Back to Journals » International Journal of Nanomedicine » Volume 14
Anti-osteoclastogenic effect of epigallocatechin gallate-functionalized gold nanoparticles in vitro and in vivo
Authors Zhu S, Zhu L, Yu J, Wang Y, Peng B ![]()
Received 9 February 2019
Accepted for publication 3 May 2019
Published 8 July 2019 Volume 2019:14 Pages 5017—5032
DOI https://doi.org/10.2147/IJN.S204628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Shenting Zhu,* Lingxin Zhu,* Jingjing Yu, Yanqing Wang, Bin Peng
State Key Laboratory Breeding Base of Basic Science of Stomatology (Hubei-MOST) & Key Laboratory of Oral Biomedicine Ministry of Education, School and Hospital of Stomatology, Wuhan University, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Background: Epigallocatechin gallate (EGCG), the major anti-inflammatory compound in green tea, has been shown to suppress osteoclast (OC) differentiation. However, the low aqueous solubility of EGCG always leads to poor bioavailability, adverse effects, and several drawbacks for clinical applications.
Purpose: In this study, we synthesized EGCG-capped gold nanoparticles (EGCG-GNPs) to solve the drawbacks for clinical uses of EGCG in bone destruction disorders by direct reduction of HAuCl4 in EGCG aqueous solution.
Methods and Results: The obtained EGCG-GNPs were negatively charged and spherical. Theoretical calculation results suggested that EGCG was released from GNPs in an acidic environment. Cellular uptake study showed an obviously large amount of intracellular EGCG-GNPs without cytotoxicity. EGCG-GNPs exhibited better effects in reducing intracellular reactive oxygen species levels than free EGCG. A more dramatic anti-osteoclastogenic effect induced by EGCG-GNPs than free EGCG was observed in lipopolysaccharide (LPS)-stimulated bone marrow macrophages, including decreased formation of TRAP-positive multinuclear cells and actin rings. Meanwhile, EGCG-GNPs not only suppressed the mRNA expression of genetic markers of OC differentiation but also inhibited MAPK signaling pathways. Furthermore, we confirmed that EGCG-GNPs greatly reversed bone resorption in the LPS-induced calvarial bone erosion model in vivo, which was more effective than applying free EGCG, specifically in inhibiting the number of OCs, improving bone density, and preventing bone loss.
Conclusion: EGCG-GNPs showed better anti-osteoclastogenic effect than free EGCG in vitro and in vivo, indicating their potential in anti-bone resorption treatment strategy.
Keywords: epigallocatechin gallate, gold nanoparticles, bone marrow macrophages, lipopolysaccharide, calvarial bone
Introduction
Excessive bone resorption leads to disorders such as postmenopausal osteoporosis, rheumatoid arthritis, periodontitis, and inflammatory osteolysis.1 Normally, alveolar bone is constantly reconstructed by means of the balanced activities of osteoclasts (OCs) and osteoblasts. However, OC activity is increased in the presence of pro-inflammatory cytokines produced by inflammatory cells and lipopolysaccharide (LPS) produced by bacteria. LPS is an important component of the outer membrane of Gram-negative bacteria and is capable of inducing bone resorption. LPS plays an important role in bone loss by initiating a local host response that involves recruitment of inflammatory cells, production of prostanoids and cytokines, elaboration of lytic enzymes, and activation of OC formation or function.2,3 Experimental evidence has shown that LPS-mediated inflammation is highly dependent on reactive oxygen species (ROS)4,5 and associated signaling pathways, such as the p38 mitogen-activated protein kinase (p38), c-Jun N-terminal protein kinase (JNK), and extracellular signal-regulated kinase (ERK) cascades in bone marrow macrophages (BMMs).6,7 Other studies revealed that LPS induces RANK signaling and cyclooxygenase 2 expression in RAW264.7 osteoclastogenesis. In animal models, LPS-induced alveolar bone resorption is substantially mediated by tumor necrosis factor receptor signaling and prostaglandin E2 production.8 Above all, LPS is a critical pathogenic factor in inflammatory bone diseases. To date, effective therapy against LPS-induced bone destruction is limited to antibiotics and surgery, which may cause undesirable complications and serious surgical trauma. Accordingly, numerous biological compounds targeting modulation of the above signaling pathways involved in OC differentiation have been found to ameliorate bone damage. Therefore, improving drugs to effectively heal and regenerate LPS-induced osteolysis is a major goal for treating chronic inflammatory bone diseases.
Habitual tea drinkers are reported to be at reduced risk of hip fractures and to have an increased bone mineral density. The main active compound of green tea is (–)-epigallocatechin gallate (EGCG), which can effectively scavenge ROS, such as superoxide anion radicals, hydrogen peroxide, hydroxyl radicals, peroxyl radicals, singlet oxygen, and peroxynitrite.9,10 EGCG can attenuate autoimmune arthritis by modulating Th17/Treg.11 EGCG can also ameliorate experimental arthritis,12 prevent osteoporosis in mice, and exert a therapeutic effect on periodontitis in rats.13 These effects may have a connection with blocking receptor activator of NF-κB ligand (RANKL)-induced nuclear factor kappa B (NF-κB),14–16 the MAPK pathway, and proteasome-dependent degradation17–19 in OC precursors. Moreover, EGCG has a wide range of pharmacological properties, including immune-modulating, anti-inflammatory, antimicrobial, antioxidant, antimutagenic, and anticancer properties.9,20,21 However, EGCG has several drawbacks for clinical applications due to its low aqueous solubility, which leads to poor bioavailability and adverse effects.22
In recent years, a number of researchers have shown that gold nanoparticles (GNPs) can be synthesized with drugs absorbed or conjugated onto the particle surface.23,24 As nanocarriers, they have the advantages of low toxicity, easy synthesis, large surface area, excess loading of pharmaceuticals, and enhanced permeability and retention effect.25,26 Most GNP carriers are easily taken up by cells through endocytic mechanisms.27–29 Tran et al developed a methotrexate (MTX)–GNP complex by binding MTX, a chemotherapeutic agent for treating a variety of neoplasms, to GNP via its carboxyl group. By exploiting GNP as a drug carrier, MTX is efficiently transported into tumor cells and exerts cytotoxic activity.30 Wang et al conjugated doxorubicin onto the surface of GNPs with a poly(ethylene glycol) spacer via an acid-labile linkage, which can release doxorubicin in response to the pH of acidic organelles following endocytosis to achieve enhanced drug accumulation and retention in multidrug-resistant cancer cells.31 Conners et al utilized bisphosphonate-functionalized GNP to treat osteoporotic bone diseases.32 Moreover, Sul et al demonstrated that GNPs inhibited RANKL and reduced ROS levels in BMMs.33 GNPs have shown favorable bioavailability and promising advantages as nanocarriers for drug delivery.34,35 Tea catechins have been reported to augment the cellular uptake of nanoparticles.36
Based on the above studies, we synthesized EGCG-capped GNPs (EGCG-GNPs) to facilitate the delivery of EGCG by engulfment of nanoparticles in a single step. Different from traditional methods, this green nanotechnology introduced no other chemicals.37 This method can be an optimal solution to clinical applications of EGCG, improving its poor bioavailability and decreasing its adverse effects. The inhibitory effects of free EGCG and EGCG-GNPs on LPS-induced OC formation in vitro and bone destruction in vivo were demonstrated and compared. The corresponding mechanisms were investigated afterward.
Materials and methods
Reagents and mice
Fetal bovine serum (FBS), penicillin, streptomycin, and α-modified Eagle’s medium (α-MEM) were obtained from Gibco BRL (Gaithersburg, USA). Recombinant murine soluble RANKL and macrophage colony-stimulating factor (M-CSF) were purchased from Peprotech (London, UK). EGCG (98%) for cell, (Escherichia coli serotype 055: B5) LPS, sodium tetrachloroaurate (III) dihydrate (99%), FITC-labeled phalloidin and 4′,6-diamidino-2-phenylindole (DAPI), human serum albumins (HSA), were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). EGCG for mice was obtained from Aladdin® (Shanghai, China). Primary antibodies against total JNK, phosphorylated JNK, ERK1, phosphorylated ERK1, p38, and phosphorylated p38 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). BABL/c mice (male; 18–20 g body weight; 5 weeks of age) were purchased from the Experimental Animal Center of Hubei Province, Wuhan, China. All mice were housed under specific pathogen-free conditions. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of Wuhan University (China).
Preparation and characterization of nanoparticles
EGCG working solution was prepared by continuously stirring EGCG powder in deionized (DI) water for 15 min at room temperature. Subsequently, 0.1 mL of 0.2% EGCG working solution was added dropwise into 1.0 mL HAuCl4 solution (4.0 and 1.0 mM) at 25°C with magnetic stirring. The color of the solution gradually changed from pale yellow to deep red within 2 min. After the complete color change, 0.1 mL of 0.2% EGCG solution was added, and stirring was continued for 10 min to ensure complete reaction and absorption. The samples were noted as 0.2EGCG-4.0 HAuCl4 and 0.2EGCG-1.0 HAuCl4. The absorption spectra of the samples were investigated with a UV-vis spectrophotometer (UV-2550, Shimadzu. Kyoto, Japan). The mean particle sizes and zeta potential values of EGCG-GNPs were determined by dynamic light-scattering technique (Zetasizer Nano ZSP, Malvern, UK). For transmission electron microscopy (TEM) observation, the samples were prepared by drop casting the nanoparticle suspensions over a copper grid and allowed to air dry at room temperature. Then, the morphology and structure of EGCG-GNPs were examined using a JEM-2100 electron microscope. For Fourier-transform infrared (FT-IR) measurements, the nanoparticles were freeze dried and diluted with potassium bromide in a ratio of 1:100. Then, the spectra of dried powder EGCG-GNPs were recorded over the range of 400–4,000 cm−1 on an FT-IR instrument (FTIR5700, Thermo Fisher Scientific, Waltham, MA, USA).
Determination of the loading capacity and EGCG release in vitro
Hydrochloric acid solution (1.0 mM) was added dropwise to EGCG-GNPs colloid until the red solution became colorless and transparent. Then the solution were centrifuged at 1,000 rpm for 5 min. The amount of free EGCG in the supernatant was determined in triplicate samples using the Folin-Ciocalteu colorimetric assay method. The loading capacity were calculated using the following formula:
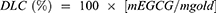
where mEGCGis the mass of EGCG that absorbed by GNP and mgoldis the mass of gold.
The in vitro EGCG release from EGCG-GNP was studied by the dialysis method. Dialysis tubing (benzoylated; 1,200 MW cut off; Sigma-Aldrich) was washed in running water for 4 h to remove glycerin, soaked in PBS for 10 min and then rinsed thoroughly in the buffer solution. One mL of EGCG-GNP (0.2EGCG-1.0GNPs) was placed in the dialysis bag and dialyzed against 10 mL of PBS buffer at pH values of 7.4 and 6.0. Control tubing, containing pure EGCG, was prepared and dialyzed. The dialysis process was performed under stirring at 37°C and away from bright light. At appropriate time intervals, 1 mL of the outer aqueous solution was withdrawn for analysis and immediately replaced by an equal volume of fresh release buffer. The cumulative amount of EGCG released was analyzed by the Folin–Ciocalteu colorimetric assay.38,39 The accumulative release percentage of EGCG (RE%) was expressed according to the equation:

where Ct is the amount of drug released from the beginning to the time t, and C0 is the total amount of drug in the solution. The data represent the average of three different analyses carried out for each sample.
In vitro stability test
In vitro stability of the EGCG-GNPs was tested in the presence of NaCl, HSA, and their mixtures. One mL of EGCG-GNPs solution was added to glass vials containing 5 ml of 150 mM NaCl (the total ion concentration of human blood is around 150 mM), 4.0% HSA (the reference range for HSA concentrations in serum is approximately 3.5–5.0%), and both 150 mM NaCl and 4.0% HSA, respectively. The stability and identity of the GNPs were measured by recording UV absorbance at 1 h as well as after 16 h. The absorption band at 530 nm confirmed the retention of nanoparticles in all the above mixtures.
Density functional theory (DFT) calculation
The GNP surface models were generated using the (111) cleavage planes of the crystal structure of bulk gold.40,41 The surfaces possessed three layers, with each layer consisting of approximately 15–20 gold atoms. Next, the citrate anion, EGCG anion, and neutral EGCG were added with an idealized starting geometry. The sodium ions were placed next to the anion oxygen to make the whole system neutral. Geometry optimizations were performed via the Perdew, Burke, and Ernzerhof functional,42 with the inclusion of dispersion correction term. The COSMO solvation model43 with water as solvent was used to account for the solvation effect. Double numerical plus polarization basis sets were used for all atoms, and the core electrons of Au were replaced by DFT semi-core pseudopots. The gold surface and organic adsorbate were also optimized separately at the same level to calculate the binding energy. All calculations were performed using the DMol3 module of Materials Studio program.44
Cell preparation and cell viability assay
Primary cultured BMMs were used in subsequent experiments. Monocytes were isolated from the tibiae of 6-week-old mice as previously described. After overnight incubation in complete α-MEM containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 ng/mL M-CSF at 37°C in a humidified atmosphere under 5% CO2, nonadherent cells were collected and further cultured until 80% confluence was achieved. For biological experiments and analyses, the adherent cells were harvested, collected by centrifugation at 1,000 rpm for 5 min, and then seeded in well plates before in vitro biological evaluation. For cell viability assay, the cells were stimulated with fresh medium containing serially diluted EGCG or EGCG-GNPs for 24, 48, and 72 h. Then, cells were measured with Cell Counting Kit-8 (CCK8; Dojindo, Japan). The results were represented as percentages of the control according to the absorbance at 450 nm by a 96-well microplate reader (BioTek Instruments Inc., Winooski, VT, USA).
Intracellular uptake determined by TEM
Primary cultured BMMs were cultured in a 6-well culture plate until 80–90% confluence was achieved before being exposed to 10 μM EGCG-GNPs for 30 min. After the media were removed, the cells were washed with fresh PBS and fixed with 2.5% glutaraldehyde. Intracellular uptake of EGCG-GNPs was observed by TEM (T700, Hitachi, Tokyo, Japan).
OC differentiation in vitro
Previously isolated BMMs (4×104 per well) were seeded in 24-well culture plates and incubated in complete culture medium with 100 ng/mL RANKL for 36 h and 1 μg/mL LPS for another 5 days. During the process, different concentrations of EGCG or EGCG-GNPs supplements were added. Cells were fixed with 4% paraformaldehyde for 15 min and stained for 30 min at 37°C in the dark via a leukocyte acid phosphatase assay kit (Sigma-Aldrich). TRAP-positive multinucleated cells with more than three nuclei were counted under a light microscope.
Fibrous actin (F-actin) ring fluorescence staining
The fixed cells were permeabilized with 0.1% Triton–PBS for 5 min and washed thrice with PBS for 10 min. After blocking with 1% bovine serum album for 1 h, cells were stained with FITC-labeled phalloidin for 40 min and then with DAPI for 5 min at 37°C in the dark, followed by washing thrice again. Finally, F-actin ring formation was observed with a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).45
Measurement of intracellular ROS
BMMs (2×106 per well) were plated into 6-well plates in complete α-MEM in triplicate in the presence or absence of 1 μg/mL LPS, with or without EGCG (10 μM) or EGCG-GNPs (10 μM) for 6 h. Cells were washed thrice, collected in PBS supplemented with 2% FBS, and incubated with Krebs–Ringer solution containing 20 μM 2′,7′-dichlorofluorescein diacetate (DCFH-DA) for 20 min on ice in the dark (Beyotime Institute of Biotechnology, Shanghai, China), followed by washing thrice again. Intracellular ROS was detected using an Accuri C6 Flow Cytometer (BD Biosciences, San Jose, CA, USA). The relative fluorescence intensity of DCFH-DA was monitored in the FL-1 channel (excitation at 480 nm and emission at 530 nm), which represented intracellular ROS levels.
Quantitative real-time PCR
BMMs (2×106 per well) were seeded in 6-well culture plates, and total cellular RNA was extracted from BMMs by TRIzol Reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA). Aliquots (1 μg) of RNA were reverse transcribed to cDNA (20 μL) with oligo dT and ReverTraAce (Toyobo, Osaka, Japan). Quantitative real-time PCR was performed as previously described.44 Specific primer sequences for PCR were designed as follows: NFATc1, 5′-CGTAACTCTGTCTTTCTAACCTTAAGCTC-3′ (forward) and 5′-GTGAT GACCCCAGCATGCACCAGTCACAG-3′ (reverse); TRAP, 5′-TGACAAGAGGTTCCAGGA-3′ (forward) and 5′-AGCCAGGACAGCTGAGTG-3′ (reverse); OSCAR, 5′-CTGCTGGTAACGG ATCAGCTCCCCAGA-3′ (forward) and 5′-CCAAGGAGCCAGAACCTTCGAAACT-3′ (reverse); MMP-9, 5′-TCCAGTACCAAGACAAAGCCTA-3′ (forward) and 5′-TTGCACTGCACGGTTGA A-3′ (reverse); and GAPDH, 5′-AACGGATTTGGTCGTATTGGG-3′ (forward) and 5′CAGGGGTG CTAAGCATTGG-3′ (reverse). GAPDH as an internal control was used to control differences in amplification of starting messenger RNA concentrations.
Western blot analysis
The total protein of BMMs was extracted as described previously.46 It was diluted with 4× SDS–PAGE sample loading buffer (Bioworld, Atlanta, GA, USA) and denatured at 95°C for 10 min. The protein concentration was measured using a microplate spectrophotometer (Benchmark Plus, Bio-Rad Laboratories Inc., Hercules, CA, USA) at a wavelength of 595 nm. An equal quantity of protein (40 μg) was electrophoresed on 10% (w/v) SDS-PAGE mini-gels until specific antibody proteins were separated from one another according to their size. The proteins on gels were then transferred onto polyvinylidene fluoride (Merck Millipore, Billerica, MA, USA) membranes. Immunoblots were blocked with nonfat dry milk in Tris-buffered saline-Tween-20 (TBST) buffer at room temperature for 1 h and cultured with corresponding primary antibodies at different dilutions (JNK and phospho-JNK, 1:500; p38 and phospho-p38, 1:400; ERK and phospho-ERK, 1:200) overnight at 4°C. After washing with TBST thrice, the blots were incubated with horseradish peroxidase conjugated secondary antibody (Pierce, Rockford, IL, USA) for 1 h at room temperature. The membranes were developed by Western Bright ECL (Advansta, San Jose, CA, USA) and converted to visualized images.
LPS-mediated calvarial bone erosion experiments
Female 5-week-old mice were randomly assigned to four groups: PBS control, LPS, LPS with EGCG, and LPS with EGCG-GNPs. EGCG (dose: 15 mg/kg) and EGCG-GNPs (dose: 15 mg/kg) were intraperitoneally injected daily for 8 days. On the second day, 30 min after intraperitoneal injection, LPS (10 mg/kg body weight) or the PBS vehicle was injected subcutaneously over the mice calvaria under light anesthesia daily for the following 7 days.47 Injectable solutions of each treatment dose in physiological saline were prepared every day. No deaths were observed during the whole experiment. Mice were sacrificed 7 days after injection. The whole calvariae were removed and fixed in 4% paraformaldehyde for 2 days. X-ray images of calvariae were taken with in vivo DXS PRO (Bruker Corporation, Massachusetts, USA). Changes in the trabecular bone were scanned with microcomputed tomography (micro-CT; Scanco Medical AG, Bassersdorf, Switzerland) at 20 μm resolution. A series of planar transverse gray-value images was acquired to reconstruct the 3D images. For histological analysis, the calvarial bone was stained with hematoxylin and eosin (H&E) to show the erosion of bone and TRAP to identify OCs.
Statistical analysis
All values are expressed as mean ± SD. Statistical analysis was performed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Multiple comparisons were analyzed using one-way analysis of variance followed by the Student–Newman–Keul tests.
Results and discussion
Characterization and DFT calculation of EGCG-loaded GNPs
EGCG is a polyphenol containing eight orthophenolic hydroxyls and serves as a bidentate ligand that forms complex with Au3+ ions and can effectively reduce the chelated Au3+ ions to GNPs. EGCG was not only used as a reducing agent but also as a stabilizing agent for preparing the EGCG-GNPs because of the acidity of the polyphenol (the pKa of EGCG is 7.75). Compared with traditional ways to load drug on GNPs, we utilized EGGC to successfully reduce gold to corresponding GNPs and load polyphenols on the particles in a single step, a process that fulfills all criteria of green nanotechnology as no “man-made” chemical other than gold acids were used (Figure 1A).
 |
Figure 1 (A) Schematic diagram of synthetic methods. The right side shows the two-step method, and the left side shows the single-step method. (B) Nanoparticle size distribution from DLS measurements. (C) TEM images. (D) Zeta potential distribution from DLS measurements. (E) UV-vis absorption spectrum. (F) FT-IR spectrum.Abbreviations: DLS, dynamic light scattering; TEM, transmission electron microscopy; UV-vis, Ultraviolet–visible spectroscopy. |
UV-vis absorption spectra of GNPs dispersed in water generally exhibited the surface plasmon resonance (SPR) at approximately 540 nm, confirming the formation of spheroid nanoparticle in solution. High concentrations of HAuCl4 (4 mM) facilitated the formation of large sizes of nanoparticles, indicated by the red shift of the SPR absorption bands of the EGCG-GNPs (Figure 1E). The EGCG-GNPs demonstrating strong SPR absorption were selected for the subsequent measurement of dynamic light scattering (DLS). The particle size accorded with normal distribution, with 35.6 nm average diameter (Figure 1B). The zeta potential of EGCG-GNPs was −19.5 mV (Figure 1D), indicating that the nanoparticles are surrounded by the negatively charged EGCG. The high-resolution TEM analysis of an individual EGCG-GNPs clearly shows that the particle sizes of the GNPs were approximately 30 nm with a spherical shape (Figure 1C), which was smaller than those measured by DLS. TEM provides the size of nanoparticles in dried form, whereas DLS indicates the hydrodynamic diameter that includes core plus any molecule attached or adsorbed on the surface.48 By comparing the infrared spectra of EGCG molecules and EGCG-GNPs, we can judge whether the EGCG molecules are successfully loaded onto the GNP surface. The FT-IR spectrum of EGCG exhibited absorption bands around 3,000 cm−1 (O-H and C-H stretching), 1,705 cm−1 (C=O stretching), 1,450–1,620 cm−1 (benzene ring stretching), 1,435 cm−1 (C-H bending), and 1,100–1,300 cm−1 (C-O stretching). These peaks can also be observed in the spectrum of EGCG-GNPs (Figure 1F). These results indicated that the GNPs are successfully coated by EGCG.
The geometries of citrate anion, EGCG anion, and neutral EGCG on the Au(111) surface were optimized by DFT. For citrate anion, the most stable geometry optimized was found to have three carboxylate groups bonded to four Au atoms, as illustrated by the schematic interaction diagram in the bottom of Figure 2A. The corresponding binding energy for citrate anion is 53.5 kcal/mol. Meanwhile, the EGCG anion is mainly bonded to one Au atom via its ionic phenolic oxygen. Three other phenolic hydroxyl groups have weak interactions with Au atoms. The binding energy for EGCG anion is 47.5 kcal/mol (Figure 2B), which is comparable with that of citrate anion. Compared with the anion EGCG, the neutral EGCG only has weak interactions with the Au surface; thus, the binding energy is considerably small (25.0 kcal/mol) (Figure 2C). Therefore, the acidic environment, which promotes the neutral EGCG formation, will facilitate the release of EGCG from GNPs. As the low pH microenvironment usually occurs at the inflammatory locus or within the endosome, EGCG molecule will most likely be released at these locations.
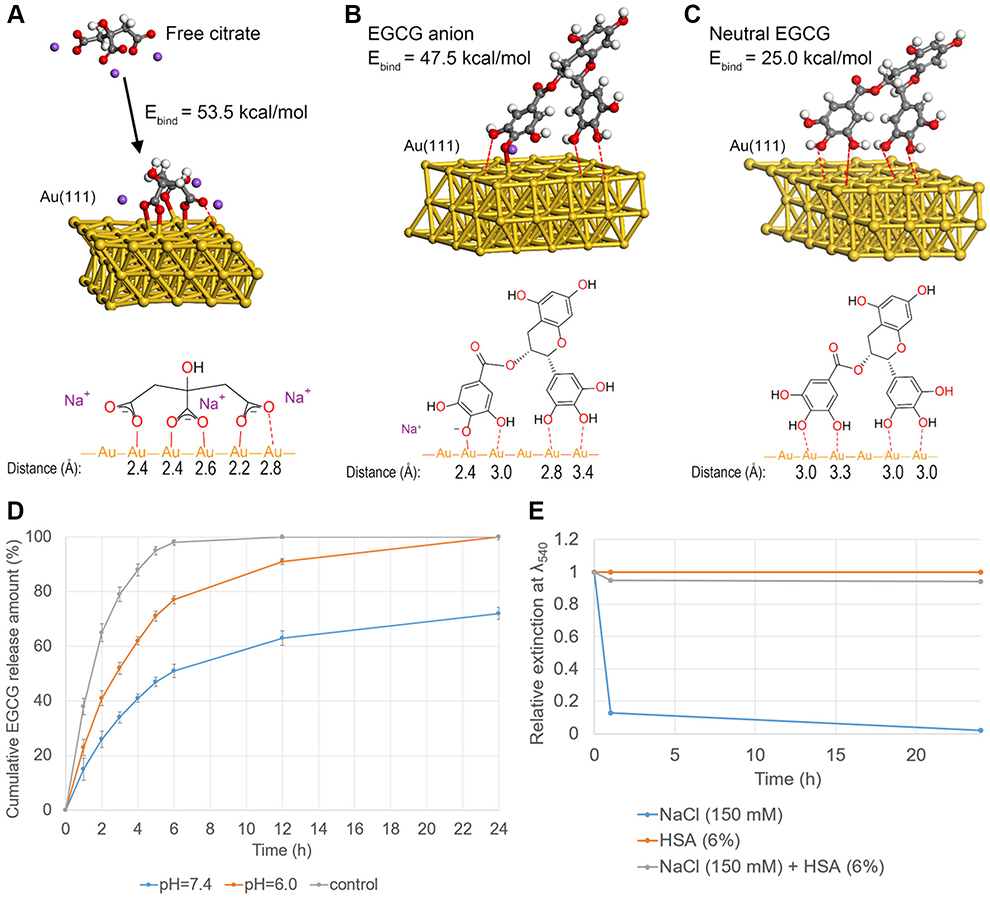 |
Figure 2 DFT-optimized ligand geometries on the Au(111) model surfaces and in vitro release assay. (A) Free citrate and citrate absorbed on the gold surface. (B) EGCG anion absorbed on the gold surface. (C) Neutral EGCG absorbed on the gold surface. (D) In vitro release of EGCG in PBS (pH 7.4 and pH 6.0) for EGCG-GNP and free EGCG solution. (E) Relative extinction at wavelength 540 nm of EGCG-GNP in 150 mM NaCl, 6% HSA, and their mixture measured at 1 and 24 h.Abbreviations: DFT, density functional theory; PBS, phosphate buffered saline; HSA, human serum albumin. |
The loading capacity, drug release and stability in vitro
Folin–Ciocalteu colorimetric assay revealed the loading capacity for 0.2EGCG-1.0GNP is 8.1±2.6%. The in vitro antioxidants released from EGCG-GNPs at different pH values are presented in Figure 2D. At pH 7.4, about 60% EGCG was released in first 6 h, then 15% EGCG was gradually released during the following 18 h. The remaining 25% EGCG was still absorbed on GNPs by 24 h. At pH 6.0, the release speed was faster. About 80% EGCG was released in 6 h, and the remaining EGCG was gradually released during the following 18 h. These results indicate that low pH value can accelerate the release of EGCG as predicted by theoretical calculation and approximately 25% EGCG was chemically bonded to the GNP surface.
Blood plasma is mostly water (up to 95% by volume), and contains 6–8% dissolved proteins (eg, serum albumins, globulins, and fibrinogen) and ∼150 mM electrolytes (Na+, Ca2+, Mg2+, HCO3−, Cl−, etc.). To estimate EGCG-GNPs stability in blood plasma, we built a simple blood plasma model made up of 6% HSA to represent proteins and 150 mM NaCl to represent electrolytes. The stability of nanoparticles was characterized by SPR absorption at 540 nm (Figure 2E). The relative extinction coefficient (to EGCG-GNPs in pure water at time 0) of EGCG-GNPs in pure NaCl solution dropped to near 0.1 in the first hour and continued dropping to near 0 at 24 h, whereas absorption spectra of in HSA solution remained unchanged after 24 h. As for HAS + NaCl mix solution, the absorption dropped to 95% in the first hour and remain unchanged for the following 23 h. These results suggest absorption of HSA onto the nanoparticle surface, which stabilizes EGCG-GNPs against electrolytes in blood plasma.
Intracellular uptake of EGCG-GNPs by bmms without inducing cytotoxic effect
To confirm the viability of BMMs at different concentrations of EGCG and EGCG-GNPs, CCK8 assays at different incubation time intervals were performed. EGCG displayed no obvious effect on the viability of BMMs when its concentration was below 20 μM and exhibited cytotoxicity at the concentration of 25 μM (Figure 3A). The EGCG-GNPs exhibited no cytotoxicity after treatment with various concentrations after 1, 2, and 3 days (Figure 3B). EGCG-GNPs had a low cytotoxicity compared to free EGCG. The toxicity of EGCG is often due to its pro-oxidative action when used at high concentration and to its strong interaction with the cellular membrane. The employment of nanoparticles probably avoids this phenomenon by increasing its internalization.49,50 These results indicated that the prepared EGCG-GNPs can be used as cytocompatible drugs.
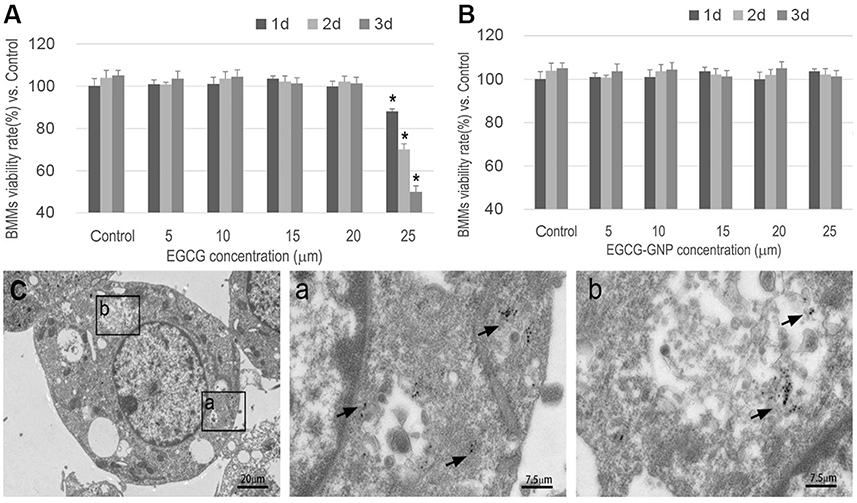 |
Figure 3 Viability of BMMs in the presence of (A) EGCG and (B) EGCG-GNPs at different concentrations. The results are mean ± SD of triplicate experiments: *P<0.05, significant differences compared with control. (C) Intracellular uptake images of BMMs treated with EGCG-GNPs as confirmed by TEM (black arrows). Enlarged views of C (scale bar =20 μm) are presented in a and b (scale bar =7.5 μm). Abbreviations: BMM, bone marrow-derived macrophages; TEM, transmission electron microscopy. |
TEM was used to observe the intracellular uptake of EGCG-GNPs. Figure 3C shows the presence of EGCG-GNPs in the BMMs. Clearly, EGCG-GNPs were effectively engulfed into the BMMs after incubation with EGCG-GNPs for 30 min, and GNPs were surrounded by membrane structures. This result indicates that the cell engulfed the particles effectively and formed the acidic endosomes, which can lead to EGCG release in the cell, as suggested by DFT calculation results.
Effect of various concentrations of EGCG and EGCG-GNPs on LPS-induced osteoclastogenesis
To verify the effect of EGCG and EGCG-GNPs, BMMs were stimulated with 100 ng/mL RANKL for 36 h and another 5 days with 1 μg/mL LPS. Together with the stimulator, various concentrations of EGCG or EGCG-GNPs were added. According to the results of CCK8, EGCG was used at a concentration below 20 μM to compare the inhibition effect of osteoclastogenesis in the absence of cytotoxicity.
Cells stained purple were TRAP positive and were used as OC markers.51 Under light microscopy, TRAP-positive OCs containing more than three nuclei were counted. One of the largest TRAP-positive OCs at 5 days of each group is shown in Figure 4A. The number of stained TRAP-positive OCs in the EGCG group and in the EGCG-GNPs group decreased compared with that of the control group in a dose-dependent manner. Specifically, EGCG has no obvious effect on OC formation at 5 μM but can inhibit the formation of TRAP-positive OCs at higher concentrations (10 and 15 μM) (P<0.05). Meanwhile, 5 μM EGCG-GNPs reduced nearly 30% TRAP-positive OC formation (P<0.05), 70% at 10 μM (P<0.01), and 90% at 15 μM (P<0.01), which was much more efficient than the EGCG groups. This result indicates that the GNPs can effectively deliver EGCG into the cells and strongly inhibit OC differentiation of OC precursors.
 |
Figure 4 BMMs were stimulated with 100 ng/mL RANKL for 36 h and another 5 days with 1 μg/mL LPS. Together with the stimulator, various concentrations of EGCG or EGCG-GNPs were added. (A) TRAP assay after treatment with EGCG and EGCG-GNPs. Cells stained purple were TRAP-positive. TRAP-positive OCs containing more than three nuclei were counted and shown in the lower left bar graph. (B) Effects of EGCG and EGCG-GNPs on LPS-induced actin formation in BMMs. Cells were stained with FITC-labeled phalloidin (green) and with DAPI (blue). The number of OCs exhibiting ring-like F-actin sealing zones was quantified in the lower left bar graph. Scale bar =200 μm. The results are mean ± SD of triplicate experiments: *P<0.05, **P<0.01, significant difference compared with the LPS group. Abbreviations: RANKL, acceptor activator of nuclear factor kappa-B ligand; LPS, lipopolysaccharides; TRAP, tartrate-resistant acid phosphatase; OC, osteoclast. |
Actin ring is a unique cytoskeletal structure, allowing for bone resorption.52 The effect of EGCG and EGCG-GNPs on actin ring formation was confirmed by fluorescent staining (Figure 4B). LPS stimulated large actin ring formation, whereas EGCG-GNPs significantly suppressed actin ring formation in a dose-dependent manner. This phenomenon was more obvious than that of the EGCG-treated group. EGCG-GNPs at 15 μM showed almost no actin formation compared with the other groups. These results were consistent with TRAP assay. From these results, we suggested that the EGCG-GNPs can effectively inhibit LPS-induced osteoclastogenesis.
EGCG-GNPs and EGCG strongly attenuated intracellular ROS
ROS plays an important role in LPS-induced OC formation as intracellular signaling molecules.53 We examined intracellular ROS levels by using the ROS Detection Kit (Beyotime Institute of Biotechnology). Flow cytometry analysis using fluorescent ROS probe revealed that LPS-mediated intracellular ROS induction was attenuated by the treatment of 15 μM EGCG or EGCG-GNPs (Figure 5A and B). EGCG-GNPs showed a more potent inhibitory effect on intracellular ROS than EGCG. These results suggested that EGCG-GNPs are better intracellular ROS scavengers than EGCG in LPS-mediated osteoclastogenesis.
 |
Figure 5 Flow cytometry analysis of intracellular ROS in BMMs. (A) LPS-induced intracellular ROS. The data shown are representative of three independent experiments performed in triplicate. (B) Quantitative analysis for percentage of ROS positive in samples. Percentage of ROS-positive population judged by the threshold in A was calculated from triplicated samples. *P<0.05 vs LPS alone, *P<0.05 between samples. Abbreviations: BMM, bone marrow-derived macrophages; LPS, lipopolysaccharides; ROS, reactive oxygen species. |
EGCG and EGCG-GNPs downregulated osteoclastogenic genes and transcriptional regulators
OC activity is closely associated with the LPS-induced mRNA expression levels of OC-related genes, such as MMP9, NFATc1, TRAP, and cathepsin K.54 Our study showed a remarkable increase in the expression of these genes in response to LPS. However, treatment with EGCG and EGCG-GNPs significantly downregulated the expression of those four genes in response to LPS. EGCG and EGCG-GNPs remarkably decreased NFATc1 mRNA expression at nearly all time points (P<0.01) (Figure 6B). For the EGCG group, TRAP and MMP9 mRNA expression was markedly decreased at 24 h (P<0.01), but no change was observed at 12 h (Figure 6C and A). Only EGCG-GNPs have an inhibitory effect on cathepsin K and MMP9 mRNA expression at 12 h (P<0.05) (Figure 6D). In particular, EGCG-GNPs showed the lowest mRNA expression levels for all markers. These results correlated well with the prior results for TRAP-positive multinuclear cell generation.
 |
Figure 6 Gene expression of (A) MMP9, (B) NFATc1, (C) TRAP, and (D) cathepsin K relative to blank (1.0 expression, not shown in the figure) in the presence of EGCG (15 μM) or EGCG-GNPs (15 μM). *P<0.05, **P<0.01, significant difference compared between groups. Abbreviations: LPS, lipopolysaccharides; MMP9, matrix metallopeptidase 9; NFATc1, nuclear factor of activated T-cells cytoplasmic 1; TRAP, tartrate-resistant acid phosphatase. |
EGCG and EGCG-GNPs inhibited LPS-induced signaling pathways
Except for decreased intracellular ROS, the inhibitory mechanism of EGCG-GNPs on OC differentiation was further investigated by Western blot analysis. NF-κB plays a key role in regulating the immune response to infection. RELA, also known as transcription factor p65, is one of the most essential transcription factors of the NF-κB family. The phosphorylation of p65 plays a key role in regulating NF-κB activation and function. Subsequent to NF-κB nuclear translocation, p65 undergoes site-specific post-translational modifications to further enhance the NF-κB function as a transcription factor.55 Sul et al described that GNPs are involved in OC differentiation of BMMs through NF-κB signaling.33 Protein expression levels of p65 and phosphorated p65 (p-p65) in the presence of EGCG and EGCG-GNPs are shown in Figure 7B. Gray-value analysis of p65 and p-p65 revealed that both EGCG and EGCG-GNPs inhibit p65 and p-p65 expression (Figure 7C). The EGCG-GNP group exhibited the lowest protein expression except for the nontreatment group.
 |
Figure 7 MAPK and NF-κB signaling was related to the EGCG-GNP-inhibited OC differentiation. (A and B) Protein expression levels of phosphorated p38, ERK, JNK, and p65 in the presence of EGCG and EGCG-GNPs were detected by Western blot analysis. (C) Four groups of gray-value analysis of p65 and p-p65 protein expression. Abbreviations: LPS, lipopolysaccharides; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; ERK, extracellular regulated protein kinases; JNK, c-Jun N-terminal kinases. |
The activation/inactivation levels of MAPKs (p38, ERK, JNK), which regulate OC differentiation, were measured at 24 h. Active forms of the respective molecules’ expression levels were confirmed by their corresponding phosphorylated-antibodies. The results showed that the EGCG–GNPs exhibited a more substantial decrease in p-p38, p-ERK, and p-JNK than that in EGCG. This indicated that both EGCG and EGCG-GNPs inhibit these signaling pathways by interacting with BMMs (Figure 7A).
Administration of EGCG and EGCG-GNPs protected against LPS-induced mouse calvarial bone erosion in vivo
To further examine the anti-osteoclastogenesis effect and prevention of pathological osteolysis of EGCG-GNPs, we established a mouse calvarial model by injecting LPS subcutaneously over the mouse calvaria for 7 days. All groups, then were injected with EGCG or EGCG-GNPs. According to previous studies of pharmacokinetic behavior of GNPs,56–58 the negative charge EGCG-GNPs were coated by serum proteins (also confirmed by a stability test in 3.2) after entering the blood stream, to facilitate being engulfed by the macrophage (opsonization). These serum coated nanoparticles then entered tissues with a leaky endothelial wall including liver, spleen, bone marrow, tumor, and the inflammation site. Finally, the macrophages from the mononuclear phagocyte system (MPS), such as Kupffer cells in the liver, and osteoclasts in bone, contribute the major sequestration of nanoparticles.59 After 7 days, all groups were analyzed by bone mineral density detection, micro-CT scanning, and 3D reconstruction. LPS greatly decreased the bone densities, but both EGCG and EGCG-GNPs reversed the effect (Figure. 8A and B). From 3D reconstruction and calculation of micro-CT, the thickness of trabecular bone in the EGCG and EGCG-GNPs groups did not show a significant difference and was higher than that of the untreated osteolysis groups (Figure 8D). Furthermore, the bone volume (BV)/total volume (TV) ratio in EGCG-GNPs was 4.6% higher than that in the EGCG groups (Figure 8C). H&E staining showed that calvariae in the EGCG-GNPs groups were infiltrated by fewer inflamed cells compared with other groups. This result, consistent with micro-CT analysis, indicated that EGCG-GNPs had better ability to prevent inflamed cells from infiltrating and to protect bones against the interruption of bone continuity. TRAP and eosin staining identified the increasing number of multinucleated OCs in the LPS-treated calvariae (Figure 8E). These results revealed that LPS-injected calvariae had profound surface erosion. By contrast, calvariae erosion was clearly prevented or restored by EGCG and EGCG-GNPs. BV/TV indicated the fraction of a given volume of interest that is occupied by mineralized bone. BV/TV can be used to evaluate relative changes in BV density following a given treatment.
 |
Figure 8 Administration of EGCG and EGCG-GNPs protected against LPS-induced mouse calvarial bone erosion in vivo. Mice were subcutaneously injected 1 day in advance and then 30 min before the daily LPS injection for 7 days. Finally, the mice were sacrificed, and calvariae were removed for fixation. (A) X-ray images. (B). Micro-CT reconstruction images. (C and D). Microarchitecture parameters assessed by micro-CT (*P<0.05). (E) Calvariae were fixed, decalcified, dehydrated, embedded, and sectioned. H&E staining (10×) and TRAP staining (10×, 40×) were performed for each group: (a) PBS, (b) LPS, (c) EGCG, and (d) EGCG-GNPs. Arrowheads indicate OCs. Abbreviations: LPS, lipopolysaccharides; H&E, haemotoxylin and eosin; TRAP, tartrate-resistant acid phosphatase; PBS, phosphate buffered saline; OC, osteoclast; BV/TV, bone volume over total volume; Tb. Th, trabecular thickness. |
Conclusion
LPS acts as a prototypical endotoxin that binds TLR4 in many cell types, but especially in monocytes, dendritic cells, and macrophages, promoting ROS secretion. MAPKs are involved in directing cellular responses to pro-inflammatory cytokines, which induce NF-κB nuclear translocation and result in the fusion of pre-OCs to mature OCs (Figure 9).
 |
Figure 9 Schematic diagram of functional GNPs covered with EGCG and their possible mechanisms for preventing osteoclastogenesis. Abbreviations: LPS, lipopolysaccharides; CD, cluster of differentiation; TLR, Toll-like receptors; ROS, reactive oxygen species; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; ERK, extracellular regulated protein kinases; JNK, c-Jun N-terminal kinases; NFATc1, nuclear factor of activated T-cells cytoplasmic 1; TRAP, tartrate-resistant acid phosphatase; CTSK, cathepsin K. |
In this study, EGCG was used to chelate with Au3+ ions. It subsequently reduced the chelated Au3+ ions to Au atoms, leading to the formation of EGCG-GNPs, which can be used as an anti-osteoclastogenic drug. DFT calculations indicated that the binding strength of EGCG on the Au surface is much weaker if EGCG adopts the neutral form, which facilitates the release of EGCG in acidic environments. Intracellular uptake analysis showed that EGCG-GNPs can effectively be taken up into the cell and locate into the intracellular compartment. EGCG-GNPs were engulfed by endocytosis. The acidic late endosomes facilitated the release of EGCG. Then, free EGCG inhibited the OC differentiation of BMMs by suppressing intracellular ROS and the related MAPK signaling pathway. EGCG-GNPs showed remarkable stability, significantly rapid cellular uptake, and excellent in vitro antioxidant activities. Furthermore, the in vivo results of an LPS-mediated calvarial bone erosion model showed that EGCG-GNPs improved bone mineral density and prevented bone loss. Our findings suggest that the biofunctional EGCG-GNPs may have significant potential applications for use as therapeutic agents in treating inflammatory osteolysis and other bone diseases associated with excessive bone loss, such as periodontitis, rheumatoid arthritis, and postmenopausal osteoporosis.
Acknowledgments
The authors are grateful for the support by grants from the National Natural Science Foundation of China (Nos. 81371131, 81570973, and 81300872) and Nature Science Foundation of Hubei Province of China (No. 2016CFB250).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Roodman GD. Advances in bone biology: the osteoclast. Endocr Rev. 1996;17(4):308–332. doi:10.1210/edrv-17-4-308
2. Hiyari S, Atti E, Camargo PM, et al. Heritability of periodontal bone loss in mice. J Periodontal Res. 2015;50(6):730–736. doi:10.1111/jre.12258
3. Park EJ, Kim SA, Choi YM, et al. Capric acid inhibits NO production and STAT3 activation during LPS-induced osteoclastogenesis. PLoS One. 2011;6(11):e27739. doi:10.1371/journal.pone.0027739
4. Yuan L, Wu Y, Ren X, Liu Q, Wang J, Liu X. Isoorientin attenuates lipopolysaccharide-induced pro-inflammatory responses through down-regulation of ROS-related MAPK/NF-kappaB signaling pathway in BV-2 microglia. Mol Cell Biochem. 2014;386(1–2):153–165. doi:10.1007/s11010-013-1854-9
5. Srinivasan S, Koenigstein A, Joseph J, et al. Role of mitochondrial reactive oxygen species in osteoclast differentiation. Ann N Y Acad Sci. 2010;1192:245–252. doi:10.1111/j.1749-6632.2009.05377.x
6. Park H, Noh AL, Kang JH, Sim JS, Lee DS, Yim M. Peroxiredoxin II negatively regulates lipopolysaccharide-induced osteoclast formation and bone loss via JNK and STAT3. Antioxid Redox Signal. 2015;22(1):63–77. doi:10.1089/ars.2013.5748
7. Hou GQ, Guo C, Song GH, et al. Lipopolysaccharide (LPS) promotes osteoclast differentiation and activation by enhancing the MAPK pathway and COX-2 expression in RAW264.7 cells. Int J Mol Med. 2013;32(2):503–510. doi:10.3892/ijmm.2013.1406
8. Li L, Khansari A, Shapira L, Graves DT, Amar S. Contribution of interleukin-11 and prostaglandin(s) in lipopolysaccharide-induced bone resorption in vivo. Infect Immun. 2002;70(7):3915–3922. doi:10.1128/iai.70.7.3915-3922.2002
9. Rameshrad M, Razavi BM, Hosseinzadeh H. Protective effects of green tea and its main constituents against natural and chemical toxins: a comprehensive review. Food Chem Toxicol. 2017;100:115–137. doi:10.1016/j.fct.2016.11.035
10. Nakagawa H, Hasumi K, Takami M, et al. Identification of two biologically crucial hydroxyl groups of (-)-epigallocatechin gallate in osteoclast culture. Biochem Pharmacol. 2007;73(1):34–43. doi:10.1016/j.bcp.2006.09.002
11. Yang EJ, Lee J, Lee SY, et al. EGCG attenuates autoimmune arthritis by inhibition of STAT3 and HIF-1alpha with Th17/Treg control. PLoS One. 2014;9(2):e86062. doi:10.1371/journal.pone.0086062
12. Morinobu A, Biao W, Tanaka S, et al. (-)-Epigallocatechin-3-gallate suppresses osteoclast differentiation and ameliorates experimental arthritis in mice. Arthritis Rheum. 2008;58(7):2012–2018. doi:10.1002/art.23594
13. Cho AR, Kim JH, Lee DE, et al. The effect of orally administered epigallocatechin-3-gallate on ligature-induced periodontitis in rats. J Periodontal Res. 2013;48(6):781–789. doi:10.1111/jre.12071
14. Oka Y, Iwai S, Amano H, et al. Tea polyphenols inhibit rat osteoclast formation and differentiation. J Pharmacol Sci. 2012;118(1):55–64.
15. Lin RW, Chen CH, Wang YH, et al. (-)-Epigallocatechin gallate inhibition of osteoclastic differentiation via NF-kappaB. Biochem Biophys Res Commun. 2009;379(4):1033–1037. doi:10.1016/j.bbrc.2009.01.007
16. An J, Hao D, Zhang Q, et al. Natural products for treatment of bone erosive diseases: the effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int Immunopharmacol. 2016;36:118–131. doi:10.1016/j.intimp.2016.04.024
17. Yun JH, Pang EK, Kim CS, et al. Inhibitory effects of green tea polyphenol (-)-epigallocatechin gallate on the expression of matrix metalloproteinase-9 and on the formation of osteoclasts. J Periodontal Res. 2004;39(5):300–307. doi:10.1111/j.1600-0765.2004.00743.x
18. Tominari T, Matsumoto C, Watanabe K, et al. Epigallocatechin gallate (EGCG) suppresses lipopolysaccharide-induced inflammatory bone resorption, and protects against alveolar bone loss in mice. FEBS Open Bio. 2015;5:522–527. doi:10.1016/j.fob.2015.06.003
19. Lee SH, Kim BJ, Choi HJ, et al. (-)-Epigallocathechin-3-gallate, an AMPK activator, decreases ovariectomy-induced bone loss by suppression of bone resorption. Calcif Tissue Int. 2012;90(5):404–410. doi:10.1007/s00223-012-9584-7
20. Jin P, Wu H, Xu G, Zheng L, Zhao J. Epigallocatechin-3-gallate (EGCG) as a pro-osteogenic agent to enhance osteogenic differentiation of mesenchymal stem cells from human bone marrow: an in vitro study. Cell Tissue Res. 2014;356(2):381–390. doi:10.1007/s00441-014-1797-9
21. Choi SY, Song MS, Ryu PD, Lam AT, Joo SW, Lee SY. Gold nanoparticles promote osteogenic differentiation in human adipose-derived mesenchymal stem cells through the Wnt/beta-catenin signaling pathway. Int J Nanomedicine. 2015;10:4383–4392. doi:10.2147/IJN.S78775
22. Lee MJ, Maliakal P, Chen L, et al. Pharmacokinetics of tea catechins after ingestion of green tea and (-)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiol Biomarkers Prev. 2002;11(10 Pt 1):1025–1032.
23. Shukla R, Chanda N, Zambre A, et al. Laminin receptor specific therapeutic gold nanoparticles (198AuNP-EGCg) show efficacy in treating prostate cancer. Proc Natl Acad Sci U S A. 2012;109(31):12426–12431. doi:10.1073/pnas.1121174109
24. Jiang X, Musyanovych A, Röcker C, Landfester K, Mailänder V, Nienhaus GU. Specific effects of surface carboxyl groups on anionic polystyrene particles in their interactions with mesenchymal stem cells. Nanoscale. 2011;3(5):2028. doi:10.1039/c0nr00944j
25. Heo DN, Ko WK, Moon HJ, et al. Inhibition of osteoclast differentiation by gold nanoparticles functionalized with cyclodextrin curcumin complexes. ACS Nano. 2014;8(12):12049–12062. doi:10.1021/nn504329u
26. Tautzenberger A, Kovtun A, Ignatius A. Nanoparticles and their potential for application in bone. Int J Nanomedicine. 2012;7:4545–4557. doi:10.2147/IJN.S34127
27. Granja A, Frias I, Neves AR, Pinheiro M, Reis S. Therapeutic potential of epigallocatechin gallate nanodelivery systems. Biomed Res Int. 2017;2017:5813793. doi:10.1155/2017/5813793
28. Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Adv Drug Deliv Rev. 2008;60(11):1307–1315. doi:10.1016/j.addr.2008.03.016
29. Levy R, Shaheen U, Cesbron Y, See V. Gold nanoparticles delivery in mammalian live cells: a acritical review. Nano Rev. 2010;1(1):4889. doi: 10.3402/nano.v1i0.4889.
30. Tran NTT, Wang TH, Lin CY, Tai Y. Synthesis of methotrexate-conjugated gold nanoparticles with enhanced cancer therapeutic effect. Biochem Eng J. 2013;78:175–180. doi:10.1016/j.bej.2013.04.017
31. Wang F, Wang YC, Dou S, Xiong MH, Sun TM, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. doi:10.1021/nn200007z
32. Conners CM, Bhethanabotla VR, Gupta VK. Concentration-dependent effects of alendronate and pamidronate functionalized gold nanoparticles on osteoclast and osteoblast viability. J Biomed Mater Res B Appl Biomater. 2017;105(1):21–29. doi:10.1002/jbm.b.33527
33. Sul OJ, Kim JC, Kyung TW, et al. Gold nanoparticles inhibited the receptor activator of nuclear factor-kappab ligand (RANKL)-induced osteoclast formation by acting as an antioxidant. Biosci Biotechnol Biochem. 2010;74(11):2209–2213. doi:10.1271/bbb.100375
34. Li JJ, Kawazoe N, Chen G. Gold nanoparticles with different charge and moiety induce differential cell response on mesenchymal stem cell osteogenesis. Biomaterials. 2015;54:226–236. doi:10.1016/j.biomaterials.2015.03.001
35. Han G, Ghosh P, Rotello VM. Functionalized gold nanoparticles for drug delivery. Nanomedicine (Lond). 2007;2(1):113–123. doi:10.2217/17435889.2.1.113
36. Lu YC, Luo PC, Huang CW, Leu YL. Augmented cellular uptake of nanoparticles using tea catechins_ effect of surface modification on nanoparticle–cell interaction. Nanoscale. 2014;6(17):10297–10306. doi:10.1039/c4nr00617h
37. Mukherjee S, Ghosh S, Das DK, et al. Gold-conjugated green tea nanoparticles for enhanced anti-tumor activities and hepatoprotection–synthesis, characterization and in vitro evaluation. J Nutr Biochem. 2015;26(11):1283–1297. doi:10.1016/j.jnutbio.2015.06.003
38. Liang J, Cao L, Zhang L, Wan XC. Preparation, characterization, and in vitro antitumor activity of folate conjugated chitosan coated EGCG nanoparticles. Food Sci Biotechnol. 2014;23(2):569–575. doi:10.1007/s10068-014-0078-4
39. Dehshahri S, Wink M, Afsharypuor S, Asghari G, Mohagheghzadeh A. Antioxidant activity of methanolic leaf extract of moringa peregrina. Fiori Res Pharm Sci. 2012;2(8):111–118.
40. Teobaldi G, Zerbetto F. Adsorption of organic molecules on gold electrodes. J Phys Chem C. 2007;111(37):13879–13885. doi:10.1021/jp074017g
41. Al-Johani H, Abou-Hamad E, Jedidi A, et al. The structure and binding mode of citrate in the stabilization of gold nanoparticles. Nat Chem. 2017;9(9):890–895. doi:10.1038/nchem.2752
42. Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868. doi:10.1103/PhysRevLett.77.3865
43. Delley B. The conductor-like screening model for polymers and surfaces. Mol Simul. 2006;32(2):117–123. doi:10.1080/08927020600589684
44. Delley B. From molecules to solids with the DMol3 approach. J Chem Phys. 2000;113(18):7756–7764. doi:10.1063/1.1316015
45. Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol. 2010;594:57–72. doi:10.1007/978-1-60761-411-1_4
46. Liu S, Zhu L, Zhang J, Yu J, Cheng X, Peng B. Anti-osteoclastogenic activity of isoliquiritigenin via inhibition of NF-kappaB-dependent autophagic pathway. Biochem Pharmacol. 2016;106:82–93. doi:10.1016/j.bcp.2016.03.002
47. Zhang J, Zhu L, Yan P, Peng B. Effect of BioAggregate on receptor activator of nuclear factor-kappa B ligand-induced osteoclastogenesis from murine macrophage cell line in vitro. J Endod. 2015;41(8):1265–1271. doi:10.1016/j.joen.2015.03.021
48. Domingos RF, Baalousha MA, Ju-Nam Y, et al. Characterizing manufactured nanoparticles in the environment: multimethod determination of particle sizes. Environ Sci Technol. 2009;43(19):7277–7284. doi:10.1021/es900249m
49. Cristina M, Paolo M, Gianluca F, et al. A Poloxamer-407 modified liposome encapsulating epigallocatechin-3-gallate in the presence of magnesium: characterization and protective effect against oxidative damage. Int J of Pharmaceutics. 2018;552:225–234. doi:10.1016/j.ijpharm.2018.10.004
50. Li G, Chen Y, Hou Z, et al. Pro-oxidative activities and dose–response relationship of (−)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: a comparative study in vivo and in vitro. Carcinogenesis. 2010;31:902–910. doi:10.1093/carcin/bgq039
51. Khan UA, Hashimi SM, Bakr MM, Forwood MR, Morrison NA. Foreign body giant cells and osteoclasts are TRAP positive, have podosome-belts and both require OC-STAMP for cell fusion. J Cell Biochem. 2013;114(8):1772–1778. doi:10.1002/jcb.24518
52. Lee EJ, Kim JL, Gong JH, Park SH, Kang YH. Inhibition of osteoclast activation by phloretin through disturbing alphavbeta3 integrin-c-Src pathway. Biomed Res Int. 2015;2015:680145.
53. Kanzaki H, Shinohara F, Kajiya M, Kodama T. The Keap1/Nrf2 protein axis plays a role in osteoclast differentiation by regulating intracellular reactive oxygen species signaling. J Biol Chem. 2013;288(32):23009–23020. doi:10.1074/jbc.M113.478545
54. Li L, Sapkota M, Gao M, Choi H, Soh Y, Macrolactin F. Inhibits RANKL-mediated osteoclastogenesis by suppressing Akt, MAPK and NFATc1 pathways and promotes osteoblastogenesis through a BMP-2/smad/Akt/Runx2 signaling pathway. Eur J Pharmacol. 2017;815:202–209. doi:10.1016/j.ejphar.2017.09.015
55. Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734. doi:10.1038/nri910
56. De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials. 2008;29(12):1912–1919. doi:10.1016/j.biomaterials.2007.12.037
57. Terentyuk GS, Maslyakova GN, Suleymanova LV, et al. Circulation and distribution of gold nanoparticles and induced alterations of tissue morphology at intravenous particle delivery. J Biophotonics. 2009;2(5):292–302. doi:10.1002/jbio.200910005
58. Tsoi KM, MacParland SA, Ma XZ, et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15(11):1212–1221. doi:10.1038/nmat4718
59. Michelle L, Choyke PL, Kobayashi H. Clearance properties of nano-sized particles and molecules as imaging agents: considerations and caveats. Nanomedicine. 2008;3(5):703–717. doi:10.2217/17435889.3.5.703
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.