Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 18
Antifibrotic Strategies Targeting Phosphodiesterase-4 in Idiopathic Pulmonary Fibrosis: Molecular Mechanisms and Clinical Translation
Authors Ma Q, Zhou S, Zhi X, Luo C, Zhu Z, Zhang X, Liu X ![]()
Received 30 September 2025
Accepted for publication 5 February 2026
Published 16 February 2026 Volume 2026:18 570975
DOI https://doi.org/10.2147/CPAA.S570975
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Quan Ma,1,2 Shixin Zhou,3 Xiaodong Zhi,3 Caifeng Luo,4 Zhongbo Zhu,1 Xuhui Zhang,5 Xiping Liu1
1School of Basic Medical Sciences, Gansu University of Traditional Chinese Medicine, Lanzhou, 730000, People’s Republic of China; 2Department of Lung Diseases, Affiliated Hospital of Gansu University of Traditional Chinese Medicine, Lanzhou, 730000, People’s Republic of China; 3College of Integrative Medicine, Gansu University of Traditional Chinese Medicine, Lanzhou, 730000, People’s Republic of China; 4Gaolan Branch Hospital, Affiliated Hospital of Gansu University of Traditional Chinese Medicine, Lanzhou, 730200, People’s Republic of China; 5The Third Affiliated Hospital of Gansu University of Traditional Chinese Medicine (The First People’s Hospital of Bai Yin City), Bai Yin, 730900, People’s Republic of China
Correspondence: Xuhui Zhang, The Third Affiliated Hospital of Gansu University of Traditional Chinese Medicine (The First People’s Hospital of Bai Yin City), Bai Yin, 730900, People’s Republic of China, Email [email protected] Xiping Liu, Gansu University of Traditional Chinese Medicine, Lanzhou, 730000, People’s Republic of China, Email [email protected]
Abstract: Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive interstitial lung disease characterised by irreversible fibrosis of the lung parenchyma and a steady decline in respiratory function. Its pathogenesis remains incompletely understood, and despite advances in diagnosis and disease management, therapeutic options remain limited and largely palliative. Emerging evidence suggests that phosphodiesterase-4 (PDE4) inhibitors may represent a novel therapeutic approach in IPF through modulation of cyclic adenosine monophosphate–dependent signalling pathways. Preclinical and clinical studies indicate that PDE4 inhibition can attenuate key pathological processes implicated in IPF, including macrophage-driven inflammatory responses, dysregulated epithelial repair, and fibroblast proliferation and differentiation. Through these mechanisms, PDE4 inhibitors demonstrate combined anti-inflammatory and antifibrotic effects in experimental models of lung fibrosis. Among this class, the PDE4B-selective inhibitor nerandomilast has shown encouraging signals of efficacy in clinical trials of IPF, supporting continued investigation of subtype-selective targeting strategies. Other PDE4 inhibitors, including roflumilast, rolipram, and structurally novel derivatives such as 2-arylbenzofurans, have also demonstrated antifibrotic activity, predominantly in preclinical studies. This review synthesises current evidence on the role of PDE4 signalling in IPF pathogenesis and critically evaluates the pharmacological rationale, therapeutic potential, and translational challenges of PDE4 inhibitors in the treatment of IPF. Future perspectives, including subtype-selective inhibition and optimised drug delivery strategies, are discussed as potential avenues to improve efficacy and tolerability in this patient population.
Keywords: phosphodiesterase, phosphodiesterase 4 inhibitor, idiopathic pulmonary fibrosis, nerandomilast
Introduction
Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing interstitial lung disease characterised by progressive scarring of the lung parenchyma, with its pathology marked by irreversible structural destruction and functional decline caused by abnormal extracellular matrix (ECM) deposition.1 Epidemiological surveys have shown a rising global incidence of IPF worldwide,2 while the median survival following diagnosis remains less than five years, underscoring its poor prognosis.3 Current evidence supports a polygenic-environmental interaction model, in which long-term environmental exposures (tobacco smoke, occupational dust) and genetic susceptibility act synergistically to drive disease initiation and progression.4
The pathogenic cascade is often described as being initiated by repeated microinjury to alveolar epithelial cells (AECs), leading to disruption of the balance between the endoplasmic reticulum stress response and mitochondrial homeostasis, and ultimately to failure of epithelial repair programmes.5 However, it is increasingly recognised that fibrotic remodelling reflects complex interactions among epithelial cells, immune cells, mesenchymal cells, and the extracellular matrix, rather than a single linear pathway.6 Damaged epithelial cells release pro-inflammatory cytokines such as IL-6 and TNF-α, which activate alveolar macrophages to secrete pro-fibrotic mediators including TGF-β and PDGF. These factors, in turn, promote fibroblast activation and the accumulation of myofibroblasts (MyoFBs), thereby establishing a self-perpetuating inflammation-fibrosis cycle.7 MyoFBs can arise from multiple cellular sources, including resident fibroblasts and other mesenchymal precursor populations.8
Molecular pathology studies have further revealed a networked interaction of multidimensional molecular events, including redox imbalance (abnormal accumulation of reactive oxygen species), cell cycle arrest via p16/p21 activation, impaired autophagy, and epithelial–mesenchymal transition (EMT). These events collectively mediate irreversible lung structural remodelling.9,10 Clinically, IPF typically manifests with progressive exertional dyspnoea, non-expectorative cough and generalised fatigue, which together cause substantial declines in quality of life (QOL). Notably, more than 60% of patients with IPF have at least one comorbidity, including pulmonary hypertension, ischaemic heart disease and pulmonary malignancy.11
Despite advances in clinical management, significant therapeutic challenges remain. The current standard of care relies primarily on two antifibrotic agents, nintedanib and pirfenidone, together with supportive interventions such as oxygen therapy and pulmonary rehabilitation.12 Both nintedanib, a multi-receptor tyrosine kinase inhibitor,13 and pirfenidone, which acts through multiple antifibrotic mechanisms,14 have been shown to slow disease progression, reducing the annual decline in lung function by approximately 50%. However, neither drug reverses established fibrosis or restores lung architecture, and both are limited by adverse events (such as liver function abnormalities and phototoxic reactions) as well as inadequate efficacy in addressing comorbidities.15 This therapeutic dilemma highlights the urgent need to develop novel therapeutic paradigms.
In recent years, intervention strategies targeting the phosphodiesterase 4 (PDE4) signalling pathway have attracted considerable research interest.16 As a key enzyme regulating the metabolism of the second messenger cyclic adenosine monophosphate (cAMP), PDE4 contributes to fibrosis by modulating immune cell activation and matrix remodelling. PDE4 is one of several phosphodiesterase isoforms involved in cAMP regulation.17 PDE4 inhibitors act by increasing intracellular cAMP levels, thereby suppressing inflammatory mediator release, inhibiting TGF-β signalling and modulating fibroblast differentiation phenotypes.18 Notably, this class of drugs has demonstrated dual anti-inflammatory and anti-fibrotic efficacy in fibrosis-related disease models such as systemic sclerosis and chronic obstructive pulmonary disease. However, their long-term benefit-risk profile in the treatment of IPF require validation through large-scale multicentre clinical trials.
Pathogenesis of IPF
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease driven by complex interactions among genetic susceptibility, environmental exposures, and dysregulated tissue repair responses.19 Rather than a single linear pathway, IPF is increasingly conceptualised as a self-perpetuating cycle in which repetitive epithelial injury and impaired regeneration promote persistent inflammatory signalling, fibroblast activation, and pathological extracellular matrix (ECM) remodelling. In this context, immune dysregulation contributes to disease initiation and persistence, particularly through macrophage–fibroblast crosstalk and cytokine-mediated amplification loops that sustain profibrotic signalling.20–22 Although this section focuses on IPF, many of the mechanisms summarised below are shared across lung fibrosis more broadly and are commonly examined using experimental fibrosis models; IPF is discussed specifically where human disease evidence and clinical outcomes are available.
Imbalance of Immune Homeostasis
IPF has historically been described as an “atypical inflammatory fibrotic disease” dominated by myofibroblast proliferation and ECM deposition; however, accumulating evidence indicates that immune microenvironment disturbances contribute to both disease onset and progression.20 IPF lung tissues have been reported to exhibit persistent activation of alveolar macrophages with enhanced production of profibrotic mediators, altered T-cell homeostasis including disruption of the Th17/Treg balance, and aberrant B-cell activation with increased autoantibody positivity.21 These immune perturbations may reinforce fibrosis through cell–cell interactions, such as macrophage–fibroblast signalling, and cytokine-driven feedback loops involving mediators including IL-13 and CCL18.21,22 Single-cell approaches further support immune cell state remodelling during IPF progression, including expansion of macrophage populations with profibrotic transcriptional signatures and changes in T-cell functional states.22
Macrophage-Centred Profibrotic Signalling
Macrophages play a central role in linking inflammatory injury to progressive fibrosis and represent a key immune node in IPF pathobiology.23 Early inflammatory macrophage activation has been associated with epithelial barrier disruption and increased production of pro-inflammatory mediators, including TNF-α, IL-1β, and IL-6, whereas later profibrotic macrophage states are characterised by elevated secretion of mediators such as TGF-β and PDGF that promote fibroblast activation and ECM synthesis.22,23 Single-cell transcriptomic studies have described profibrotic macrophage subsets and lipid-associated metabolic signatures in IPF lungs, suggesting that immunometabolic adaptations may contribute to persistent matrix remodelling.22 In addition, macrophage-derived extracellular vesicles have been reported to participate in epithelial–mesenchymal signalling and fibroblast activation in experimental systems.24
Other Innate and Adaptive Immune Contributors
Beyond macrophages, additional immune cell populations have been implicated in shaping the profibrotic microenvironment. Neutrophil-associated processes, including the formation of neutrophil extracellular traps, have been reported in bronchoalveolar lavage fluid and linked to epithelial injury and fibroblast activation in experimental studies.25–28 Mast cells, T-cell subset imbalance involving Th17/Treg dysregulation, and aberrant B-cell activation with tertiary lymphoid structure formation and autoantibody production have also been described in IPF, with proposed roles in sustaining cytokine signalling and fibroblast responses.19,29–38 γδ T cells have additionally been reported to contribute through cytokine secretion and interactions within broader immune networks in fibrotic lung tissue.39–42
Immunometabolic Reprogramming
In parallel with immune cell dysregulation, the IPF microenvironment exhibits metabolic remodelling that may amplify inflammatory and mesenchymal signalling. Reported alterations include increased glycolytic flux, altered glutamine utilisation, and enhanced fatty-acid oxidation pathways that influence macrophage activation states and fibroblast contractility. Preclinical studies targeting selected metabolic nodes have shown attenuation of fibrotic endpoints, supporting immunometabolic reprogramming as a contributor to disease progression.30,43 (Figure 1).
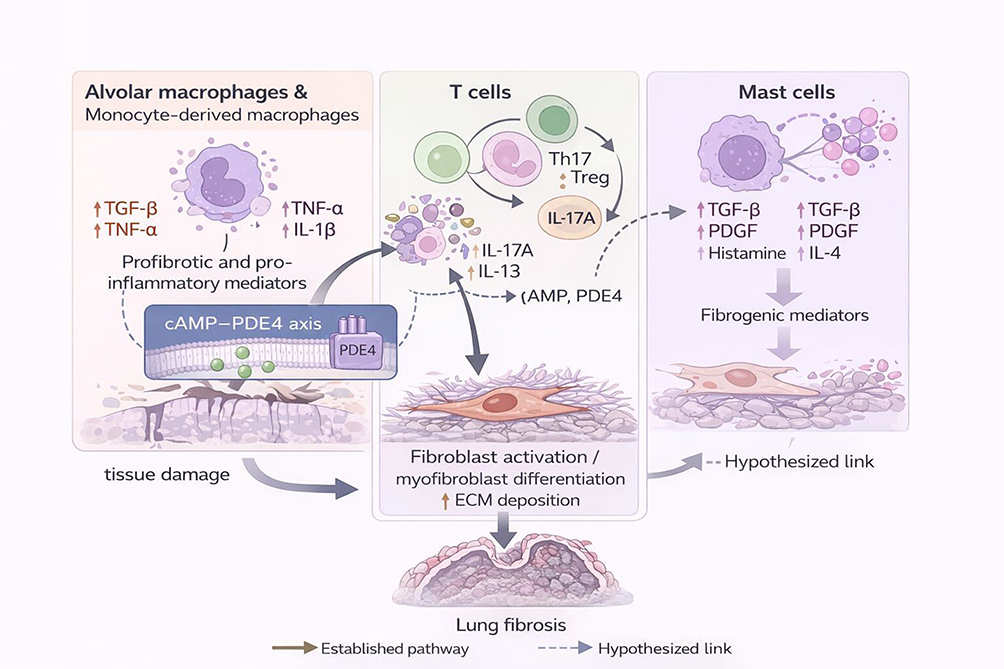 |
Figure 1 Immune mechanisms contributing to IPF pathogenesis. Schematic overview of immune cell populations implicated in idiopathic pulmonary fibrosis, including alveolar macrophages, T-cell subsets, and mast cells. These cells contribute to a profibrotic microenvironment through cytokine and mediator release, promoting fibroblast activation, myofibroblast differentiation, and extracellular matrix deposition. The cyclic adenosine monophosphate (cAMP)–phosphodiesterase 4 (PDE4) axis is depicted as a modulatory signalling node influencing immune activation states and downstream fibrotic responses. Solid arrows indicate established pathways, whereas dashed arrows denote hypothesised or emerging links. |
Collectively, these immune and immunometabolic disturbances contribute to a profibrotic ecosystem characterised by persistent cytokine signalling, altered intercellular communication, and fibroblast activation. Importantly, many of these pathways converge on intracellular second-messenger systems, particularly cyclic adenosine monophosphate (cAMP), which regulates inflammatory mediator release, immune cell activation states, and fibroblast behaviour.44 Given that phosphodiesterases control cAMP degradation, dysregulated phosphodiesterase activity may represent a mechanistically relevant link between immune microenvironment alterations and progressive fibrotic remodelling, providing a rationale for targeting the PDE4 signalling pathway in IPF.45
Dysregulation of Tissue Repair Mechanisms
The progression of IPF is closely associated with abnormalities in tissue repair and regenerative responses, in which persistent injury signals, maladaptive innate immune activation, and impaired regeneration may reinforce a self-sustaining cycle of fibrosis. Clinical cohort studies have reported that the pulmonary microenvironment in IPF is characterised by ongoing low-grade inflammatory activity, including increased NET-related deposits, evidence of prolonged NLRP3 inflammasome activation, and elevated IL-1β/IL-18 levels.46
Epithelial Regeneration Disorders
Defective alveolar epithelial regeneration is considered a key contributor to IPF pathobiology. Dysregulation of the Wnt/β-catenin pathway has been associated with impaired AT2 cell proliferative capacity through mechanisms involving altered transcriptional regulation, including aberrant TCF4 activity and reduced Axin2 expression. In experimental organoid systems, treatment with the β-catenin/CBP interaction inhibitor ICG-001 has been reported to improve epithelial regenerative responses.40 Senescent alveolar epithelial cells, reported to be increased in IPF compared with controls, may contribute to fibroblast activation by releasing SASP mediators (eg, IL-6, MMP3, PAI-1) and by altering immune cell activation states, thereby sustaining profibrotic signalling.47 Single-cell metabolomics studies have further suggested that impaired mitochondrial quality control in senescent epithelial cells may contribute to altered energy homeostasis through pathways involving AMPK and mTORC1 signalling.47
Abnormal Fibroblast Activation
Fibroblast activation and expansion of α-smooth muscle actin-positive myofibroblasts are hallmarks of IPF progression. Aberrant mechanosensing and sustained activation of integrin-linked signalling, including β1-associated pathways and YAP activity, have been implicated in maintaining myofibroblast phenotypes in experimental settings.48 Activated fibroblasts have also been reported to exhibit metabolic reprogramming, including increased glycolytic flux (with upregulated HK2 and LDHA) and enhanced glutamine utilisation, with mTORC1-4EBP1 signalling proposed as a maintenance axis.49 In intervention studies, the GLS1 inhibitor CB-839 has been reported to attenuate collagen-related endpoints, with proposed mechanisms involving altered α-ketoglutarate availability and downstream epigenetic regulation. Spatial transcriptomic observations have further described co-localisation of profibrotic signalling with ECM stiffness gradients within fibrotic lesions, supporting a model in which biochemical and biomechanical cues jointly reinforce fibroblast activation.
Together, inflammatory mediators and dysregulated repair responses intersect with intracellular signalling nodes that are regulated by cyclic adenosine monophosphate (cAMP), which can influence immune activation states, senescence-associated signalling, and fibroblast effector functions.50 As PDE4 is a major regulator of cAMP degradation in inflammatory and structural cells, targeting PDE4 provides a mechanistically plausible strategy to modulate interconnected profibrotic pathways relevant to IPF.
Disruption of Microenvironmental Homeostasis
Microenvironmental dysregulation in IPF involves both altered biomechanical properties of the lung parenchyma and remodelling of intercellular signalling networks.51 Excess ECM accumulation can enhance mechanotransduction pathways in epithelial and mesenchymal compartments, including integrin-FAK-associated signalling and YAP/TAZ activation, which may further reinforce profibrotic programmes. Single-cell intercellular analyses have reported aberrant epithelial–fibroblast signalling circuits in IPF tissues, including interactions involving IL-11-expressing epithelial populations and PDGFRα⁺ fibroblasts, with miR-21-5p implicated as a candidate mediator in some models. These interactions have been proposed to contribute to LOXL2-associated collagen cross-linking and sustained TGF-β/Smad3 signalling.52 Spatial metabolomics studies have also reported altered glutamine–glutamate patterns within fibrotic lesions, with mTORC1-4EBP1 signalling suggested as a potential axis supporting metabolic adaptation in the fibrotic microenvironment (Figure 2).
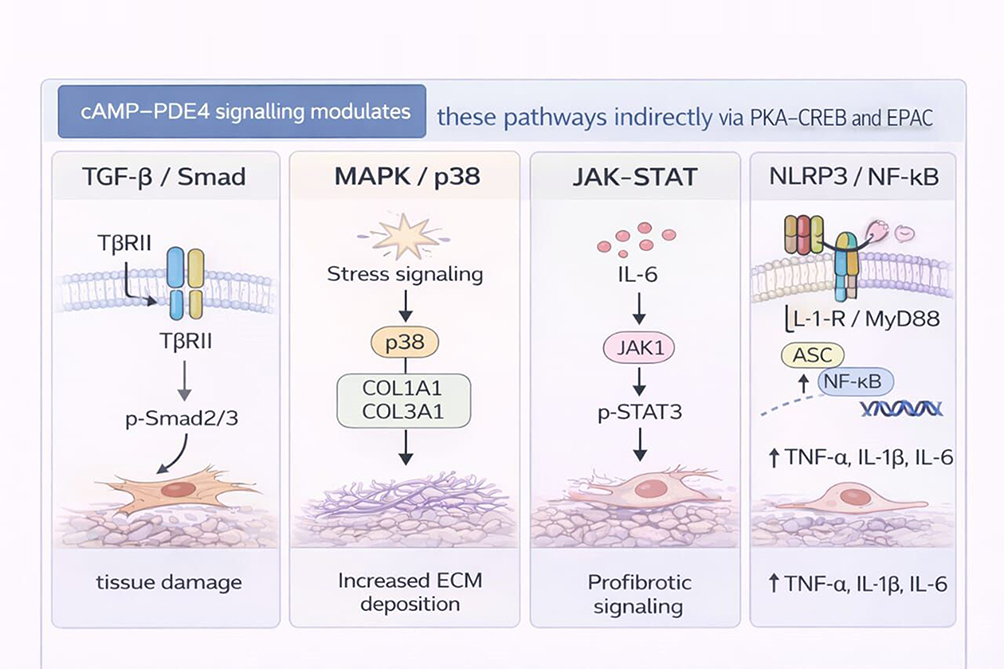 |
Figure 2 Dysregulation of inflammatory signalling pathways in IPF. Key profibrotic and inflammatory signalling pathways implicated in IPF, including TGF-β/Smad, MAPK/p38, JAK–STAT, and NLRP3/NF-κB cascades. Activation of these pathways promotes fibroblast activation, extracellular matrix accumulation, and sustained inflammatory signalling. cAMP–PDE4 signalling is shown as an upstream modulatory mechanism that indirectly influences pathway intensity through PKA-CREB and EPAC-dependent processes. Solid arrows represent established signalling events, and dashed arrows represent proposed or context-dependent mechanisms. |
Key Signalling Axes in IPF
TGF-β signalling is widely regarded as a central regulatory pathway in pulmonary fibrosis.53 Canonical Smad signalling, including TβRII-dependent phosphorylation of Smad2/3, promotes fibroblast-to-myofibroblast differentiation and ECM gene expression, while non-canonical pathways such as MAPK/p38 and PI3K/AKT/mTOR signalling have been implicated in profibrotic transcriptional and metabolic regulation. Microregional or compartmentalised activation of TGF-β signalling has been reported in progressive fibrotic lung tissues, and cross-talk with Wnt/β-catenin and Notch signalling has been proposed to contribute to the persistence of profibrotic microenvironments.54
Innate immune signalling pathways also contribute to inflammatory amplification and epithelial injury in IPF. The NLRP3 inflammasome–IL-1β axis has been reported to promote maturation of IL-1β through ASC/caspase-1/NLRP3 complex activation, with downstream IL-1R/MyD88 signalling linked to NF-κB pathway activation and pyroptosis-related processes in experimental contexts. In clinical analyses, activation markers of this pathway have been reported to correlate with lung function decline in IPF cohorts.22
The JAK–STAT system, particularly sustained STAT3 activation, has been implicated in fibrosis through transcriptional regulation of profibrotic mediators, modulation of immune regulatory programmes, and interactions with cellular metabolism. STAT3 activity has been linked to increased expression of TGF-β1-associated programmes, epigenetic regulation involving HDAC3-dependent effects on FoxO1 accessibility, and metabolic shifts that may enhance glutamine utilisation in fibroblasts.55 Preclinical studies have reported that selective JAK1 inhibition can reduce IL-6 levels in bronchoalveolar lavage fluid, supporting continued evaluation of pathway-targeted strategies (Figure 3).
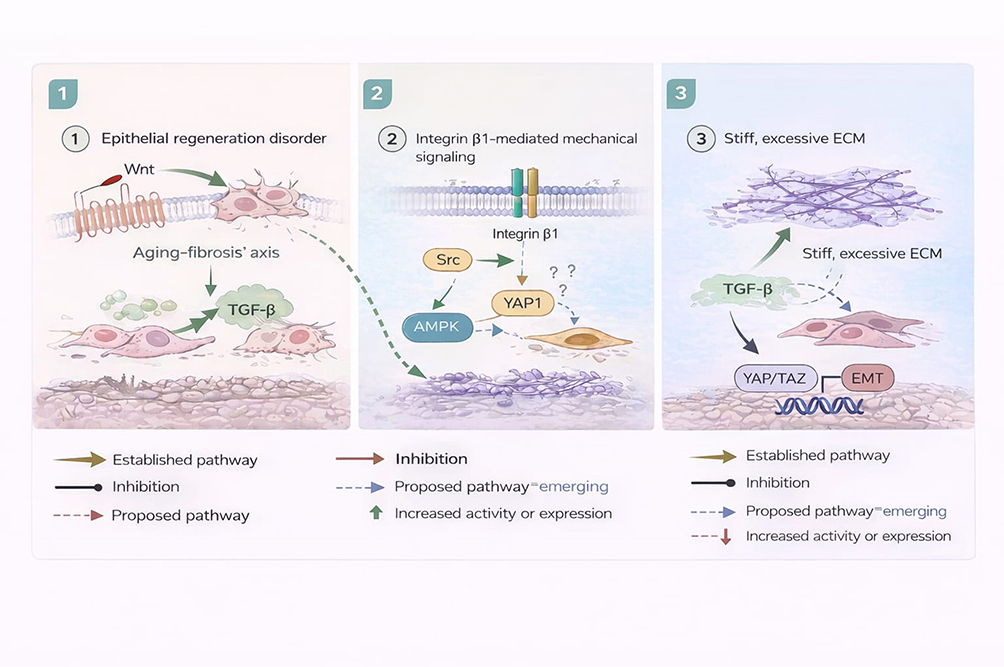 |
Figure 3 Dysregulation of tissue repair and microenvironmental homeostasis in IPF. Mechanisms underlying defective epithelial regeneration, aberrant fibroblast activation, and extracellular matrix stiffening in IPF. Alterations in Wnt/β-catenin signalling, integrin-mediated mechanotransduction, and YAP/TAZ activation contribute to impaired tissue repair and progressive fibrosis. Established pathways are distinguished from proposed or emerging mechanisms, highlighting the convergence of biomechanical and biochemical signals in sustaining the fibrotic microenvironment. |
Phosphodiesterase 4 Regulates IPF Pathogenesis
Phosphodiesterase 4 (PDE4), a key enzyme regulating cyclic adenosine monophosphate (cAMP) signalling, is involved in IPF pathogenesis through modulation of inflammatory signalling and fibroproliferative responses.56 PDE4 inhibition increases intracellular cAMP and activates downstream protein kinase A (PKA) signalling, which can suppress pro-inflammatory mediator release and reduce profibrotic signalling activity in relevant cellular and preclinical models.50
At the immune regulation level, PDE4 inhibition has been reported to modulate innate and adaptive immune responses, including macrophage activation states and inflammatory cytokine production, thereby reducing the profibrotic microenvironment that sustains fibroblast activation.57 At the tissue remodelling level, PDE4 inhibition has been associated with attenuation of fibroblast activation and extracellular matrix accumulation in experimental settings, supporting its potential as an anti-fibrotic strategy in lung fibrosis.56
Proposed or emerging mechanisms. In addition to these core cAMP-mediated actions, PDE4 inhibition has been suggested to influence additional downstream pathways related to TGF-β signalling, epithelial injury responses, and cellular metabolism. However, several of these mechanisms remain proposed and require further validation in IPF-specific models and clinical studies.57
Phosphodiesterase 4
The cyclic nucleotide phosphodiesterase (PDE) superfamily comprises 11 families (PDE1 to PDE11) that regulate intracellular signalling by catalysing the hydrolysis of cyclic nucleotide second messengers, principally cAMP and cGMP.45 Members of this superfamily share a conserved catalytic core and regulatory regions that influence subcellular localisation, assembly into signalling complexes, and responsiveness to post-translational modification.45 The biological diversity across PDE families and isoforms contributes to compartmentalised cyclic nucleotide signalling, enabling fine regulation of cell-type-specific responses.
Cyclic nucleotide signalling plays a central role in biological processes relevant to fibrotic lung disease, including inflammatory responses, epithelial repair, fibroblast activation, and metabolic regulation.58 PDE4 isoforms are cAMP-specific hydrolases and are expressed in multiple cell populations relevant to lung inflammation and remodelling, including macrophages, T lymphocytes, fibroblasts, and epithelial cells. Pharmacological inhibition of PDE4 increases intracellular cAMP, which can activate downstream pathways such as PKA–CREB and EPAC–Rap1 signalling.59 In experimental systems, PDE4 inhibition has been associated with suppression of inflammatory mediator expression (including TNF-α and IL-6), enhancement of epithelial barrier-associated programmes, and attenuation of profibrotic signalling linked to pathways such as TGF-β/Smad3 and Wnt/β-catenin.59–61 PDE4 inhibition has also been reported to increase anti-inflammatory programmes, including CREB-associated regulation of IL-10 expression.60,61 Emerging studies further suggest that PDE4 inhibitors may influence immunometabolic features of fibrotic microenvironments; however, these effects remain model-dependent and require further validation in IPF-relevant experimental systems and clinical contexts.
Phosphodiesterase 4 Inhibitors
Clinical development of PDE4 inhibitors has expanded across inflammatory and immune-mediated diseases, with efficacy demonstrated in selected Th2-driven inflammatory conditions and autoimmune disorders.62 In fibrosis-relevant experimental models, PDE4 inhibition has been associated with anti-inflammatory and antifibrotic effects through cAMP-dependent pathways including PKA and EPAC signalling. However, clinical development of systemic PDE4 inhibitors has been constrained by dose-limiting gastrointestinal adverse events, including nausea and vomiting, reported in dose-escalation settings. These adverse effects have been linked to PDE4 isoform distribution and central mechanisms, and have motivated efforts to optimise selectivity and delivery strategies.63
Accordingly, several approaches have been explored to improve therapeutic index, including subtype-selective inhibitors with increased activity toward PDE4B, inhaled formulations designed to increase lung exposure relative to systemic exposure, and multi-target strategies combining PDE4 inhibition with additional antifibrotic mechanisms.63 Candidate approaches relevant to fibrotic lung disease include inhaled PDE4 inhibitors (eg, GSK256066), dual-mechanism inhibitors (eg, PDE4 and tyrosine kinase inhibition), and subtype-selective strategies aimed at improving tolerability while maintaining biological activity (Figure 4).63
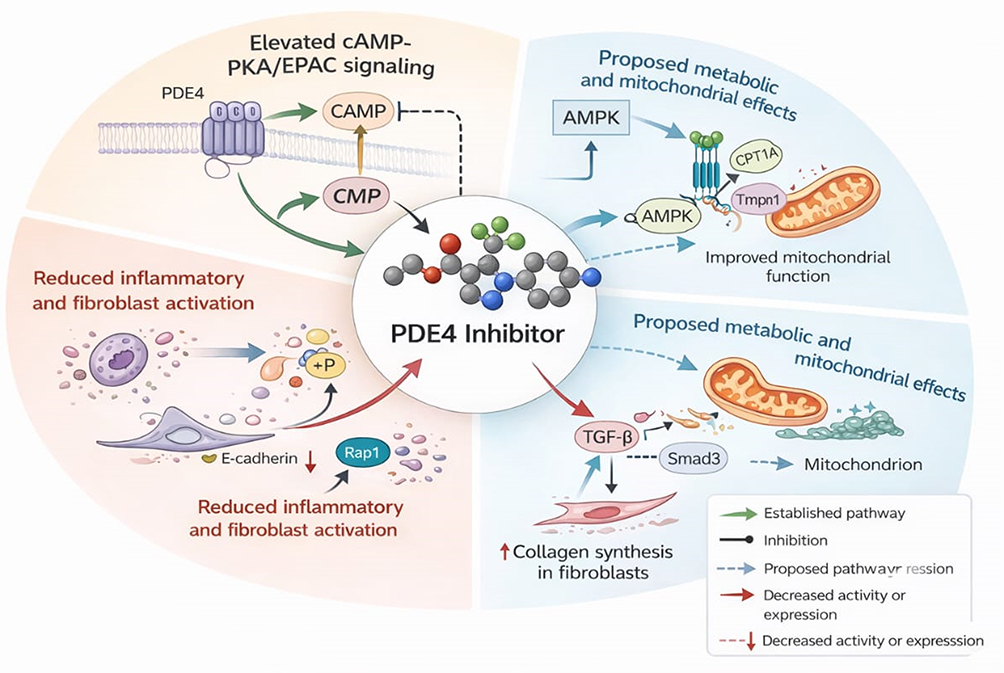 |
Figure 4 Mechanistic framework of PDE4 inhibition in fibrotic lung disease. Proposed mechanisms by which PDE4 inhibition modulates inflammatory and fibrotic processes in the lung. Inhibition of PDE4 increases intracellular cAMP levels, activating PKA and EPAC signalling to suppress inflammatory mediator production, attenuate fibroblast activation, and modulate profibrotic TGF-β/Smad signalling. Potential metabolic and mitochondrial effects are depicted as emerging mechanisms. Subtype-selective inhibition of PDE4B, relative to PDE4D, is illustrated to reflect efforts to optimise therapeutic efficacy while limiting central nervous system–related adverse effects. Solid arrows denote established effects, and dashed arrows indicate proposed mechanisms. |
Nerandomilast
Nerandomilast (BI 1015550) is a PDE4 inhibitor with preference for PDE4B and has been developed for fibrotic lung diseases including IPF and progressive pulmonary fibrosis (PPF).64 Its proposed therapeutic rationale is based on PDE4B-biased inhibition leading to increased intracellular cAMP signalling and downstream modulation of inflammatory and fibrotic pathways relevant to lung fibrosis.64 Nerandomilast has progressed through late-stage clinical development programmes.
Across clinical studies, nerandomilast has been associated with gastrointestinal adverse events, including nausea, diarrhoea, and related symptoms, consistent with known class effects of PDE4 inhibition. Interpretation of tolerability relative to other PDE4 inhibitors should be cautious in the absence of head-to-head trials, given differences in study populations, dosing regimens, and trial design. The reduced activity of nerandomilast against PDE4D relative to PDE4B has been proposed as a potential contributor to tolerability in relevant settings, but this hypothesis requires careful interpretation alongside clinical trial data.64
The first-in-human evaluation (NCT04419506) used a randomised, double-blind, placebo-controlled dose-escalation design.65 Healthy male volunteers received single ascending doses or multiple ascending doses, and an extended cohort included patients with IPF treated for 12 weeks. Treatment-emergent adverse events were reported, with headache and gastrointestinal symptoms among the most frequent events. Most events were mild to moderate, no treatment-related serious adverse events were reported, and drug exposure was higher in the IPF cohort than in healthy volunteers, suggesting potential disease-related influences on pharmacokinetics.65
A Phase II study (NCT05321069) used a multicentre, randomised, double-blind, placebo-controlled design in patients with confirmed IPF and evaluated change in forced vital capacity (FVC) over the treatment period.66 Results suggested differences in FVC trajectories across subgroups with and without background antifibrotic therapy. Treatment-emergent adverse events were common and predominantly gastrointestinal, and most events were mild to moderate.66 These findings supported continued evaluation in confirmatory trials.
International multicentre Phase III trials in IPF and PPF were conducted as randomised, double-blind, placebo-controlled studies with treatment durations of at least 52 weeks and stratification by background antifibrotic therapy.67 Across studies, nerandomilast was associated with a reduced rate of FVC decline compared with placebo over the observation period, including among participants receiving background antifibrotic therapy.67 Overall adverse event rates were broadly comparable across groups, while discontinuations due to adverse events showed dose-related patterns. Gastrointestinal events, particularly diarrhoea, were among the leading reasons for discontinuation.67 Collectively, these findings support continued evaluation of nerandomilast as a therapeutic option in fibrotic lung disease, while highlighting tolerability considerations relevant to PDE4 inhibition.
Preclinical pharmacodynamic studies reported activity of BI 1015550 in bleomycin-induced and silica-induced models of pulmonary fibrosis in mice, including reductions in lung hydroxyproline and improvement in structural indices.68 In vitro experiments have suggested attenuation of TGF-β1-induced profibrotic gene expression and modulation of fibroblast signalling, with additive effects reported alongside nintedanib in selected assays.68 Molecular docking analyses have proposed isoform-selective binding features that may contribute to reduced inhibitory activity against PDE4D relative to PDE4B, but such structural interpretations should be considered supportive rather than definitive predictors of clinical tolerability.68
Roflumilast
Roflumilast (Daxas®) is an oral PDE4 inhibitor that can be used to reduce the risk of acute exacerbations of chronic obstructive pulmonary disease (COPD). Its potential relevance to fibrotic mechanisms has been explored in experimental studies. Metabolic profiling based on nuclear magnetic resonance (NMR) spectroscopy revealed significant changes in lung tissue metabolism following bleomycin-induced pulmonary fibrosis in mice. The analysis showed that amino acid metabolites, including proline and glycine, were significantly accumulated in fibrotic lungs compared with control animals, while intermediates associated with the fatty acid β-oxidation pathway were significantly reduced. These metabolic disturbances suggest that the fibrotic process is characterised by a shift from energy-producing lipid metabolism to biosynthetic processes.69 Roflumilast intervention (1 mg/kg per day) was reported to reverse these metabolic disturbances, resulting in a reduction of proline levels, a key precursor of collagen synthesis, while elevating glutathione reductase activity to improve oxidative stress status.
In an allogeneic lung transplantation model (B10.D2→BALB/c), roflumilast inhibited the gene expression of cytokines and pro-fibrotic mediators (IL-6, IL-1β, TGF-β, CTGF) and elevated the proportion of CD4⁺ FoxP3⁺ regulatory T-cell infiltration compared to the control group.70 In C57BL/6J mice, prophylactic administration (5 mg/kg per day) reduced lung hydroxyproline content and decreased the right ventricular hypertrophy index. Transcriptome analyses showed reduced expression of key fibrosis-related genes (COL1A1, EDN1, etc.) and a decrease in the concentration of the lipid peroxidation marker MDA.71
Recent studies have also reported that roflumilast can inhibit the formation of neutrophil extracellular traps (NETs). In neutrophils from cystic fibrosis (CF) patients, NET production was inhibited and histone H3 citrullination levels were significantly reduced.72 In a model of Pseudomonas aeruginosa chronic airway infection, treated mice showed decreased free DNA content in bronchoalveolar lavage fluid and increased body weight recovery compared to controls, suggesting modulation of innate immunity in an infection-associated setting. Collectively, these findings suggest that PDE4 inhibition may influence pathways linked to inflammation, oxidative stress, and fibrotic remodelling in experimental models, although clinical relevance for lung fibrosis requires further confirmation.
Mangosteen Derivatives
Huang’s research team73 reported a novel class of phosphodiesterase 4 (PDE4) inhibitors based on structural modification of α-mangostin. Among these, compound 18a exhibited oral bioavailability and target selectivity in preclinical evaluation. The derivative showed a half-inhibitory concentration of 4.2 nmol/L against PDE4 isoforms, and molecular docking analysis reported differential binding characteristics compared with the clinical drug roflumilast. Toxicological evaluation reported that no adverse effects such as central vomiting were observed in the 10 mg/kg dose group administered by gavage to rats, whereas the positive control rolipram triggered gastrointestinal toxicity at an equivalent dose (1 mg/kg).73
In a bleomycin-induced rat model of lung fibrosis, 18a (10 mg/kg, once daily) administered orally inhibited collagen deposition. Improvement in lung histopathological scores was reported to be statistically comparable to that of high-dose pirfenidone (150 mg/kg). The study further suggested that, by optimising physicochemical parameters such as lipophilicity and ionisation constants, this compound may reduce central nervous system side effects reported with conventional PDE4 inhibitors while maintaining PDE4 inhibitory activity.73 These findings support 18a as an early preclinical candidate with translational potential, but current evidence remains limited to preclinical models, and validation in well-designed clinical studies with meaningful endpoints is required.73
2-Arylbenzofurans
Wang’s research team74 designed and synthesised a class of small molecule compounds targeting PDE4 based on a 2-arylbenzofuran backbone. The lead molecule, L13, showed nanomolar inhibitory activity against PDE4 in vitro. In a bleomycin-induced mouse model of lung fibrosis, L13 (10 mg/kg) was reported to improve lung histopathological features and reduce extracellular matrix deposition. In the same experimental setting, the antifibrotic effect was reported to be comparable to that of pirfenidone (300 mg/kg). Structure–activity relationship analyses suggested that introduction of aryl substituents to optimise molecular rigidity and hydrophobicity enhanced interaction with the PDE4 catalytic domain and improved plasma stability and oral bioavailability. The authors further reported that optimisation of physicochemical properties, including the lipid–water partition coefficient, may reduce off-target risk while maintaining enzyme inhibitory activity.74
These findings support 2-arylbenzofuran derivatives as early preclinical PDE4-directed candidates in experimental lung fibrosis models. However, evidence remains limited to preclinical studies, and further pharmacology, safety, and clinical evaluation are needed before translational conclusions can be drawn.
Rolipram
Pharmacological studies by Wu et al75 reported that rolipram, a prototypical PDE4 inhibitor, elevates intracellular cAMP by inhibiting PDE4-mediated cAMP hydrolysis. In experimental settings, rolipram has been used as a reference compound to demonstrate anti-inflammatory effects and to explore antifibrotic signalling pathways linked to cAMP elevation.75
However, rolipram is not widely used clinically due to tolerability limitations, and its primary value in this context is as a mechanistic tool compound rather than a practical therapeutic option.
Aminophylline
In a study by Suat Ekin et al,76 theophylline was evaluated in a bleomycin-induced rat model of pulmonary fibrosis. The authors reported that bleomycin exposure was associated with oxidative stress and inflammatory activation, including reduced antioxidant enzyme activity and increased malondialdehyde levels, alongside activation of NF-κB signalling and increased IL-6 expression. Theophylline treatment (75 mg/kg) was reported to restore antioxidant markers, reduce lipid peroxidation, and attenuate inflammatory signalling, with improvement in histopathological findings.76
As a non-selective agent with multiple pharmacologic actions, theophylline and aminophylline are best interpreted as providing supportive mechanistic context rather than serving as selective PDE4-targeted antifibrotic strategies.
AA6216
Matsuhira’s group77 used a bleomycin-induced mouse model of lung fibrosis to evaluate the regulation of segregated-nucleus-containing atypical monocytes (SatMs) by a novel compound, AA6216 (10 mg/kg per day, orally), compared with the clinical antifibrotic drug nintedanib (100 mg/kg per day, orally). The experimental design included continuous dosing from pre-treatment to day 8. Pathological analyses showed reduced accumulation of SatMs in the lung parenchyma in the AA6216-treated group, whereas SatM numbers were increased in the nintedanib-treated group under the conditions reported. In an in vitro bone marrow-derived SatM culture system, AA6216 inhibited TNF-α secretion in a concentration-dependent manner.77
These findings suggest that AA6216 may modulate lung fibrosis through SatM-associated mechanisms, including effects on monocyte accumulation and inflammatory mediator production. However, current evidence is limited to preclinical models, and additional studies are required to confirm efficacy, clarify mechanism, and establish relevance to clinical lung fibrosis (Table 1).77
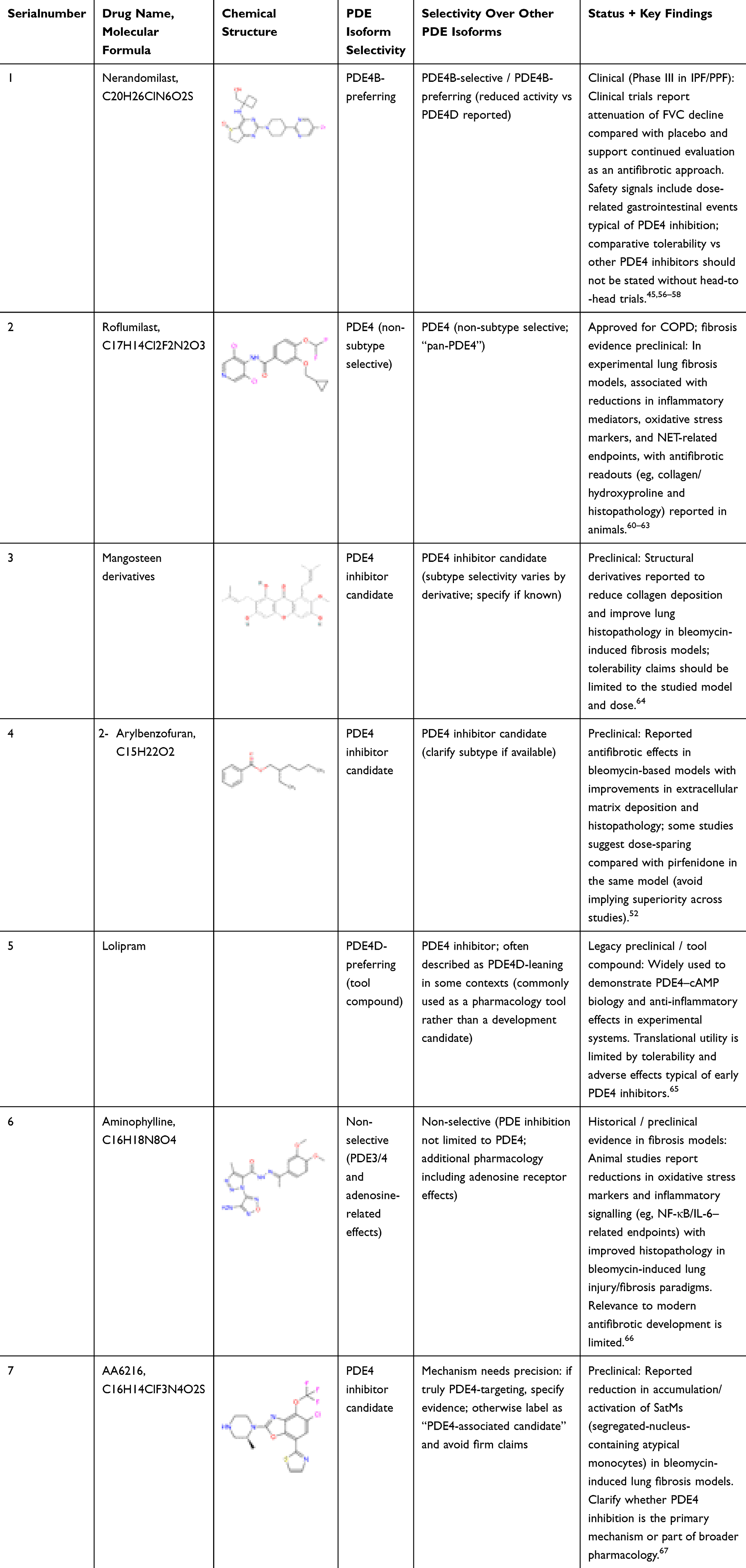 |
Table 1 Phosphodiesterase 4 (PDE4) Inhibitors Under Development for the Treatment of IPF and Inflammatory Diseases |
Discussion
The pathogenesis of lung fibrosis, including idiopathic pulmonary fibrosis (IPF), involves a complex network of interactions among genetic background, environmental stimuli, and aberrant injury and repair responses. The disease is clinically characterised by progressive and largely irreversible deterioration of lung function, and median survival after diagnosis is commonly reported in the range of 3 to 5 years. Although the available standard therapies include antifibrotic agents such as nintedanib and pirfenidone, their principal benefit is to slow disease progression rather than reverse established fibrosis. In response to this therapeutic dilemma, phosphodiesterase (PDE) inhibition, particularly targeting PDE4 as a major cAMP-hydrolysing enzyme in relevant immune and structural cells, has attracted attention because it may offer combined anti-inflammatory and antifibrotic effects.
In this paper, we explored the potential mechanisms of PDE4 inhibition through the cAMP-PKA-CREB and EPAC signalling axes. By elevating intracellular cAMP, PDE4 inhibitors can suppress the production of pro-inflammatory mediators such as TNF-α and IL-1β through cAMP-dependent signalling. They may also influence profibrotic pathways, including TGF-β-associated fibroblast activation and myofibroblast differentiation, thereby providing multidimensional intervention in fibrotic remodelling. Because many of these pathways are context dependent, and evidence quality varies across experimental systems, mechanistic conclusions should be interpreted with appropriate caution.
At the translational level, the PDE4B-selective inhibitor nerandomilast has advanced the furthest in clinical development. In phase II studies, nerandomilast showed signals consistent with attenuation of lung function decline, including changes in FVC over the treatment period. Larger phase III programs were subsequently designed to evaluate longer-duration outcomes and to define safety and tolerability in broader patient populations. Mechanistic studies have suggested that subtype selectivity may reduce centrally mediated adverse effects observed with earlier PDE4 inhibitors, and may differentially modulate immune and mesenchymal cell phenotypes relevant to fibrotic progression. Importantly, direct comparative conclusions against other PDE4 inhibitors cannot be made in the absence of head-to-head randomised trials.
In parallel, several additional PDE4-directed strategies remain at an earlier stage. Roflumilast and other PDE4 inhibitors have shown antifibrotic signals in experimental models, including effects linked to inflammation, oxidative stress, and metabolic pathways. However, tolerability, dosing constraints, and uncertainty in clinical translation remain key considerations. Similarly, structurally novel candidates such as α-mangostin-derived PDE4 inhibitors and 2-arylbenzofuran derivatives have demonstrated antifibrotic-associated readouts in preclinical models. These findings are hypothesis generating and support continued medicinal chemistry and mechanistic refinement, but they require further validation in well-designed studies using clinically relevant endpoints. Overall, these advances reflect a shift from broad PDE4 inhibition toward approaches emphasising improved selectivity, tissue targeting, and an improved therapeutic index, which may help address persistent limitations in current antifibrotic therapy.
Summary
Lung fibrosis, including idiopathic pulmonary fibrosis (IPF), is a chronic progressive interstitial lung disease characterised by a multidimensional pathological network involving dysregulation of the inflammatory microenvironment and activation of fibrotic remodelling processes. Based on the role of phosphodiesterase 4 (PDE4) in regulating cAMP signalling in immune and structural cells, PDE4 modulation has translational potential for interstitial lung remodelling diseases.
Clinical pharmacology studies in non-pulmonary indications have shown that PDE4 inhibitors, including apremilast and roflumilast, can provide anti-inflammatory efficacy with a safety profile that is generally manageable in appropriately selected patients. These agents may be relevant to lung fibrosis by modulating inflammatory pathways that have been implicated in fibrotic progression, including effects on Th17-associated signalling and other cytokine networks. However, the extent to which these mechanisms translate into clinically meaningful antifibrotic benefit in IPF remains to be established.
Translational research should prioritise evaluating the therapeutic potential of PDE4 inhibitors in pulmonary fibrosis. Randomised controlled trials are needed to confirm their impact on clinically relevant endpoints, including longitudinal lung function measures such as FVC decline. In parallel, pharmacokinetic and pharmacodynamic optimisation, including approaches that reduce centrally mediated adverse effects, will be important for improving the therapeutic index. At the basic research level, further study of subtype-selective PDE4 inhibitors, for example PDE4B-preferring strategies, may help clarify how cAMP signalling influences immune cell phenotypes, including macrophage activation states, within fibrotic lesions. In addition, exploring sequential or combination regimens of PDE4 inhibitors with existing antifibrotic agents (nintedanib and pirfenidone) may be informative, but such approaches require careful evaluation of safety, tolerability, and additive benefit in well-designed trials.
With ongoing development of inhaled PDE4 inhibitors and other lung-targeted delivery strategies, local drug delivery may reduce systemic exposure while increasing drug concentration in lung tissue. If supported by robust clinical evidence, these approaches could contribute to more individualised treatment strategies for lung fibrosis, including IPF.
Abbreviations
AECs, Alveolar Epithelial Cells; AT1/AT2, Alveolar Type I / Type II Epithelial Cells; BALF, Bronchoalveolar Lavage Fluid; cAMP, Cyclic Adenosine Monophosphate; COPD, Chronic Obstructive Pulmonary Disease; ECM, Extracellular Matrix; EMT, Epithelial-Mesenchymal Transition; EndMT, Endothelial-Mesenchymal Transition; FAO, Fatty Acid Oxidation; FVC, Forced Vital Capacity; FEV1, Forced Expiratory Volume in 1 Second; GI, Gastrointestinal; IL-6/IL-1β, Interleukin-6 / Interleukin-1 Beta; IPF, Idiopathic Pulmonary Fibrosis; MCs, Mast Cells; MMP-9, Matrix Metalloproteinase 9; MPO, Myeloperoxidase; MyoFBs, Myofibroblasts; NETs, Neutrophil Extracellular Traps; NF-κB, Nuclear Factor Kappa B; NLRP3, NLR Family Pyrin Domain Containing 3; OXPHOS, Oxidative Phosphorylation; PDE4, Phosphodiesterase 4; PDGF, Platelet-Derived Growth Factor; PPF, Progressive Pulmonary Fibrosis; QOL, Quality of Life; SAEs, Serious Adverse Events; SASP, Senescence-Associated Secretory Phenotype; SatMs, Non-classical Monocyte Subpopulations; STAT3, Signal Transducer and Activator of Transcription 3; TEAEs, Treatment-Emergent Adverse Events; TGF-β, Transforming Growth Factor Beta; Th17/Treg, T Helper 17 Cells / Regulatory T Cells; TIMP-1, Tissue Inhibitor of Metalloproteinase 1; TLR9, Toll-Like Receptor 9; TLS, Tertiary Lymphoid Structures; TNF-α, Tumor Necrosis Factor Alpha; TRAEs, Treatment-Related Adverse Events; YAP/TAZ, Yes-Associated Protein / Transcriptional Coactivator with PDZ-Binding Motif.
Data Sharing Statement
No data were used in the study described in the article.
Author Contributions
Ma Quan and Zhou Shixin contributed to the conception of the review, literature search, data interpretation, and drafting of the original manuscript. Zhi Xiaodong, Luo Caifeng, and Yanlin Wu contributed to figure design, data visualisation, and critical revision of the manuscript for important intellectual content. Zhongbo Zhu contributed to study supervision, conceptual guidance, and critical review of the manuscript. Xuhui Zhang contributed to manuscript revision, intellectual oversight, and refinement of clinical and translational content. Xiping Liu contributed to conceptualisation of the study, overall project coordination, and critical revision of the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (No.82260889) and the Natural Science Foundation of Gansu Province (No.24JRRD002).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that appear likely to influence the work reported here.
References
1. Maher TM. Interstitial lung disease: a review. JAMA. 2024;331(19):1655–18. doi:10.1001/jama.2024.3669
2. Maher TM, Bendstrup E, Dron L, et al. Global incidence and prevalence of idiopathic pulmonary fibrosis. Respir Res. 2021;22(1):197. doi:10.1186/s12931-021-01791-z
3. Griese M, Kurland G, Cidon M, et al. Pulmonary fibrosis may begin in infancy: from childhood to adult interstitial lung disease. thorax. 2024;79(12):1162–1172. doi:10.1136/thorax-2024-221772
4. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952. doi:10.1016/S0140-6736(17)30866-8
5. Sun ZH, He WY, Meng HW, Ji ZH, Qu JX, Yu GY. Lactate activates er stress to promote alveolar epithelial cells apoptosis in pulmonary fibrosis. Respir Res. 2024;25(1):401. doi:10.1186/s12931-024-03016-5
6. Lederer DJ, Martinez FJ. Idiopathic Pulmonary Fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra1705751
7. Huang QQ, Chen Y, Shen SR, et al. Klotho antagonizes pulmonary fibrosis through suppressing pulmonary fibroblasts activation, migration, and extracellular matrix production: a therapeutic implication for idiopathic pulmonary fibrosis. Aging (Albany NY). 2020;12(7):5812–5831. doi:10.18632/aging.102978
8. Zabihi M, Shahriari Felordi M, Lingampally A, Bellusci S, Chu X, El Agha E. Understanding myofibroblast origin in the fibrotic lung. Chin Med J Pulm Crit Care Med. 2024;2(3):142–150. doi:10.1016/j.pccm.2024.08.003
9. Zhou Y, Ling TT, Shi WH. Current state of signaling pathways associated with the pathogenesis of idiopathic pulmonary fibrosis. Respir Res. 2024;25(1):245. doi:10.1186/s12931-024-02878-z
10. Margaritopoulos GA, Lasithiotaki I, Antoniou KM. Toll-like receptors and autophagy in interstitial lung diseases. Eur J Pharmacol. 2017;808:28–34. doi:10.1016/j.ejphar.2016.09.032
11. King CS, Nathan SD. Idiopathic pulmonary fibrosis: effects and optimal management of comorbidities. Lancet Respir Med. 2017;5(1):72–84. doi:10.1016/S2213-2600(16)30222-3
12. Bai X, Nie P, Lou Y, et al. Pirfenidone is a renal protective drug: mechanisms, signalling pathways, and preclinical evidence. Eur J Pharmacol. 2021;911:174503. doi:10.1016/j.ejphar.2021.174503
13. Richeldi L, Du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–2082. doi:10.1056/NEJMoa1402584
14. Ko IG, Hwang L, Jin -J-J, et al. Pirfenidone improves voiding function by suppressing bladder fibrosis in underactive bladder rats. Eur J Pharmacol. 2024;977:176721. doi:10.1016/j.ejphar.2024.176721
15. Sun XY, Wang HG, Zhan X, et al. Comparison of the safety profiles for pirfenidone and nintedanib: a disproportionality analysis of the us food and drug administration adverse event reporting system. Front Pharmacol. 2024;15:1256649. doi:10.3389/fphar.2024.1256649
16. Kawamatawong T. Phosphodiesterase-4 inhibitors for non-copd respiratory diseases. Front Pharmacol. 2021;12:518345. doi:10.3389/fphar.2021.518345
17. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. doi:10.1124/pr.58.3.5
18. Wójcik-Pszczoła K, Chłoń-Rzepa G, Jankowska A, et al. A novel, pan-pde inhibitor exerts anti-fibrotic effects in human lung fibroblasts via inhibition of tgf-β signaling and activation of camp/pka signaling. Int J Mol Sci. 2020;21(11):4008. doi:10.3390/ijms21114008
19. Tiwari P, Verma S, Washimkar KR, Mugale MN. Immune cells crosstalk pathways, and metabolic alterations in idiopathic pulmonary fibrosis. Int Immunopharmacol. 2024;135:112269. doi:10.1016/j.intimp.2024.112269
20. Yang HZ, Cui B, Liu HZ, et al. Targeting tlr2 attenuates pulmonary. J Immunol. 2009;182(1):692–702. doi:10.4049/jimmunol.182.1.692
21. Yin YQ, Peng F, Situ HJ, et al. Construction of prediction model of inflammation related genes in idiopathic pulmonary fibrosis and its correlation with immune microenvironment. Front Immunol. 2022;13:1010345. doi:10.3389/fimmu.2022.1010345
22. Chen WC, Yu WK, Vyf S, Hsu HS, Yang KY. Nlrp3 inflammasome activates endothelial-to-mesenchymal transition via focal adhesion kinase pathway in bleomycin-induced pulmonary fibrosis. Int J Mol Sci. 2023;24(21):15813. doi:10.3390/ijms242115813
23. Ge ZL, Chen Y, Ma LK, Hu FJ, Xie LB. Macrophage polarization and its impact on idiopathic pulmonary fibrosis. Front Immunol. 2024;15:1444964. doi:10.3389/fimmu.2024.1444964
24. Yan LY, Su YF, Hsia I, et al. Delivery of anti-microrna-21 by lung-targeted liposomes for pulmonary fibrosis treatment. Mol Ther Nucleic Acids. 2023;32:36–47. doi:10.1016/j.omtn.2023.02.031
25. Lodge K, Nakanishi S, Yazbeck L, Guck J, Molyneaux PL, Cowburn AS Neutrophils in idiopathic pulmonary fibrosis have a distinct biomechanical phenotype of systemic activation that correlates with disease severity.
26. Du XL, Ma Z, Xing YQ, et al. Identification and validation of potential biomarkers related to oxidative stress in idiopathic pulmonary fibrosis. Immunobiology. 2024;229(5):152791. doi:10.1016/j.imbio.2024.152791
27. Alekseeva LA, Sen’kova AV, Sounbuli K, Savin IA, Zenkova MA, Mironova NL. Pulmozyme ameliorates lps-induced lung fibrosis but provokes residual inflammation by modulating cell-free DNA composition and controlling neutrophil phenotype. Biomolecules. 2025;15(2):298. doi:10.3390/biom15020298
28. Su QY, Feng YY, Guo J, et al. Pirfenidone alleviates interstitial lung disease in mice by inhibiting neutrophil extracellular trap formation and nlrp3 inflammasome activation. Clin Exp Immunol. 2025;219(1):uxaf019. doi:10.1093/cei/uxaf019
29. Siddhuraj P, Jönsson J, Alyamani M, et al. Dynamically upregulated mast cell cpa3 patterns in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Front Immunol. 2022;13:924244. doi:10.3389/fimmu.2022.924244
30. Wang S, Yu HM, Liu S, Liu YQ, Gu X. Regulation of idiopathic pulmonary fibrosis: a cross-talk between tgf-β signaling and micrornas. Front Med. 2024;11:1415278. doi:10.3389/fmed.2024.1415278
31. Otaki N, Motomura Y, Terooatea T, et al. Activation of ilc2s through constitutive ifnγ signaling reduction leads to spontaneous pulmonary fibrosis. Nat Commun. 2023;14(1):8120. doi:10.1038/s41467-023-43336-6
32. Minnis P, Kane R, Lumsden R, Whitty S, Donnelly SC, Keane MP. S120 serum microrna profiles in ipf patients–biomarkers or potential therapeutic targets? thorax. 2015;70(Suppl 3):
33. Galati D, De Martino M, Trotta A, et al. Peripheral depletion of nk cells and imbalance of the treg/th17 axis in idiopathic pulmonary fibrosis patients. Cytokine. 2014;66(2):119–126. doi:10.1016/j.cyto.2013.12.003
34. Van Geffen C, Deißler A, Quante M, Renz H, Hartl D, Kolahian S. Regulatory immune cells in idiopathic pulmonary fibrosis: friends or foes? Front Immunol. 2021;12:663203. doi:10.3389/fimmu.2021.663203
35. Nie YJ, Wu SH, Xuan YH, Yan G. Role of il-17 family cytokines in the progression of ipf from inflammation to fibrosis. Mil Med Res. 2022;9(1):21. doi:10.1186/s40779-022-00382-3
36. Ali MF, Egan AM, Shaughnessy GF, et al. Antifibrotics modify b-cell–induced fibroblast migration and activation in patients with idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2021;64(6):722–733. doi:10.1165/rcmb.2020-0387OC
37. Neys SFH, Heukels P, Van Hulst JAC, et al. Aberrant b cell receptor signaling in naïve b cells from patients with idiopathic pulmonary fibrosis. Cells. 2021;10(6):1321. doi:10.3390/cells10061321
38. Li J, Chen XH, Zhang BH, Wang CL. Circ_0035796 depletion inhibits transforming growth factor-β1-induced pulmonary fibrosis in a mir-150-5p/l1cam-dependent manner. Autoimmunity. 2023;56(1):2250099. doi:10.1080/08916934.2023.2250099
39. Okuno D, Sakamoto N, Akiyama Y, et al. Two distinct mechanisms underlying γδ t cell-mediated regulation of collagen type i in lung fibroblasts. Cells. 2022;11(18):2816. doi:10.3390/cells11182816
40. Han D, Gong HY, Wei Y, et al. Hesperidin inhibits lung fibroblast senescence via il-6/stat3 signaling pathway to suppress pulmonary fibrosis. Phytomedicine. 2023;112:154680. doi:10.1016/j.phymed.2023.154680
41. Wu JZ, Gong LP, Li YJ, et al. Sgk1 aggravates idiopathic pulmonary fibrosis by triggering h3k27ac-mediated macrophage reprogramming and disturbing immune homeostasis. Int J Biol Sci. 2024;20(3):968–986. doi:10.7150/ijbs.90808
42. Zhang Y, Lu YB, Zhu WJ, et al. Leech extract alleviates idiopathic pulmonary fibrosis by tgf-β1/smad3 signaling pathway. J Ethnopharmacol. 2024;324:117737. doi:10.1016/j.jep.2024.117737
43. Gao H, Sun ZY, Hu XX, et al. Identification of glycolysis-related gene signatures for prognosis and therapeutic targeting in idiopathic pulmonary fibrosis. Front Pharmacol. 2025;16:1486357. doi:10.3389/fphar.2025.1486357
44. Insel PA, Murray F, Yokoyama U, et al. cAMP and Epac in the regulation of tissue fibrosis. Br J Pharmacol. 2012;166(2):447–456. doi:10.1111/j.1476-5381.2012.01847.x
45. Kolb M, Crestani B, Maher TM. Phosphodiesterase 4B inhibition: a potential novel strategy for treating pulmonary fibrosis. Eur Respir Rev. 2023;32(167):220206. doi:10.1183/16000617.0206-2022
46. Sivakumar P, Ammar R, Thompson JR, et al. Integrated plasma proteomics and lung transcriptomics reveal novel biomarkers in idiopathic pulmonary fibrosis. Respir Res. 2021;22(1):273. doi:10.1186/s12931-021-01860-3
47. Wu WS, Wang Z, Zhang HY, Zhang XJ, Tian H. Circgrhpr inhibits aberrant epithelial-mesenchymal transformation progression of lung epithelial cells associated with idiopathic pulmonary fibrosis. Cell Biol Toxicol. 2024;40(1):7. doi:10.1007/s10565-024-09839-8
48. Parimon T, Yao CF, Stripp BR, Noble PW, Chen P. Alveolar epithelial type ii cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci. 2020;21(7):2269. doi:10.3390/ijms21072269
49. Sharma P, Alizadeh J, Juarez M, et al. Autophagy, apoptosis, the unfolded protein response, and lung function in idiopathic pulmonary fibrosis. Cells. 2021;10(7):1642. doi:10.3390/cells10071642
50. Sun MY, Sun YY, Feng ZR, et al. New insights into the hippo/yap pathway in idiopathic pulmonary fibrosis. Pharmacol Res. 2021;169:105635. doi:10.1016/j.phrs.2021.105635
51. Yan PS, Liu JY, Li ZW, et al. Glycolysis reprogramming in idiopathic pulmonary fibrosis: unveiling the mystery of lactate in the lung. Int J Mol Sci. 2023;25(1):315. doi:10.3390/ijms25010315
52. Schick MA, Schlegel N. Clinical Implication of Phosphodiesterase-4-Inhibition. Int J Mol Sci. 2022;23(3):1209. doi:10.3390/ijms23031209
53. He AD, He LZ, Chen TW, Li XJ, Cao C. Biomechanical properties and cellular responses in pulmonary fibrosis. Bioengineering. 2024;11(8):747. doi:10.3390/bioengineering11080747
54. Guo JS, Fang YS, Jiang FX, et al. Neohesperidin inhibits tgf-β1/smad3 signaling and alleviates bleomycin-induced pulmonary fibrosis in mice. Eur J Pharmacol. 2019;864:172712. doi:10.1016/j.ejphar.2019.172712
55. Kim J, Jeon S, Kang SJ, et al. Lung-targeted delivery of tgf-β antisense oligonucleotides to treat pulmonary fibrosis. J Control Release. 2020;322:108–121. doi:10.1016/j.jconrel.2020.03.016
56. Sun ZR, Yang ZZ, Wang MM, et al. Paraquat induces pulmonary fibrosis through wnt/β-catenin signaling pathway and myofibroblast differentiation. Toxicol Lett. 2020;333:170–183. doi:10.1016/j.toxlet.2020.08.004
57. Jia MJ, Liu YM, Liu J, et al. Xuanfei baidu decoction ameliorates bleomycin-elicited idiopathic pulmonary fibrosis in mice by regulating the lung-gut crosstalk via ifnγ/stat1/stat3 axis. Phytomedicine. 2024;135:155997. doi:10.1016/j.phymed.2024.155997
58. Sisson TH, Christensen PJ, Muraki Y, et al. Phosphodiesterase 4 inhibition reduces lung fibrosis following targeted type II alveolar epithelial cell injury. Physiol Rep. 2018;6(12):e13753. doi:10.14814/phy2.13753
59. Zebda R, Paller AS. Phosphodiesterase 4 inhibitors. J Am Acad Dermatol. 2018;78(3):S43–S52. doi:10.1016/j.jaad.2017.11.056
60. Yougbare I, Keravis T, Lugnier C. Ncs 613, a pde4 inhibitor, by increasing camp level suppresses systemic inflammation and immune complexes deposition in kidney of mrl/lpr lupus-prone mice. Biochim Biophys Acta Mol Basis Dis. 2021;1867(3):166019. doi:10.1016/j.bbadis.2020.166019
61. Wang S, Yang GF, Zhang K, et al. Structural optimization of moracin m as novel selective phosphodiesterase 4 inhibitors for the treatment of idiopathic pulmonary fibrosis. Bioorg Chem. 2024;149:107474. doi:10.1016/j.bioorg.2024.107474
62. Sanz MJ, Cortijo J, Morcillo EJ. Pde4 inhibitors as new anti-inflammatory drugs: effects on cell trafficking and cell adhesion molecules expression. Pharmacol Ther. 2005;106(3):269–297. doi:10.1016/j.pharmthera.2004.12.001
63. Reininger D, Herrmann FE, Nickolaus P, et al. Pde4b inhibitor bi1015550 improves lung fibrosis, modulates transcriptome in bleomycin-treated rats, and attenuates fibrotic markers in human lung fibroblasts and epithelial cells. Eur Respir Soc. 2023;62(suppl 67):PA1863.
64. Li H, Zuo JP, Tang W. Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol. 2018;9:1048. doi:10.3389/fphar.2018.01048
65. Reininger D, Fundel‐Clemens K, Mayr CH, et al. Pde4b inhibition by nerandomilast: effects on lung fibrosis and transcriptome in fibrotic rats and on biomarkers in human lung epithelial cells. Br J Pharmacol. 2024;181(23):4766–4781. doi:10.1111/bph.17303
66. Maher TM, Schlecker C, Luedtke D, Bossert S, Zoz DF, Schultz A. Phase I studies of bi 1015550, a preferential phosphodiesterase 4b inhibitor, in healthy males and patients with idiopathic pulmonary fibrosis. ERJ Open Res. 2022;8(4):00240–2022. doi:10.1183/23120541.00240-2022
67. Maher T, Assassi S, Azuma A, et al. Pos1329 design of a Phase III, randomised, placebo-controlled trial of bi 1015550 in patients with progressive fibrosing interstitial lung disease. Ann Rheum Dis. 2023;82(Supplement 1):1014–1015. doi:10.1136/annrheumdis-2023-eular.897
68. Richeldi L, Azuma A, Cottin V, et al. Design of a phase iii, double-blind, randomised, placebo-controlled trial of bi 1015550 in patients with idiopathic pulmonary fibrosis (fibroneer-ipf). BMJ Open Respir Res. 2023;10(1):e001563. doi:10.1136/bmjresp-2022-001563
69. Herrmann FE, Hesslinger C, Wollin L, Nickolaus P. Bi 1015550 is a pde4b inhibitor and a clinical drug candidate for the oral treatment of idiopathic pulmonary fibrosis. Front Pharmacol. 2022;13:838449. doi:10.3389/fphar.2022.838449
70. Milara J, Morcillo E, Monleon D, Tenor H, Cortijo J. Roflumilast prevents the metabolic effects of bleomycin-induced fibrosis in a murine model. PLoS One. 2015;10(7):e0133453. doi:10.1371/journal.pone.0133453
71. Kim SW, Lim JY, Rhee CK, et al. Effect of roflumilast, novel phosphodiesterase-4 inhibitor, on lung chronic graft-versus-host disease in mice. Exp Hematol. 2016;44(5):332–341.e334. doi:10.1016/j.exphem.2016.02.002
72. Song SR, Fu ZL, Guan RJ, et al. Intracellular hydroxyproline imprinting following resolution of bleomycin-induced pulmonary fibrosis. Eur Respir. 2022;59(5):2100864. doi:10.1183/13993003.00864-2021
73. Totani L, Amore C, Piccoli A, et al. Type-4 phosphodiesterase (pde4) blockade reduces netosis in cystic fibrosis. Front Pharmacol. 2021;12:702677. doi:10.3389/fphar.2021.702677
74. Huang YY, Deng JH, Tian YJ, et al. Mangostanin derivatives as novel and orally active phosphodiesterase 4 inhibitors for the treatment of idiopathic pulmonary fibrosis with improved safety. J Med Chem. 2021;64(18):13736–13751. doi:10.1021/acs.jmedchem.1c01085
75. Wu LK, Xu L, Chen Y, et al. Rolipram plays an anti-fibrotic effect in ligamentum flavum fibroblasts by inhibiting the activation of erk1/2. BMC Musculoskelet Disord. 2021;22(1):818. doi:10.1186/s12891-021-04712-9
76. Ekin S, Yildirim S, Akkoyun MB, et al. Theophylline attenuates bleomycin-induced oxidative stress in rats: the role of il-6, nf-κb, and antioxidant enzymes. Braz J Pharm Sci. 2022;58:e20804.
77. Matsuhira T, Nishiyama O, Tabata Y, et al. The phosphodiesterase 4 inhibitor aa6216 suppresses activity of fibrosis-specific macrophages. Biochem Biophys Rep. 2021;28:101118.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.