediated endocytosis. Other inhibitors, such as chlorpromazine, amiloride, and filipin, had only a weak inhibitory effect.
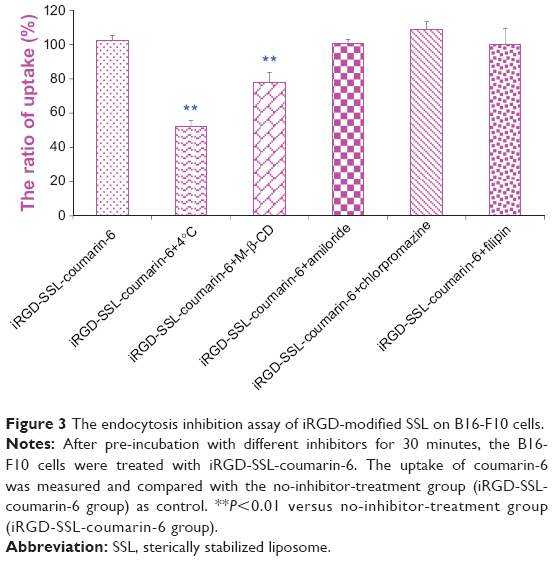 | Figure 3 The endocytosis inhibition assay of iRGD-modified SSL on B16-F10 cells. |
Real-time confocal microscopic analysis
Figure 4, A and B show that iRGD-SSL-coumarin-6 was internalized into B16-F10 cells faster and exhibited higher fluorescence intensity at every time compared with SSL-coumarin-6. In addition, coumarin-6 was also rapidly absorbed into the B16-F10 cells (Figure S2).
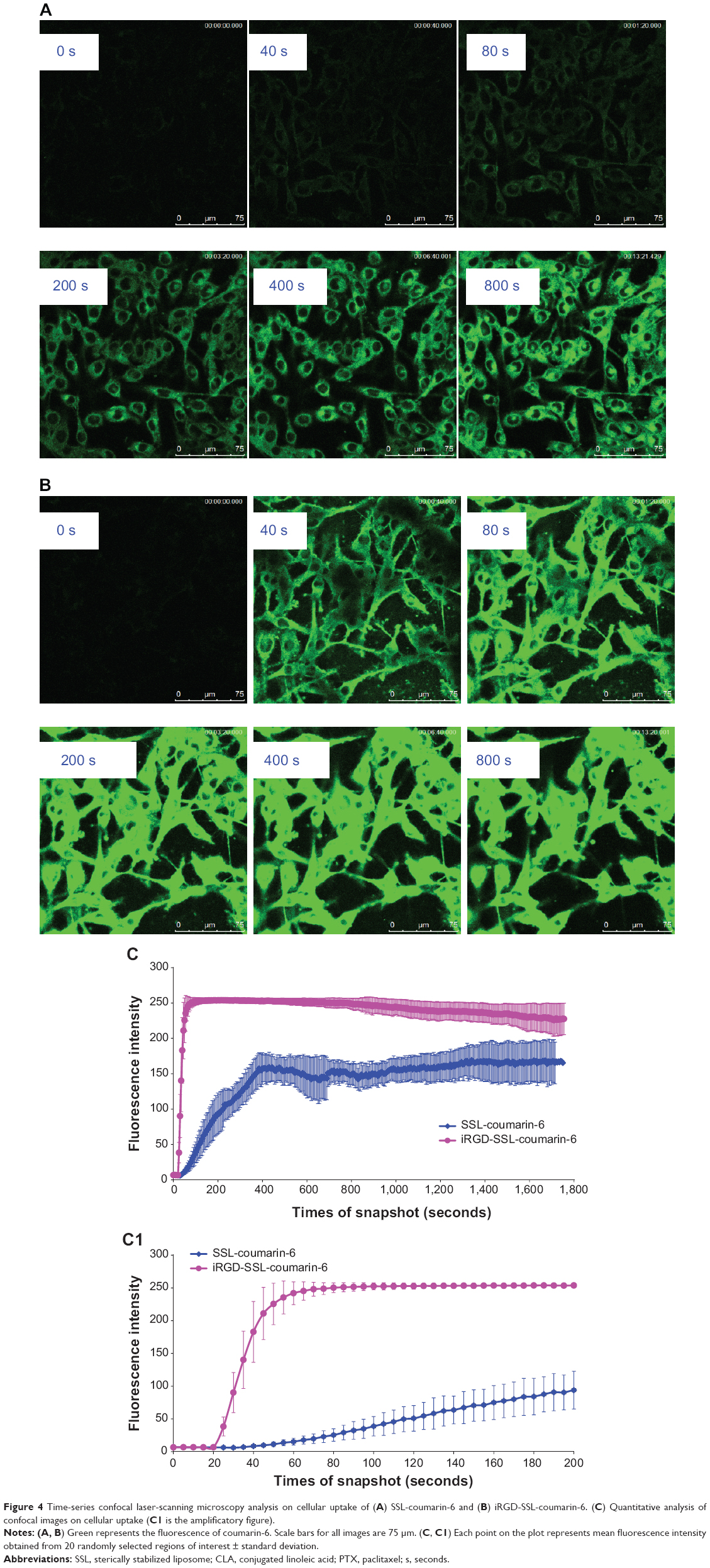 | Figure 4 Time-series confocal laser-scanning microscopy analysis on cellular uptake of (A) SSL-coumarin-6 and (B) iRGD-SSL-coumarin-6. (C) Quantitative analysis of confocal images on cellular uptake (C1 is the amplificatory figure). |
To further investigate the internalization speed and degree of iRGD-SSL-coumarin-6 and SSL-coumarin-6, a quantitative analysis of the images was conducted. Twenty ROIs were randomly selected to determine the fluorescence intensity. As shown in Figure 4C, iRGD-SSL-coumarin-6 showed greater internalization in B16-F10 cells at every time point compared with SSL-coumarin-6. We found that the fluorescence intensity of iRGD-SSL-coumarin-6 internalized in B16-F10 cells at the 80-second time point was almost 100% compared with that at the end of the test (Figure 4C[1]). On the other hand, only about 30% was found in SSL-coumarin-6 groups.
In vitro cellular uptake of iRGD-SSL-CLA-PTX
The uptake of CLA-PTX by B16-F10 cells in the iRGD-SSL-CLA-PTX-, SSL-CLA-PTX-, and CLA-PTX-solution groups is shown in Figure 5. The cellular uptake of CLA-PTX in the iRGD-SSL-CLA-PTX group was significantly higher than that in the SSL-CLA-PTX- and CLA-PTX-solution groups (P<0.01) after 2, 4, and 6 hours’ incubation. These results indicated that the increased cellular uptake of CLA-PTX in the iRGD-SSL-CLA-PTX-treated group was 1.9-, 2.4-, or 2.1-fold compared with that in the CLA-PTX group after 2, 4, or 6 hours’ incubation, respectively. For SSL-CLA-PTX groups, there was 1.2-, 1.5-, or 1.3-fold increased uptake compared with that in CLA-PTX group after 2, 4, or 6 hours’ incubation, respectively.
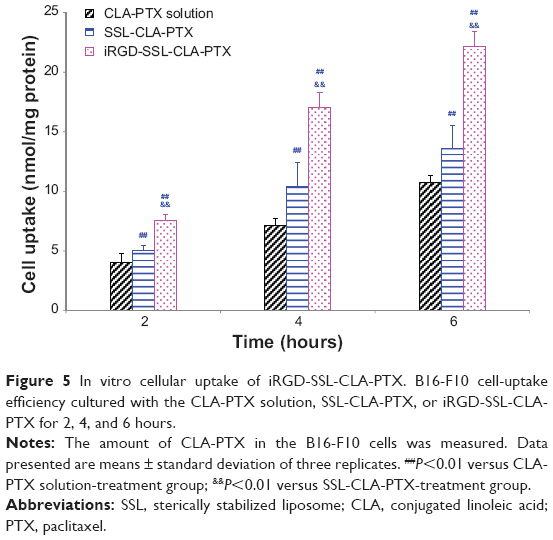 | Figure 5 In vitro cellular uptake of iRGD-SSL-CLA-PTX. B16-F10 cell-uptake efficiency cultured with the CLA-PTX solution, SSL-CLA-PTX, or iRGD-SSL-CLA-PTX for 2, 4, and 6 hours. |
In vitro cytotoxicity
B16-F10 cells were used to evaluate the cytotoxicity of iRGD-SSL-CLA-PTX in comparison with SSL-CLA-PTX and CLA-PTX solutions. IC50 values are given in Table 2. The IC50 value of iRGD-SSL-CLA-PTX was similar to that of CLA-PTX (P>0.05). The IC50 value of SSL-CLA-PTX was significantly higher than that of iRGD-SSL-CLA-PTX and CLA-PTX (P<0.01).
 | Table 2 Cytotoxicity of iRGD-SSL-CLA-PTX for B16-F10 cells (means ± standard deviation, n=3) |
Biodistribution
The concentration of CLA-PTX in the tumor tissues and excised organs (heart, liver, spleen, lung, kidney, and brain) of tumor-bearing mice treated with CLA-PTX solution, SSL-CLA-PTX, and iRGD-SSL-CLA-PTX is shown in Figure 6. The calculated area under the curve for hours 0–24 (AUC0–24) is shown in Table 3. The recovery efficiency of CAL-PTX in organs was in the range of 81%–94%. A targeting effect of iRGD-SSL-CLA-PTX was observed in tumor tissues compared with the SSL-CLA-PTX and CLA-PTX solutions. As shown in Figure 6, the CLA-PTX level in tumor tissues from iRGD-SSL-CLA-PTX-treated mice at 1 hour (1.84±0.17 μg/g) and 4 hours (1.17±0.28 μg/g) was 2.3- and 2.0-fold higher than that of CLA-PTX solution at 1 hour (0.79±0.06 μg/g) and 4 hours (0.58±0.04 μg/g). The AUC0–24 value in the tumors of the iRGD-SSL-CLA-PTX-treated mice was significantly higher than that in the SSL-CLA-PTX and CLA-PTX solution-treated groups. In addition, drug accumulation in the liver was higher than that in other tissues.
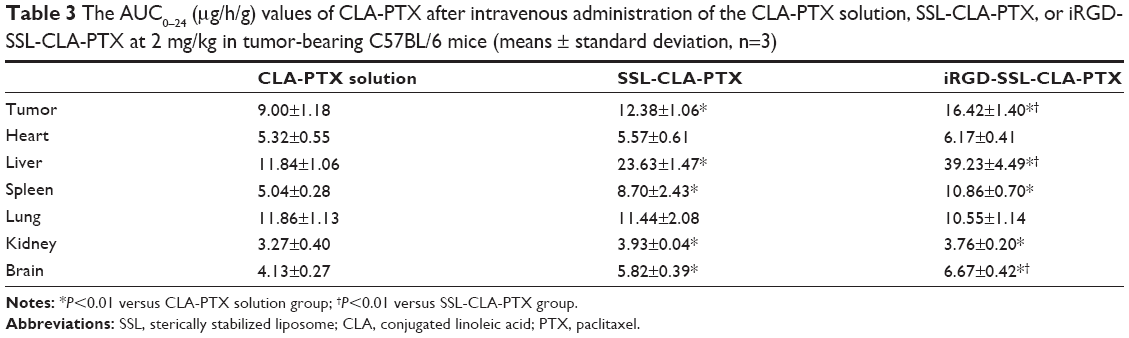 | Table 3 The AUC0–24 (μg/h/g) values of CLA-PTX after intravenous administration of the CLA-PTX solution, SSL-CLA-PTX, or iRGD-SSL-CLA-PTX at 2 mg/kg in tumor-bearing C57BL/6 mice (means ± standard deviation, n=3) |
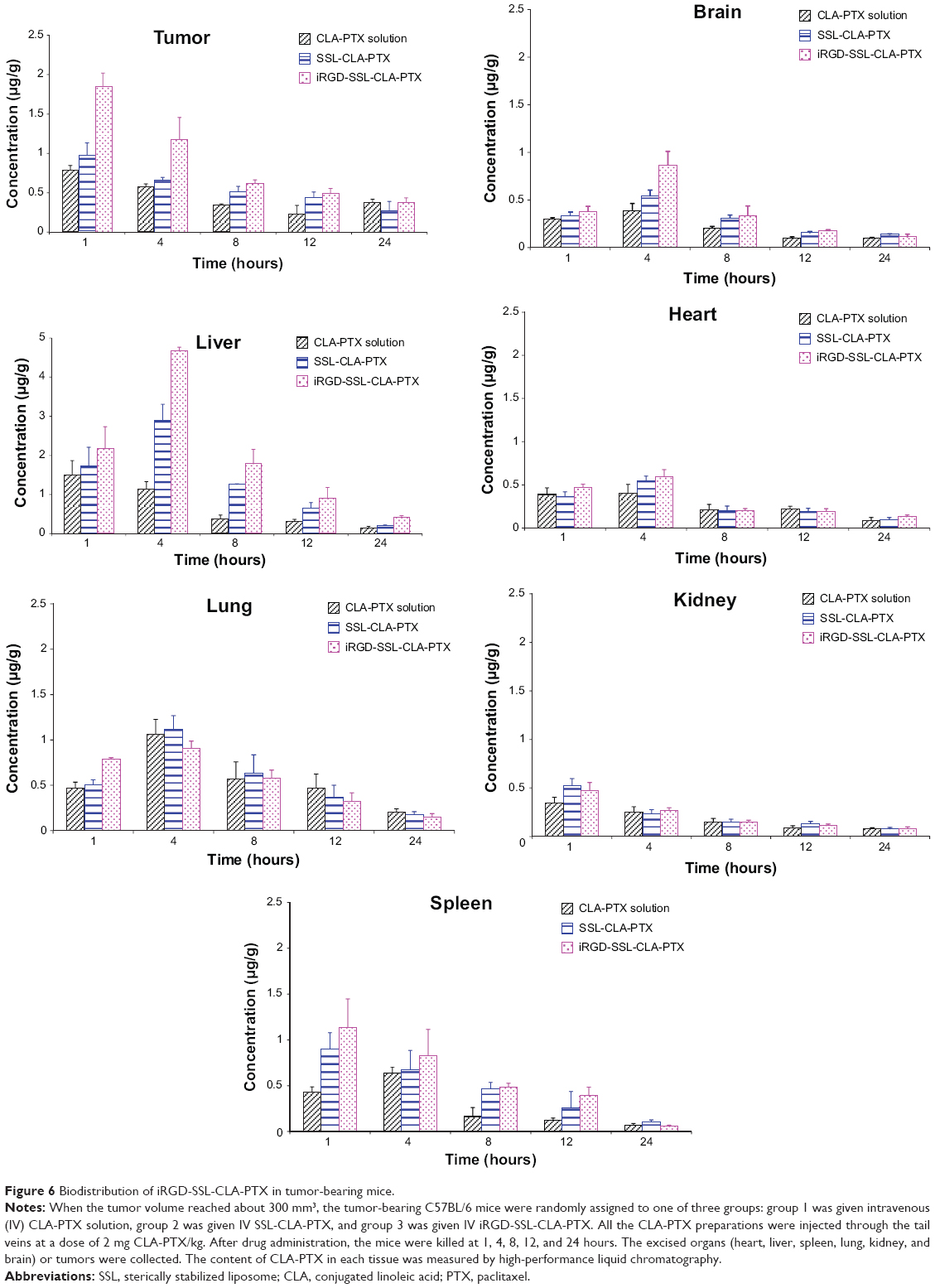 | Figure 6 Biodistribution of iRGD-SSL-CLA-PTX in tumor-bearing mice. |
In vivo antitumor activity of iRGD-SSL-CLA-PTX
As shown in Figure 7, tumor growth was significantly inhibited in all CLA-PTX treatment groups compared with the physiological saline treatment group (P<0.01). iRGD-SSL-CLA-PTX significantly inhibited the growth of B16-F10 tumors compared with the SSL-CLA-PTX or CLA-PTX solution-groups (P<0.01). The average tumor size at day 19 in the CLA-PTX-solution, SSL-CLA-PTX, and iRGD-SSL-CLA-PTX groups was 4,723±853, 3,969±495 and 1,793±355 mm3, respectively, compared with 10,054±996 mm3 in the physiological saline group (P<0.01). The corresponding tumor-growth inhibition in the SSL-CLA-PTX- and iRGD-SSL-CLA-PTX-treated groups was 52.5% and 82.7%, respectively. Figure 8 shows the Kaplan–Meier survival curve. After five administrations, the median survival time of mice treated with iRGD-SSL-CLA-PTX (34 days) was significantly longer than that of mice treated with physiological saline (24 days, P<0.01), CLA-PTX solution (28 days, P<0.01), and SSL-CLA-PTX (29 days, P<0.01), respectively.
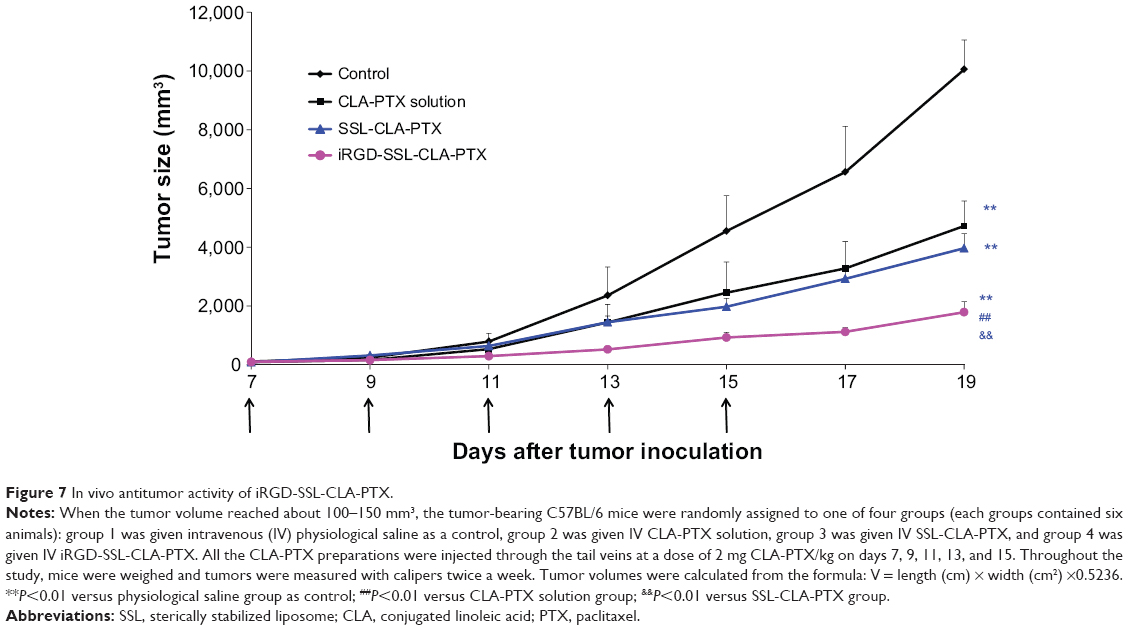 | Figure 7 In vivo antitumor activity of iRGD-SSL-CLA-PTX. |
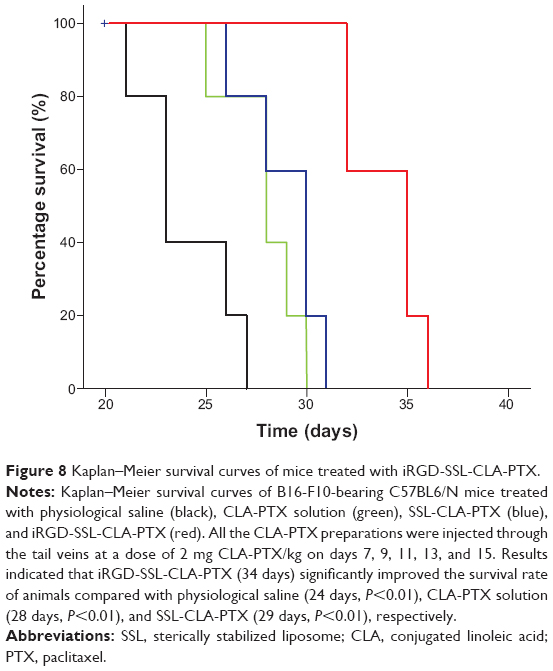 | Figure 8 Kaplan–Meier survival curves of mice treated with iRGD-SSL-CLA-PTX. |
We also evaluated the effect of tumor-cell apoptosis by TUNEL analysis staining of tumor-tissue sections. As shown in Figure 9A, tumors from the iRGD-SSL-CLA-PTX-treated group exhibited more advanced cell apoptosis compared with the control, CLA-PTX solution-, and SSL-CLA-PTX-treated groups. The calculated results are shown in Figure 9B.
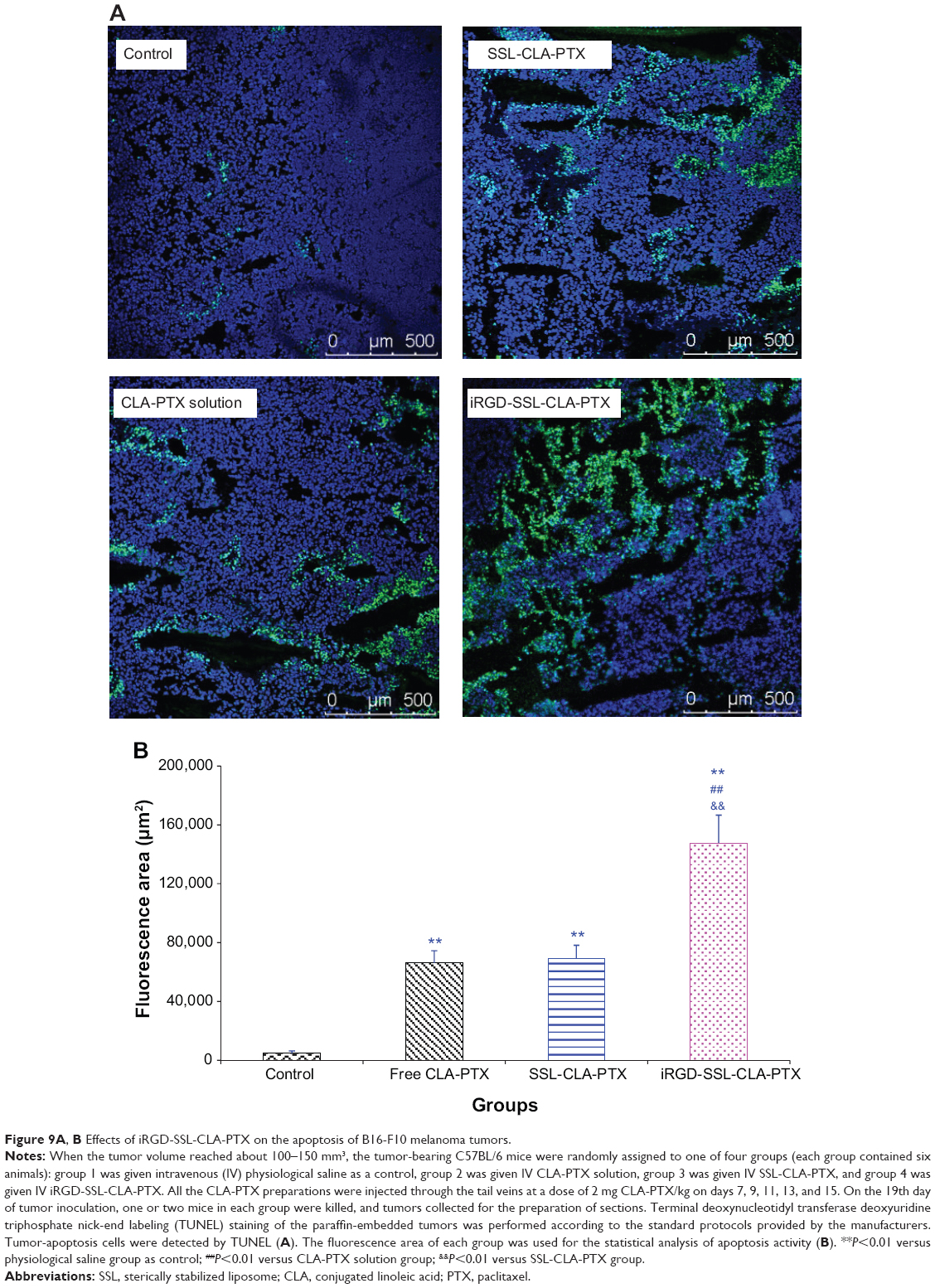 | Figure 9A, B Effects of iRGD-SSL-CLA-PTX on the apoptosis of B16-F10 melanoma tumors. |
Discussion
The in vitro cellular uptake and in vitro cytotoxicity of CLA-PTX in B16-F10 melanoma cells were demonstrated in our previous research study;23 however, the efficacy of active targeting of a modified CLA-PTX delivery system is unknown. Therefore, we designed and prepared iRGD-SSL-CLA-PTX and investigated its antitumor effect in B16-F10 melanoma cells in vitro and in vivo.
The tumor-targeting and tumor-penetrating mechanisms of iRGD have been reported.24,25 Recently, iRGD-modified delivery systems have been reported for targeted tumor therapy.28–30 Our previous results indicated that iRGD could target cells that over-express αv-integrins or the NRP-1 receptor.26 The targeting effect of iRGD-modified SSLs on B16-F10 melanoma cells has been demonstrated by our previous in vitro flow cytometry and confocal microscopy.26 In order to further confirm the targeting effect of iRGD-modified SSLs on B16-F10 melanoma cells in the present study, real-time confocal microscopic analysis was performed. The real-time uptake results indicated that iRGD-modified SSLs internalized into B16-F10 cells faster than unmodified SSLs at every time point (Figure 4). This is because ligand-modified delivery systems may prove beneficial in increasing drug exposure, due to increased target-cell uptake and target-tissue retention compared to ligand-lacking delivery systems.31 Our in vitro cellular uptake experiment also demonstrated that the iRGD-SSL-CLA-PTX was absorbed more than unmodified SSLs (Figure 5). In addition, a higher level and AUC of CLA-PTX in tumor tissue in the iRGD-SSL-CLA-PTX-treatment group was also observed in our in vivo biodistribution experiment (Figure 6). In addition, drug accumulation in the liver was higher than that in other tissues. It has been reported that iRGD-modified nanoparticles accumulate in the liver at high levels.32,33 Those researchers suggested that these nanoparticles were rapidly recognized by the phagocytic cells and the reticuloendothelial system or easily penetrated through the endothelial wall of liver fenestrae, resulting in enhanced liver uptake.
Endocytosis is a fundamental process that is used to internalize molecules and macromolecules by cells. Many nanoparticles have been customized to enter cells through endocytosis and deliver the cargo within the cells,34 such as clathrin-mediated and caveolae-mediated endocytosis, as well as rhodamine A-mediated and CDC42-mediated macropinocytosis. For example, conventional liposomes are taken up by cells via clathrin-mediated endocytosis.35,36 It has been reported that RGD-modified nanoparticles can be internalized via the receptor-mediated or clathrin-mediated endocytosis pathway.37 Therefore, understanding the cellular entry of nanoparticles has become a key factor in drug delivery. Many endocytic inhibitors have been used to characterize the endocytosis pathway involved in nanoparticles.38–42 In the present study, we selected chlorpromazine, amiloride, filipin, and M-β-CD as endocytic inhibitors to investigate the endocytosis pathway of iRGD-modified SSLs. Also, 4°C was selected to investigate the impact of energy on cellular uptake. Our results indicate that 4°C and M-β-CD significantly reduced the cellular uptake of iRGD-modified SSLs, suggesting that energy- and lipid raft-mediated endocytosis play a critical role in the cellular uptake of iRGD-modified SSLs (Figure 3). It was found that chlorpromazine, amiloride, and filipin did not reduce the cellular uptake of iRGD-modified SSLs, indicating that the clathrin-, macropinocytosis-, and caveolae-mediated endocytosis pathways are not involved in iRGD-modified SSL cellular transport (Figure 3). Our results suggest that iRGD-modified SSLs, unlike RGD-modified nanoparticles, are not internalized via the clathrin-mediated endocytosis pathway.37 Although iRGD has the RGD sequence, we speculated that the endocytosis pathway of iRGD-modified SSLs was different from that of RGD-modified nanoparticles.
It has been reported that liposomes could be efficiently accumulated in tumor because of the enhanced-permeability-and-retention effect. The addition of targeting moieties, such as antibodies or Fab fragments or small peptides and aptamers, to the surface of liposomes allows liposomes to actively target tumor cells. A lipsome is a spherical vesicle consisting of a lipid bilayer that can encapsulate various types of drugs into an inner aqueous phase or lipid bilayer.43–45 Previously, we prepared iRGD-modified SSLs containing the aqueous antitumor drug doxorubicin.26 In the present study, we prepared RGD-SSL-CLA-PTX. Our results also demonstrated the antitumor effect of iRGD-SSL-CLA-PTX (Figures 7–9).
In the present cellular uptake study, we did not observe PTX that was hydrolyzed from CLA-PTX. In addition, our biodistribution study indicated that intact CLA-PTX was identified in tumor tissue and other organs after iRGD-SSL-CLA-PTX, SSL-CLA-PTX, or CLA-PTX solution was given to tumor-bearing mice. According to these results, we suggest that the antitumor activity was due to the intact CLA-PTX, not the hydrolyzed PTX.
Conclusion
In the present study, we prepared a novel iRGD-SSL-CLA-PTX solution with the objective of investigating its in vitro and in vivo targeting and antitumor activity in B16-F10 cells. The targeting effect of iRGD-modified SSLs was demonstrated by the real-time confocal microscopy analysis and in vivo biodistribution experiments. The results of the endocytosis pathway of iRGD-modified SSLs indicated that energy- and lipid raft-mediated endocytosis played a key role in the cellular uptake of iRGD-modified SSLs. In addition, iRGD-SSL-CLA-PTX produced a very marked in vivo antitumor effect in B16-F10 tumor-bearing mice.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (81172992), the National Basic Research Program of China (973 Program 2013CB932501), and the Innovation Team of the Ministry of Education (BMU20110263).
Disclosure
The authors report no conflicts of interest in this work.
References
Long HJ. Paclitaxel (Taxol): a novel anticancer chemotherapeutic drug. Mayo Clin Proc. 1994;69(4):341–345. | ||
Zhang Z, Mei L, Feng SS. Paclitaxel drug delivery systems. Expert Opin Drug Deliv. 2013;10(3):325–340. | ||
Sato NL, Niimura S, Fujisawa N, Maeda Y. Characterization of vascular permeability-increasing component isolated from solid tumors and the effect of highly polymerized dextran sulfate on its activity. Jpn J Pharmacol. 1986;41(2):163–171. | ||
Maruyama K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv Drug Deliv Rev. 2011;63(3): 161–169. | ||
Feng X, Yuan YJ, Wu JC. Synthesis and evaluation of water-soluble paclitaxel prodrugs. Bioorg Med Chem Lett. 2002;12(22):3301–3303. | ||
Ryu BY, Sohn JS, Hess M, Choi SK, Choi JK, Jo BW. Synthesis and anti-cancer efficacy of rapid hydrolysed water-soluble paclitaxel pro-drugs. J Biomater Sci Polym Ed. 2008;19(3):311–324. | ||
Hayashi Y, Skwarczynski M, Hamada Y, Sohma Y, Kimura T, Kiso Y. A novel approach of water-soluble paclitaxel prodrug with no auxiliary and no byproduct: design and synthesis of isotaxel. J Med Chem. 2003;46(18):3782–3784. | ||
Dhanikula AB, Panchagnula R. Preparation and characterization of water-soluble prodrug, liposomes and micelles of paclitaxel. Curr Drug Deliv. 2005;2(1):75–91. | ||
Chipman SD, Oldham FB, Pezzoni G, Singer JW. Biological and clinical characterization of paclitaxel poliglumex (PPX, CT-2103), a macromolecular polymer-drug conjugate. Int J Nanomedicine. 2006;1(4):375–383. | ||
Kratz F, Abu Ajaj K, Warnecke A. Anticancer carrier-linked prodrugs in clinical trials. Expert Opin Investig Drugs. 2007;16(7):1037–1058. | ||
Li G, Liu J, Pang Y, et al. Polymeric micelles with water-insoluble drug as hydrophobic moiety for drug delivery. Biomacromolecules. 2011; 12(6):2016–2026. | ||
Yang D, Liu X, Jiang X, et al. Effect of molecular weight of PGG-paclitaxel conjugates on in vitro and in vivo efficacy. J Control Release. 2012;161(1):124–131. | ||
Zhang R, Luo K, Yang J, et al. Synthesis and evaluation of a backbone biodegradable multiblock HPMA copolymer nanocarrier for the systemic delivery of paclitaxel. J Control Release. 2013;166(1):66–74. | ||
Ceruti M, Crosasso P, Brusa P, Arpicco S, Dosio F, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing water-soluble prodrugs of paclitaxel. J Control Release. 2000;63(1–2):141–153. | ||
Greenwald RB, Choe YH, McGuire J, Conover CD. Effective drug delivery by PEGylated drug conjugates. Adv Drug Deliv Rev. 2003;55(2):217–250. | ||
Lee H, Lee K, Park TG. Hyaluronic acid-paclitaxel conjugate micelles: synthesis, characterization, and antitumor activity. Bioconjug Chem. 2008;19(6):1319–1325. | ||
Lundberg BB, Risovic V, Ramaswamy M, Wasan KM. A lipophilic paclitaxel derivative incorporated in a lipid emulsion for parenteral administration. J Control Release. 2003;86(1):93–100. | ||
Ansell SM, Johnstone SA, Tardi PG, et al. Modulating the therapeutic activity of nanoparticle delivered paclitaxel by manipulating the hydrophobicity of prodrug conjugates. J Med Chem. 2008;51(11): 3288–3296. | ||
Ma P, Rahima Benhabbour S, Feng L, Mumper RJ. 2′-Behenoyl-paclitaxel conjugate containing lipid nanoparticles for the treatment of metastatic breast cancer. Cancer Lett. 2013;334(2):253–262. | ||
Bradley MO, Webb NL, Anthony FH, et al. Tumor targeting by covalent conjugation of a natural fatty acid to paclitaxel. Clin Cancer Res. 2001; 7(10):3229–3238. | ||
Ke XY, Zhao BJ, Zhao X, et al. The therapeutic efficacy of conjugated linoleic acid-paclitaxel on glioma in the rat. Biomaterials. 2010;31(22): 5855–5864. | ||
Li D, Yang K, Li JS, et al. Antitumor efficacy of a novel CLA-PTX microemulsion against brain tumors: in vitro and in vivo findings. Int J Nanomedicine. 2012;7:6105–6114. | ||
Li JS, Yang K, Ke XY, Du R, Zhang X, Zhang Q. In vitro and in vivo antitumor efficacy of CLA-PTX on B16-F10 melanoma cells. Zhongguo Yaoxue. 2014;23(1):46–53. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009; 16(6):510–520. | ||
Sugahara KN, Teesalu T, Karmali PP, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–1035. | ||
Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. | ||
Luo LM, Huang Y, Zhao BX, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34(4):1102–1114. | ||
Tian Y, Li S, Song J, et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–2390. | ||
Gu G, Gao X, Hu Q, et al. The influence of the penetrating peptide iRGD on the effect of paclitaxel-loaded MT1-AF7p-conjugated nanoparticles on glioma cells. Biomaterials. 2013;34(21):5138–5148. | ||
Su S, Wang H, Liu X, Wu Y, Nie G. iRGD-coupled responsive fluorescent nanogel for targeted drug delivery. Biomaterials. 2013;34(13):3523–3533. | ||
van der Meel R, Vehmeijer LJ, Kok RJ, Storm G, van Gaal EV. Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev. 2013;65(10):1284–1298. | ||
Zhu Z, Xie C, Liu Q, et al. The effect of hydrophilic chain length and iRGD on drug delivery from poly(ε-caprolactone)-poly(N-vinylpyrrolidone) nanoparticles. Biomaterials. 2011;32(35):9525–9535. | ||
Wang X, Zhen X, Wang J, Zhang J, Wu W, Jiang X. Doxorubicin delivery to 3D multicellular spheroids and tumors based on boronic acid-rich chitosan nanoparticles. Biomaterials. 2013;34(19):4667–4679. | ||
Canton I, Battaglia G. Endocytosis at the nanoscale. Chem Soc Rev. 2012;41(7):2718–2739. | ||
Ziello JE, Huang Y, Jovin IS. Cellular endocytosis and gene delivery. Mol Med. 2010;16(5–6):222–229. | ||
Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem J. 2004;377(Pt 1):159–169. | ||
Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–2973. | ||
Gu G, Xia H, Hu Q, et al. PEG-co-PCL nanoparticles modified with MMP-2/9 activatable low molecular weight protamine for enhanced targeted glioblastoma therapy. Biomaterials. 2013;34(1):196–208. | ||
Xia H, Gao X, Gu G, et al. Low molecular weight protamine-functionalized nanoparticles for drug delivery to the brain after intranasal administration. Biomaterials. 2011;32(36):9888–9898. | ||
Song Q, Yao L, Huang M, et al. Mechanisms of transcellular transport of wheat germ agglutinin-functionalized polymeric nanoparticles in Caco-2 cells. Biomaterials. 2012;33(28):6769–6782. | ||
Zhang Q, Tang J, Fu L, et al. A pH-responsive α-helical cell penetrating peptide-mediated liposomal delivery system. Biomaterials. 2013; 34(32):7980–7993. | ||
Gao X, Wang T, Wu B, et al. Quantum dots for tracking cellular transport of lectin-functionalized nanoparticles. Biochem Biophys Res Commun. 2008;377(1):35–40. | ||
Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30(11):592–599. | ||
Schwendener RA. Liposomes in biology and medicine. Adv Exp Med Biol. 2007;620:117–128. | ||
Un K, Sakai-Kato K, Oshima Y, Kawanishi T, Okuda H. Intracellular trafficking mechanism, from intracellular uptake to extracellular efflux, for phospholipid/cholesterol liposomes. Biomaterials. 2012;33(32): 8131–8141. |
Supplementary materials
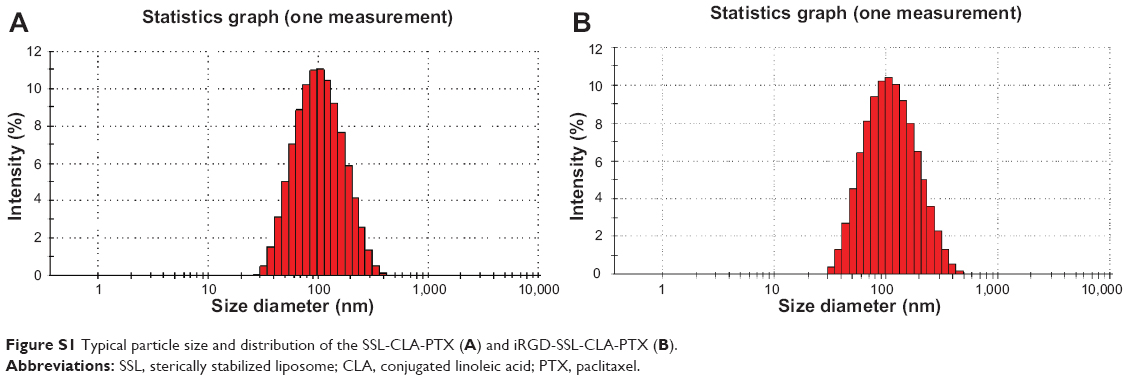 | Figure S1 Typical particle size and distribution of the SSL-CLA-PTX (A) and iRGD-SSL-CLA-PTX (B). |
 | Figure S2 Time-series confocal laser-scanning microscopy analysis on cellular uptake of coumarin-6 dimethyl sulfoxide solution. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.