Back to Journals » International Journal of Nanomedicine » Volume 11
Annonaceous acetogenins nanosuspensions stabilized by PCL–PEG block polymer: significantly improved antitumor efficacy
Authors Hong J, Li Y, Li Y, Xiao Y, Kuang H, Wang X
Received 10 March 2016
Accepted for publication 26 May 2016
Published 18 July 2016 Volume 2016:11 Pages 3239—3253
DOI https://doi.org/10.2147/IJN.S108143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jingyi Hong,1,* Yanhong Li,1,2,* Yijing Li,1 Yao Xiao,1,2 Haixue Kuang,2 Xiangtao Wang1
1Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, 2School of Pharmacy, Heilongjiang University of Chinese Medicine, Harbin, People’s Republic of China
*These authors contributed equally to this work
Abstract: Annonaceous acetogenins (ACGs) have shown superior antitumor activity against a variety of cancer cell lines, but their clinical application has been limited by their poor solubility. In this study, ACGs-nanosuspensions (NSps) were successfully prepared by a precipitation ultrasonic method using monomethoxypoly (ethylene glycol)2000–poly (ε-caprolactone)2000 (mPEG2000–PCL2000) as a stabilizer. The resultant ACGs-NSps had a mean particle size of 123.2 nm, a zeta potential of -20.17 mV, and a high drug payload of 73.68%. ACGs-NSps were quite stable in various physiological solutions, and they exhibited sustained drug release. Compared to free drug, ACGs-NSps exhibited stronger cytotoxicity against 4T1, MCF-7, and HeLa cells. An in vivo real-time biodistribution investigation after labeling with 1,1'-dioctadecyltetramethyl indotricarbocyanine iodide, a noninvasive near-infrared fluorescence probe, demonstrated that ACGs-NSps could effectively accumulate in tumor. An in vivo antitumor activity study in 4T1 tumor-bearing mice revealed that ACGs-NSps achieved much better therapeutic efficacy than the traditional dosage form (oil solution) even at 1/10 of the dose (74.83% vs 45.53%, P<0.05), demonstrating that NSp was a good dosage form for ACGs to treat cancer.
Keywords: annonaceous acetogenins, mPEG2000–PCL2000, near–infrared fluorescence, biodistribution, antitumor efficacy
Introduction
Annonaceous acetogenins (ACGs), which are an effective fraction separated from the seeds of Annonaceae species,1–3 have attracted much attention over the past 3 decades due to their strong antitumor activity and broad antineoplastic spectrum.4–6 For example, bullatacin, the major component from ACGs, displayed 104–105 times the antitumor activity of doxorubin against both the A549 (human lung carcinoma cells) and the MCF-7 (human breast cancer cells)7 cell lines and 300 times the activity of taxol in L1210 (murine leukemia cells)-bearing mice.8 The three bis-tetrahydrofuran compounds from Annona squamosa seeds showed 50% inhibitory concentration (IC50) values that were one to two orders of magnitude less than fluorouracil against SMMC 7721 (human hepatocellular carcinoma), HeLa (human cervix carcinoma), MKN-45 (human gastric cancer), and HepG2 (human hepatocellular carcinoma) cell lines.9 It was demonstrated that ACGs exerted antitumor effects via inhibition of mitochondrial complex I (NADH:ubiquinone oxidoreductase) of the electron transport chain,10,11 which might favor reversing multiple drug resistance in many tumor cell lines because multiple drug resistance is an energy-consuming process.12
In vivo tests in mice showed the antitumor effects and even some possible adverse effects of ACGs.5 It was reported that bullatacin led to liver and kidney toxicity by increasing calcium concentrations, reactive oxygen species production, and Bax expression, together with the Bax/Bcl-2 ratio in rats.13 An acute toxicity study indicated that the oral median lethal dose of ACGs for mice was 19.21 mg/kg, which was approximately five times the highest effective dose in an antitumor experimental study (4 mg/kg).14 Because of the relatively strong toxicity of ACGs, it is important to enhance drug accumulation in the tumor tissue using targeted drug delivery technology to reduce the dose and toxic effects.
However, ACGs are composed of a series of components that have similar chemical structures, such as long aliphatic chains of 35–37 carbons, an α,β-unsaturated γ-lactone ring, and 0–3 tetrahydrofuran ring(s).15–17 As long chains of aliphatic derivatives, ACGs are highly insoluble (<1 μg/mL), resulting in great difficulty in drug delivery and further study. Moreover, ACGs are mixtures, and none of their components have characteristic ultraviolet or fluorescent absorption, which would cause difficulty in formulation preparation and the quantitative analysis of ACGs in vitro and in vivo. These findings might partially explain why there have been only limited reports so far in terms of ACGs drug delivery, such as dropping pills,18 solid dispersions,19 and lipid emulsions.20 However, the drug-loading capacity of all these drug carriers was low, and there have been no further in vivo investigations. To date, nearly all the in vivo studies used oral administration of medication in the very primitive ACGs formulation, such as physical suspensions or oil solutions. No physiologically accepted dosage form for intravenous (iv) injection has been reported thus far.
For hydrophobic antitumor drug candidates, the solubility, bioavailability, and dissolution rate are important parameters for achieving in vivo efficacy. Nanoscale drug delivery systems are usually preferred when considering the dosage form, due to their increased dissolution rates and enhanced bioavailability.21 Nanosuspensions (NSps), which are well known for their large, specific surfaces and very high drug-loading capacity, seem to be a good option. They consist of nearly pure drug, and they include only small amounts of surfactants or polymeric materials for stabilization.22 By reducing drug particle size to nanometer size, the total effective surface area is increased, allowing for greater interaction with the solvent; thus, the dissolution rate would be enhanced. As a result, the active compound can reach the maximum plasma level more quickly. Moreover, NSps could also modify the pharmacokinetic profiles of drugs and thus improve their safety and efficacy.23 The reduced particle size allows for the possibility of iv administration of poorly soluble drugs without any blockade of the blood capillaries. Bullatacin, which is the major component in ACGs, was selected as an indicator for quantitative analysis to evaluate the drug-loading capacity and overall release performance of ACGs nanosuspensions (ACGs-NSps). To explore their in vivo performance, 1,1′-dioctadecyltetramethyl indotricarbocyanine iodide (DiR), a hydrophobic, near-infrared fluorescent probe, was co-encapsulated into the hydrophobic interior cores of ACGs-NSps to trace their dynamic biodistribution in tumor model mice with the aid of in vivo imaging techniques.
Materials and methods
Materials
ACGs were provided as a gift by Huang’s laboratory (Institute of Medicinal Plant Development, Beijing, People’s Republic of China). Monomethoxypoly (ethylene glycol)2000–poly (ε-caprolactone)2000 (mPEG2000–PCL2000) was purchased from Jinan Daigang Biomaterial Co., Ltd, Jinan, People’s Republic of China. Float-A-Lyzer® G2 dialysis cassette was obtained from Thermo Fisher Scientific (Waltham, MA, USA).The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was provided by Sigma-Aldrich Co., (St Louis, MO, USA). DiR was purchased from AAT Bioquest Inc. (Sunnyvale, CA, USA). Hydroxycamptothecin (HCPT) injections were supplied by Shenghe Pharm Ltd, Szechwan, People’s Republic of China. RPMI 1640 was developed by Roswell Park Memorial Institute (hence the acronym RPMI 1640); penicillin–streptomycin and fetal bovine serum were purchased from Thermo Fisher Scientific. All the other organic solvents and chemicals were of the highest commercially available grade. The water used in the experiments was deionized.
Animals and cell lines
Female Balb/c mice (6–8 weeks old, 20±2 g) were purchased from Vital River Laboratory Animal Technology Co., Ltd, Beijing, People’s Republic of China. All the animal experiments were performed in accordance with the Ethical and Regulatory Guidelines for Animal Experiments as defined by the Institute of Medicinal Plant Development, People’s Republic of China. The ethics committee of the Institute of Medicinal Plant Development (Beijing, People’s Republic of China) granted ethical approval for this study. The animals were acclimated at 25°C and a relative humidity of 70%±5% under 12 hours light–dark cycle conditions for 1 week prior to the experiments. The 4T1 (murine mammary carcinoma), HeLa (cervix carcinoma), and MCF-7 (breast carcinoma) cell lines were provided by Cell Culture Center, Institute of Basic Medical Sciences (Beijing, People’s Republic of China) and were cultured with RPMI 1640 medium (Thermo Fisher Scientific) containing 10% fetal calf serum (Thermo Fisher Scientific), penicillin (100 U/mL), and streptomycin (100 U/mL) at 37°C with 5% CO2 (Sanyo, Osaka, Japan).
Preparation of ACGs-NSps
ACGs-NSps were prepared via a precipitation–ultrasonication method with modification.24 Briefly, ACGs and mPEG2000–PCL2000 were co-dissolved in acetone, forming a mixed solution containing 18 mg/mL ACGs and 6 mg/mL mPEG2000–PCL2000. Then, 0.5 mL of the mixed solution was injected dropwise into 4 mL of distilled water at 28°C±2°C with ultrasonication at 250 W for 10 minutes (Kun Shan Ultrasonic Instruments Co., Ltd, Kunshan, People’s Republic of China), followed by evaporation of acetone under vacuum at 40°C until no organic solvent remained. The resultant ACGs-NSps were directly used for subsequent studies. To determine the concentration of ACGs-NSps, samples were dissolved in ninefold volumes of acetonitrile for the disintegration of NSps and then were filtered through a 0.22 μm filter before high-performance liquid chromatography (HPLC) analysis.
Fluorescent labeling of ACGs-NSps
DiR, a hydrophobic, near-infrared fluorescent dye, was incorporated into the hydrophobic cores of ACGs-NSps to trace their in vivo performance, such as their biodistribution. Briefly, 0.5 mL of DiR/acetone solution (0.45 mg/mL) was mixed with 0.5 mL ACGs (18 mg/mL, DiR:ACGs =1:40, weight ratio), was co-dissolved with mPEG2000–PCL2000 in acetone, and then was slowly injected into distilled water to prepare NSps as described earlier.
Size and morphology of ACGs-NSps
The mean particle size, polydispersity index (PDI), and zeta potential of ACGs-NSps were measured by dynamic light scattering (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) at 25°C. Each sample was measured in triplicate with 12 scans each.
The morphology of ACGs-NSps was observed using a JEM-1400 electron microscope (JEOL, Tokyo, Japan). One drop of the ACGs-NSps was placed on a 300-mesh copper grid, air-dried, and negatively stained with 2% (w/v) uranyl acetate for electron microscope observation.
The stability of ACGs-NSps
Stability of ACGs-NSps in various physiological solutions
ACGs-NSps (2 mg/mL) were mixed (1:1, v/v) with 1.8% NaCl, 10% glucose, and phosphate-buffered solution (PBS) (pH 7.4), respectively, to obtain an isotonic solution and then were incubated at 37°C. At specific time intervals, a 1 mL sample was removed and analyzed for size changes and particle distribution. The concentration of ACGs-NSps was determined by the HPLC method as described earlier. Each experiment was performed in triplicate.
Stability of ACGs-NSps in rat plasma
To study whether plasma components, such as enzymes and serum albumin, could interact with ACGs-NSps and induce aggregation, in vitro plasma stability research was conducted as follows: 1.0 mg/mL, 0.5 mg/mL, and 0.25 mg/mL ACGs-NSps (the concentration was determined by HPLC analysis and then diluted to the required concentration using normal saline) were mixed with rat plasma (1:4, v/v) and incubated at 37°C. At each time interval, a 1 mL sample was withdrawn and analyzed for size changes and particle distribution. Each experiment was performed in triplicate.
Chromatographic conditions for HPLC analysis of ACGs and DiR
The content of ACGs was determined by HPLC using bullatacin, which is a major component of ACGs, as an indicator. The HPLC system (Dionex Ultimate 3000, Sunnyvale, CA, USA) was equipped with an autosampler, and chromatographic separation was performed using a Symmetry C18 column (4.6×250 mm, 5 μm; Waters Corporation, Milford, MA, USA) at 30°C. The mobile phase was composed of acetonitrile and water (70/30, v/v) and ran at a flow rate of 1 mL/min. The detection wavelength was set at 210 nm (UV detector, Dionex).
For DiR analysis, the same HPLC system was used together with a fluorescence detector (Dionex) set at excitation wavelength =748 nm and emission wavelength =780 nm. The mobile phase was a mixture of acetonitrile and water (90:10, v/v) with a flow rate of 1.0 mL/min at 25°C.
In vitro drug release behavior
ACGs/DiR-NSps (4 mL, 1 mg/mL) were placed in Float-A-Lyzer dialysis cassettes (molecular weight cutoff 20 kDa; Spectrum Labs, Rancho Dominguez, CA, USA), immersed into 2 L of PBS (pH 7.4, 0.1 mol/L), and incubated at 37°C with stirring (100 rpm). ACGs physical suspensions (4 mg of ACGs were directly dispersed in 4 mL of water at 37°C with ultrasonication at 250 W for 15 minutes) were used as a control and were treated under the same condition. At specific time intervals, 50 μL samples were withdrawn from the cassettes, and the cassettes were replenished with 50 μL of fresh PBS solution. The cumulative release of both ACGs/DiR-NSps and ACGs physical suspensions was estimated by the reduction of the quantity inside the dialysis bag with the HPLC method. Briefly, the withdrawn sample was dissolved in 450 μL of acetonitrile and then was filtered through a 0.22 μm filter before HPLC analysis for the amount of ACGs and DiR. The release medium was replaced every 24 hours to ensure sink conditions. All the assays were performed in triplicate.
In vitro cytotoxicity assay
The cytotoxicity of ACGs-NSps was measured using the MTT assay against the 4T1, HeLa, and MCF-7 cell lines. Typically, 150 μL of cells (5.3×104 cells/mL) were seeded in a 96-well plate and maintained overnight at 37°C in 5% CO2. The cells were then incubated with ACGs-NSps and free ACGs solution (dissolved in dimethyl sulfoxide [DMSO], further diluted with culture medium, final concentration of DMSO ≤0.1%) at serial concentrations for 24 hours, using blank RPMI 1640 as the negative control. In addition, the cytotoxicity of mPEG2000–PCL2000 was also investigated (0.5 mL of mPEG2000–PCL2000/acetone solution [6 mg/mL] was added dropwise into 4 mL of water followed by the evaporation of acetone). Then the blank polymers were diluted to the 100 μg/mL, 10 μg/mL, and 1 μg/mL concentrations using culture medium. After that, the cells were treated with 20 μL of MTT solution (5 mg/mL in PBS) for another 4 hours. Finally, the medium was removed, and 150 μL of DMSO was added to each well to dissolve formazan crystals. The maximum absorbance was detected at 570 nm using an ELISA plate reader (Biotek, Winooski, VT, USA).
Cell inhibition rate (%) = (1−ODt/ODc) ×100% | (1) |
where optical density (OD)t is the mean OD of tested group and ODc is the mean OD of control group.
The IC50 value for ACGs-NSps was determined using GraphPad Prism software, Version 5 (GraphPad Software, Inc., La Jolla, CA, USA), by the sigmoidal dose–response variable curve-fitting method.
In vivo biodistribution study
Female Balb/c nude mice at 6 weeks of age were injected subcutaneously in the right armpit with 0.2 mL of 4T1 cells (1.0×107 cells/mL). When the size of tumors reached 300 mm3, the 4T1 tumor-bearing mice were randomly divided into three groups (five mice per group) and were intravenously injected with free DiR “solution” (0.25 mg DiR was dissolved in 0.5 mL of DMSO and diluted with saline to 10 mL before use), ACGs/DiR “solution” (10 mg of ACGs and 0.25 mg of DiR were dissolved in 1.0 mL of DMSO/Tween 80 [1:1, v/v] mixed solution and then diluted to 10 mL with saline before use), and ACGs/DiR-NSps, respectively. During the experiments, the whole-body fluorescence images were recorded at 0.5, 1, 2, 4, 8, 12, 24, 48, 72, and 96 hours postdose using the IVIS Living Image® 4.4 (Caliper Life Sciences, Hopkinton, MA, USA). At the end of the experiments, the mice were sacrificed, and their organs (heart, liver, spleen, lung, kidney, brain, and tumor) were excised and imaged. Living Image software, Version 4.2, was used for quantitative analysis. All the mice were imaged with identical instrument settings, and all the groups at each time point were under the same scale bar. The distribution of DiR in the tumor was quantified by average radiant efficiency and total photons per second per square centimeter per steradian in their radiance range (microwatts per square centimeter): [ps−1·cm−2·sr−1]/[μW·cm−2].25
In vivo antitumor activity in 4T1 tumor-bearing mice
The antitumor effect of ACGs-NSps was tested in 4T1 tumor-bearing mice. At the beginning of the experiment, 4T1 cells suspended in 0.2 mL of PBS (1.0×107 cells/mL) were injected subcutaneously into the right armpit of the mice under sterile conditions. When the tumor reached 300 mm3, the 4T1 tumor-bearing mice were randomly divided into five groups (ten mice per group) and administered ACGs-NSps (0.4 mg/kg, iv), ACGs “solution” (10 mg of ACGs dissolved in 1.0 mL of DMSO/Tween 80 [1:1, v/v] mixed solution and then diluted to 10 mL with saline before use, 0.4 mg/kg, iv), and ACGs oil solution (dissolved in soybean oil, 4 mg/kg, intragastric) every 2 days. HCPT injection (5 mg/kg) was administered intravenously via the tail vein as a positive control and saline only as the negative control. During the whole process of experimentation, the body weight and the volume of tumors were monitored every day. The mice were sacrificed after 15 days of treatment. The tumors were harvested and weighed. The in vivo tumor inhibition ratio (TIR%) was calculated as follows:
TIR% = (1−Wt/Wn) ×100% | (2) |
where Wn is the average tumor weight of negative control group and Wt is the average tumor weight of the tested group.
Statistical analysis
The statistical analysis among the experimental groups was performed using the independent-samples t-test and IBM SPSS Statistics software, Version 19 (IBM Corporation, Armonk, NY, USA). P-value <0.05 was considered statistically significant.
Results and discussion
Preparation of ACGs-NSps
Screening for the proper stabilizer
It is well known that the stabilizer plays a key role in the successful preparation of NSps. Generally, all the amphiphilic molecules are potential stabilizers. Bovine serum albumin, due to its specific binding ability to various hydrophobic drugs, also has a stabilizing effect on NSps.24 In this study, Tween 80, soybean lecithin, bovine serum albumin, D-α-tocopherol polyethylene glycol 1000 succinate, and mPEG2000–PCL2000 were first tried in our trial test (drug:stabilizer =1:1, weight ratio). The results demonstrated that all of them could stabilize the resultant ACGs-NSps with small size and relatively high drug payloads well (Figure 1A). However, ACGs-NSps prepared using mPEG2000–PCL2000 had the smallest diameter and the highest zeta potential (absolute value), suggesting better storage stability of aqueous NSps.26 mPEG–PCL with different molecular weights of block copolymer was also investigated to create ACGs-NSps in our preliminary experiment (Table S1), and it was demonstrated that mPEG2000–PCL2000 resulted in the smallest size. Therefore, mPEG2000–PCL2000 was selected as the optimal stabilizer in this study, perhaps because the hydrophobic moiety of mPEG2000–PCL2000 is most compatible with ACGs. ACGs are made up of a series of active components that have long aliphatic chains containing 35–37 carbon atoms, quite similar to PCL2000 moiety in structure.27 At the same time, the mPEG2000 section extended outward into the aqueous environment, forming a layer of hydrophilic shell to provide steric stabilization due to its long hydrophilic chain.
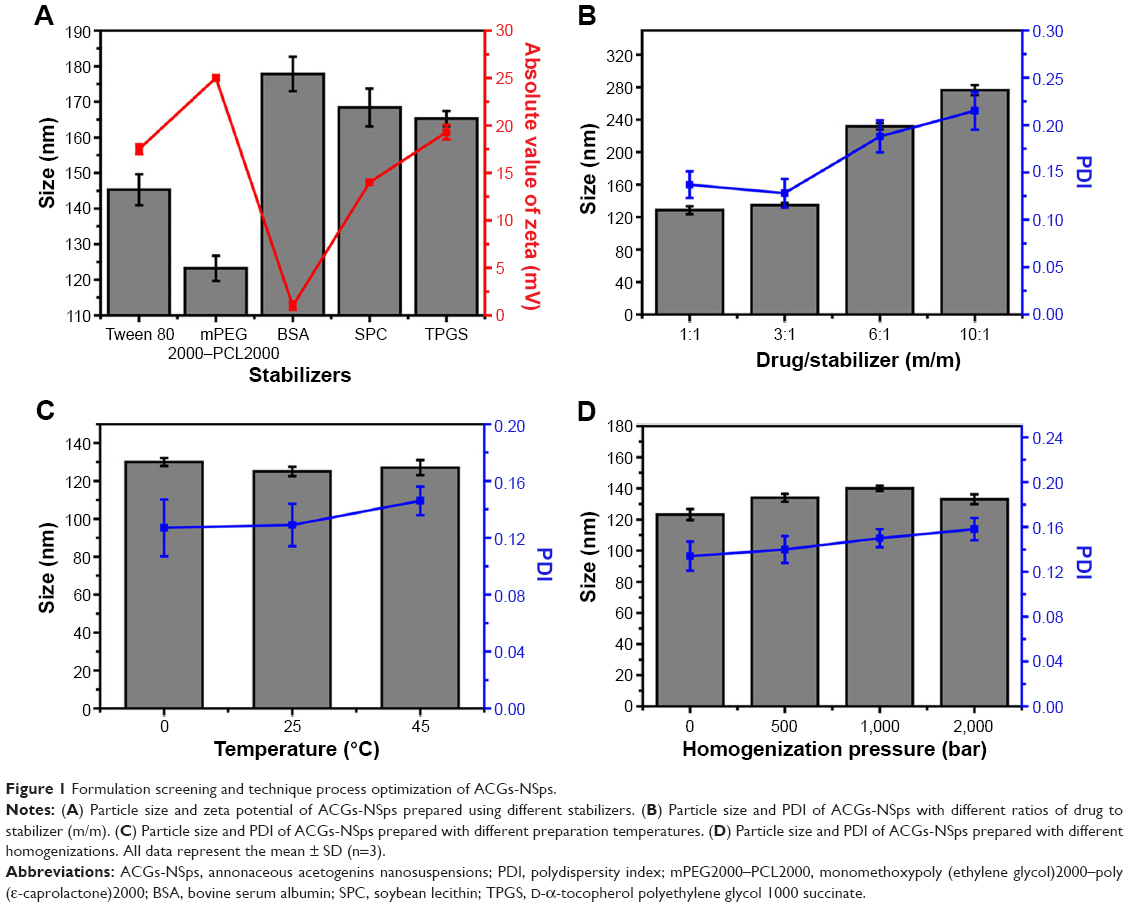 | Figure 1 Formulation screening and technique process optimization of ACGs-NSps. |
Formulation optimization
To increase the drug payload while maintaining the small size and stability, different drug–stabilizer ratios (1:1, 3:1, 6:1, 10:1, weight ratio) were also investigated for the preparation of ACGs-NSps. In our study, the preparations of these four drug–stabilizer ratios were successful. The actual drug payloads of ACGs-NSps of these four drug–stabilizer ratios (1:1, 3:1, 6:1, 10:1) were determined by HPLC to be 48.9%, 73.7%, 82.3%, and 86.6%, respectively. As seen in Figure 1B, there was little difference in the size of the obtained ACGs-NSps when the drug–stabilizer ratio was increased from 1:1 to 3:1. In this case, the amount of stabilizer was sufficient, compared to the amount of hydrophobic drug. However, when the amount of stabilizer was not sufficient to stabilize the drug well, the resultant NSps would enlarge in particle size to accommodate the relative remnant drugs.28 Therefore, the particle size of ACGs-NSps increased significantly when the drug–stabilizer ratio was elevated to 6:1 and 10:1. Larger particles tended to aggregate to a greater extent than smaller particles, thereby resulting in sedimentation with a large PDI value and a decrease in homogeneity.29 As a result, a drug–stabilizer ratio of 3:1 was chosen for the subsequent preparation of ACGs-NSps.
Technique process optimization
Based on our experience, the preparation method, temperature, and homogenization could have effects on the size and stability of the resultant NSp prepared using an antisolvent microprecipitation method.30 In the preparation method, the dropwise method was selected but not the jet stream because our preliminary experiment demonstrated that the jet stream method would lead to a large particle size with a large PDI value or precipitation due to nonuniform nucleus formation and fast crystal growth. The slow speed of the dropwise method could guarantee sufficient drug supersaturation and uniform nucleus formation.23 The investigation showed that in this case stirring was not suitable due to the very large particle size and the tendency to precipitate overnight of the resultant NSps (276.4±6.7 nm; PDI: 0.214±0.129). Compared to stirring, ultrasonication was more effective in controlling the process of nucleation and crystallization. Further, ultrasound irradiation helps to intensify mass transfer and accelerate molecular diffusion.31 Different sonication temperatures (0°C, 25°C, and 45°C) exhibited insignificant effects on the size change of the resultant ACGs-NSps (Figure 1C). In consideration of convenience, 25°C, which is close to room temperature, was chosen as the final preparation temperature. In addition, it was also found that homogenization failed to decrease the size of ACGs-NSps (Figure 1D), perhaps because the average diameter of the obtained ACGs-NSps was already small (123.2±3.5 nm), and the homogenization energy was not sufficient to break down the obtained particles further.21
Characterization of ACGs-NSps
Antisolvent precipitation is a simple but quite effective approach to prepare NSps of poorly water-soluble drugs. When the solution of drug and polymer in acetone was dropped into distilled water, a colloidal system with blue opalescence was obtained with no turbidity or observed precipitation. As shown in Table 1, the average diameter of the obtained ACGs-NSps was 123.2±3.5 nm (also shown in Figure 2A) with a small PDI value of 0.134±0.013 and the zeta potential was −20.17±3.12 mV. For the in vivo tracing of intravenously injected ACGs-NSps, DiR, a hydrophobic near-infrared fluorescent dye, was incorporated into the hydrophobic core of ACGs-NSps. The resultant ACGs/DiR-NSps were 157.9±4.83 nm in mean diameter, slightly larger than that of ACGs-NSps (Table 1; Figure 2B). Transmission electron microscopy observation (Figure 2C and D) revealed that ACGs-NSps were regular, spherical, and uniform in shape. Their size was in good agreement with that determined by dynamic light scattering.
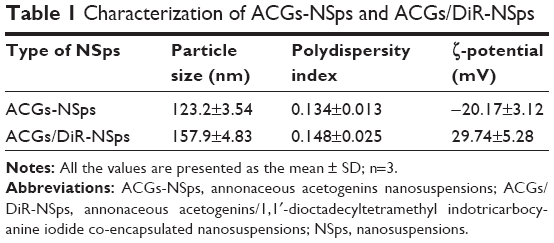 | Table 1 Characterization of ACGs-NSps and ACGs/DiR-NSps |
 | Figure 2 The particle size distribution and morphology of ACGs-NSps. |
The obtained ACGs-NSps were quite stable in PBS, normal saline, and 5% glucose (Table 2) after incubation at 37°C for 12 hours, despite a relatively larger size than that in deionized water. Therefore, the dispersion medium for ACGs-NSps and ACGs/DiR-NSps was adjusted to an isotonic solution using sodium chloride before iv administration. When different concentrations of ACGs-NSps (1 mg/mL, 0.5 mg/mL, and 0.25 mg/mL) were incubated with plasma, the particle size change of ACGs-NSps showed concentration dependence (Table 3). None of these three concentrations of ACGs-NSps displayed significant particle size enlargement or aggregation within 5 hours. But lower concentration showed smaller size with the extension of incubation time. For 0.25 mg/mL ACGs-NSps, the average size decreased from beginning at 154.8 nm to 105.6 nm after 12 hours and 92.3 nm after 24 hours. This demonstrated that the size of ACGs-NSps would not significantly increase when entering the blood circulation due to dilution by blood. Therefore, the obtained ACGs-NSps were suitable for iv administration. The zeta potential of ACGs/DiR-NSps was positive (29.74±5.28 mV) in water, but it would quickly reverse the surface charge to negative when incubated with rat plasma at 37 C, which could be attributed to the absorption of serum proteins (Table S2).
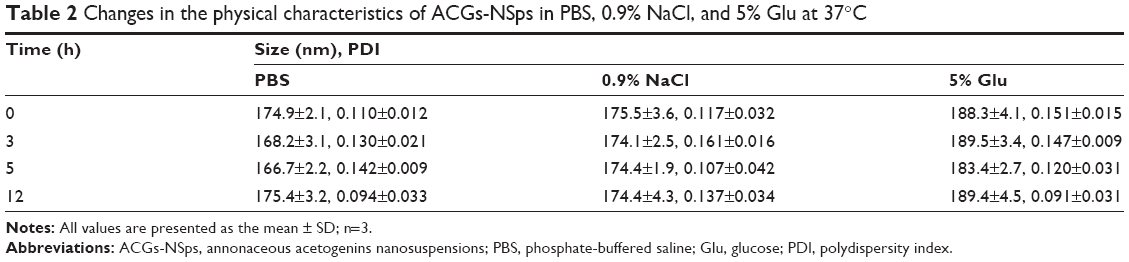 | Table 2 Changes in the physical characteristics of ACGs-NSps in PBS, 0.9% NaCl, and 5% Glu at 37°C |
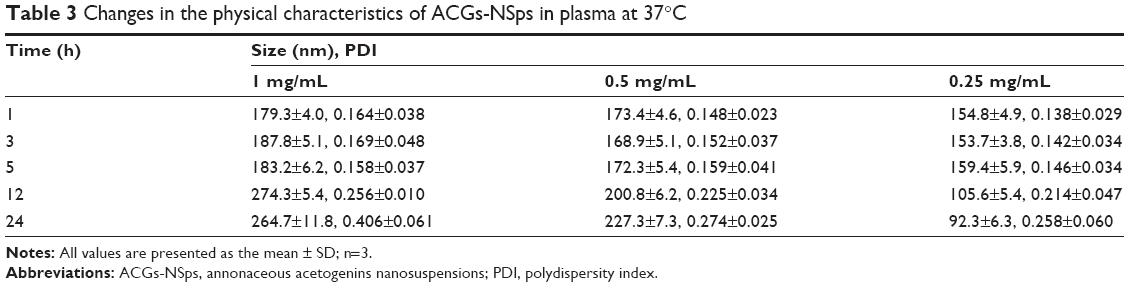 | Table 3 Changes in the physical characteristics of ACGs-NSps in plasma at 37°C |
In vitro release behavior of ACGs/DiR-NSps
The in vitro drug release from ACGs/DiR-NSps was investigated using the dialysis bag diffusion method. Bullatacin, a bis-tetrahydrofuran ACG which was the most abundant compound in ACGs (Figure S1), was used as an indicator of quantitative analysis to assess the overall release performance of ACGs from NSps, while DiR was also quantified by HPLC to analyze its release behavior from NSps.
The cumulative dissolution profiles are presented in Figure 3. It was clear that no significant burst release of drug was observed. Only 10.32% of the encapsulated drugs released over 2 hours, and the cumulative drug release reached 20.65% at 24 hours. Then, the release of ACGs-NSps followed nearly zero-order kinetics, with a steady and persistent drug release up to 80.98% by 96 hours. We also used ACGs physical suspensions (dispersed directly in deionized water) as a control and treated them under the same conditions. However, the solubility of ACGs is so poor that no drug release was detected in the dialysate outside the dialysis bag, and no reduction was detected inside the dialysis bag during the whole process of trial (data not shown). These findings demonstrated that no drug was released from ACGs physical suspensions under the same conditions. In addition, the release kinetics of DiR from ACGs/DiR-NSps was also investigated. DiR was gradually released from NSps with a half-life at ~60 hours. After 96 hours, 78.97% of DiR had been released from ACGs/DiR-NSps. This cumulative release profile of DiR was similar to that of ACGs, indicating that ACGs and DiR were released simultaneously from the NSps.
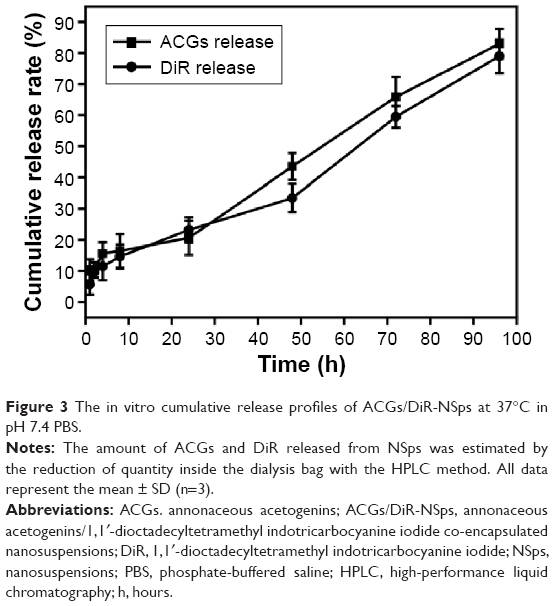 | Figure 3 The in vitro cumulative release profiles of ACGs/DiR-NSps at 37°C in pH 7.4 PBS. |
In vitro cytotoxicity assay
We analyzed the cytocompatibility of blank mPEG2000–PCL2000 toward 4T1, HeLa, and MCF-7 cells after 24 hours incubation by MTT assay. As seen in Figure 4A, blank polymer of mPEG2000–PCL2000 did not exhibit any noticeable toxicity in the tested concentration range. These results indicated that mPEG2000–PCL2000 was biocompatible and could provide a safe drug delivery system.32
 | Figure 4 The in vitro antiproliferative activity of ACGs-NSps and ACGs solution. |
The in vitro cytotoxicity of ACGs-NSps and free ACGs solution against 4T1, HeLa, and MCF-7 cells lines is shown in Figure 4B–D. It was obvious that both ACGs-NSps and ACGs solution inhibited the growth of cells in a dose-dependent manner. The results indicated that, at the same concentration of drug, ACGs-NSps could inhibit the proliferation of these three cancer cell lines more effectively. Data from Table 4 displayed, according to the IC50 values, that ACGs-NSps were 1.70, 2.12, and 2.65 times more effective than ACGs solution against 4T1, HeLa, and MCF-7, respectively. Remarkably, the IC50 values of ACGs-NSps in this study were much lower than the values reported thus far for diverse ACG compounds against a series of cancer cells,33–35 suggesting that NSps might be a good drug delivery system for ACGs for further anticancer research and therapy.
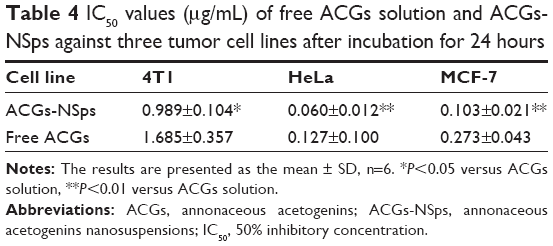 | Table 4 IC50 values (μg/mL) of free ACGs solution and ACGs-NSps against three tumor cell lines after incubation for 24 hours |
In vivo biodistribution study
Because ACGs were multiple-component effective parts obtained from natural resources and none of their components had characteristic ultraviolet or fluorescent absorption, it was almost impossible to trace the in vivo distribution of ACGs-NSps. In theory, determination of the major component (such as bullatacin) as an indicator using liquid chromatography–mass spectrometry might be a feasible means to monitor the biodistribution of ACGs after administering a high dose to experimental animals. However, as an antitumor agent, ACGs are so toxic that most of the animals would die when administered a high dose. If a relatively safe dose was administered, the drug in plasma would be undetectable after administration, not to mention the drug detection in the tumor and other organs. To solve this problem, a hydrophobic near-infrared fluorescence probe (DiR) was co-encapsulated with the ACGs for imaging to guide biodistribution. In our experiment, the in vitro release profile of DiR was simultaneous with that of ACGs (Figure 3). The synchronous release of ACGs and DiR indicated that DiR could be applied to trace the in vivo distribution behavior of ACGs.
Figure 5 demonstrated the in vivo fluorescence intensity of mice at different time points after iv administration of DiR “solution”, ACGs/DiR-NSps, and ACGs/DiR “solution” (all with the same dose of DiR). ACGs/DiR-NSps were mainly concentrated in the liver and spleen within the first 2 hours (Figure 5A). Then, increasing fluorescence began to accumulate in the tumor and reached the maximum at 48 hours; correspondingly, the fluorescence in the liver gradually receded and reached almost a similar level to that in the tumor, perhaps contributing to the slow drug (DiR) release from ACGs/DiR-NSps (Figure 3), and the temporarily trapped ACGs/DiR-NSps in the liver or spleen might be transported back into the blood circulation36,37 and then accumulate in the tumor through the enhanced permeability and retention effects.38
 | Figure 5 The in vivo biodistribution of DiR solution, ACGs/DiR-NSps, and ACGs/DiR solution in 4T1 tumor-bearing mice after intravenous administration. |
ACGs/DiR “solution” was first prepared to be used only as a control in this study because similar formulations and methods were commonly used in preparing solutions for hydrophobic drugs in pharmacological experimentation. However, as shown in Figure 5A, the biodistribution of ACGs/DiR “solution” was similar to that of ACGs/DiR-NSps, which were first concentrated in the liver and then transferred to the tumor. Moreover, the fluorescence density of ACGs/DiR “solution” in the tumor was equivalent to that in the liver at 8–12 hours and then exceeded that in the liver. ACGs/DiR “solution” mainly concentrated in the tumor with significantly higher fluorescence density than other tissues after the 24th hour (Figure 5A and C), and the elimination of ACGs/DiR “solution” accumulated in the tumor was much slower than that of ACGs/DiR-NSps (Figure 5B), resulting in a greater retention in the tumor at the end of the trial (Figure 5D). All these findings demonstrated that ACGs/DiR “solution” had even better tumor-targeted ability than ACGs/DiR-NSps.
Subsequent particle size analysis showed that ACGs/DiR “solution” was not a real solution but hybrid nanoparticles with a mean diameter smaller than 20 nm (Figure S2), which is much smaller than that of ACGs/DiR-NSps (157.9±4.83 nm; Table 1). As reported,39,40 smaller particles have better ability to escape the capture of reticuloendothelial cells, penetrating deep tumor tissues and residing in the tumor for a longer time. However, there is a lower size limit for this effect. It was reported that, in most cases, small nanoparticles ~20 nm in sizes showed fast clearance from tumor, leading to low tumor retention.31 The high tumor retention of ACGs/DiR “solution” (~20 nm) compared to ACGs/DiR-NSps (~157 nm) in our study was somewhat beyond expectation. Therefore, the in vivo therapeutic efficacy in tumor-bearing mice was investigated subsequently.
Free DiR “solution”, which was prepared by dissolving 0.25 mg DiR in 0.5 mL of DMSO and then diluting it with saline to 10 mL, although it resembled a clear solution, was proved to contain DiR nanoparticles (128.30±2.12 nm; Figure S3). However, there was little DiR “solution” accumulated in the tumor (Figure 5A), which was quite different from that of ACGs/DiR-NSps and ACGs/DiR “solution”. Considering that the percentage of DiR was very low in ACGs/DiR-NSps and ACGs/DiR “solution” (ACGs:DiR =40:1, weight ratio), it is reasonable to deduce that the fluorescence accumulation of ACGs/DiR-NSps and ACGs/DiR “solution” in the tumor was mainly due to the outcome of the in vivo biodistribution of ACGs-NSps and ACGs “solution”. The relatively high fluorescence density in the liver for all three of these formulations could be ascribed to the uptake of NSps by macrophages of the reticuloendothelial system, particularly the Kupffer cells in the liver.41
In vivo antitumor efficacy
The antitumor efficacy of different formulations of ACGs was investigated in 4T1 tumor-bearing mice using HCPT injection as a positive control and normal saline as a negative control. As shown in Figure 6A, significant tumor growth inhibition was displayed in all mice receiving ACGs. The tumor volume in the saline control group increased rapidly and reached ~3,000 mm3 at the end of the trial. ACGs-NSps (0.4 mg/kg) displayed limited tumor growth (1,400 mm3), which was even less than that of the positive control (5 mg/kg HCPT injections, 2,000 mm3). Table 5 also showed that ACGs-NSps were superior to HCPT injections regarding tumor inhibition rate (74.83% vs 49.32%, P<0.05). Even oral administration of ACGs oil solution (4 mg/kg) exhibited nearly the same level of antitumor activity as iv administration of HCPT injections (5 mg/kg) (Table 2; 45.53% vs 49.32%, P>0.05). From this point of view, ACGs were more potent than the marketed HCPT injections. The average weight of the mice receiving ACGs-NSps increased slightly, showing good tolerance and less harm (Figure 6B).
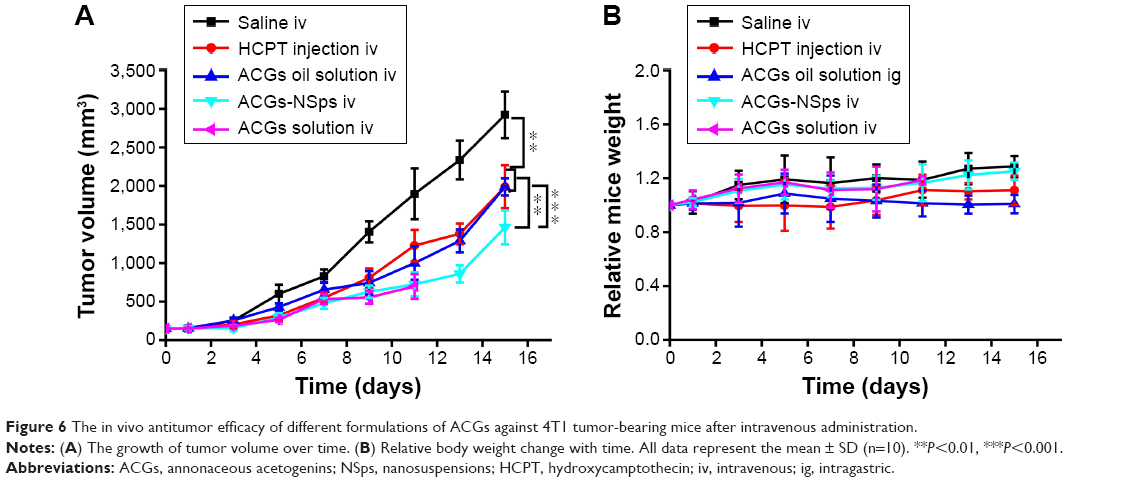 | Figure 6 The in vivo antitumor efficacy of different formulations of ACGs against 4T1 tumor-bearing mice after intravenous administration. |
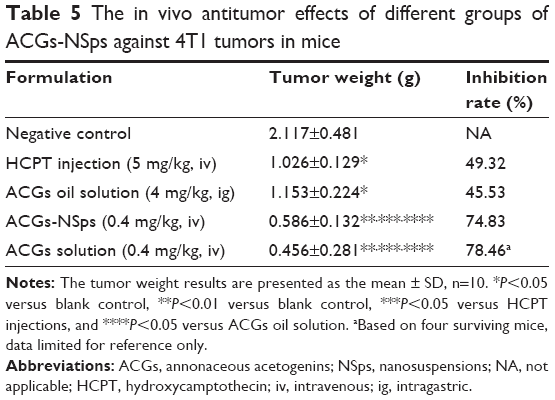 | Table 5 The in vivo antitumor effects of different groups of ACGs-NSps against 4T1 tumors in mice |
Obviously, iv administration is more effective for ACGs-NSps. Compared with oil solution by intragastric administration, ACGs-NSps exhibited notably enhanced anticancer efficacy at only 1/10 the dosage (Table 3; 74.83% vs 45.53%, P<0.05), because, as has been well discussed, high bioavailability and nanodrug delivery systems, including NSps, tend to accumulate in tumors through enhanced permeability and retention effects.38
ACGs “solution” was originally prepared to be used as a control in this study. However, based on our previous biodistribution study, ACGs “solution” appeared to have even better tumor-targeted ability than ACGs-NSps. To verify this phenomenon, in vivo antitumor efficacy was examined here in comparison. The results showed that ACGs “solution” exhibited nearly the same antitumor activity as ACGs “solution” at the same dosage (0.4 mg/kg, 78.46% vs 74.83%, P>0.05). A statistically significant difference was reported between nanoparticles with smaller sizes (<20 nm) and larger sizes (100–200 nm) after a long experimental period of >18 days.40 Therefore, the similar inhibitory rate of ACGs “solution” and ACGs-NSps might be due to the relatively short test duration. Undesirably, ACGs “solution” exhibited much stronger toxicity than ACGs-NSps. By the end of the experiment, six of ten mice in the ACGs “solution” group died, while only one of ten mice died in the ACGs-NSps group. This finding demonstrated that ACGs “solution” had more unfavorable effects than therapeutic effects in cancer treatment; thus, it was abandoned in this regard. This high toxicity might be related to the presence of 5% DMSO (v/v) and 5% Tween 80 (v/v) in the formulation of ACGs “solution”. All the above findings suggested that NSps might be a good dosage form for ACGs to treat cancer with reduced dose.
Conclusion
Herbal medicine is one of the most important resources for antitumor drugs. ACGs have received much attention due to their strong inhibition of various tumor cells. However, the poor solubility has limited their clinic application, and their multicomponent composition along with the lack of characteristic ultraviolet absorbance makes them difficult for use in in vivo analysis and biodistribution studies. As far as we know, this report was the first of ACGs-NSps, being prepared and fluorescence-labeled to overcome these problems. Using mPEG2000–PCL2000 amphiphilic block polymer, ACGs-NSps were successfully prepared with a small size and good stability, exhibiting stronger in vitro cytotoxicity than free ACGs solution against 4T1, HeLa, and MCF-7 cells. In vivo antitumor studies in 4T1 tumor-bearing mice demonstrated that ACGs-NSps could effectively accumulate in the tumor and achieve much better therapeutic efficacy than traditional dosage forms (oil solution) even at 1/10 the dose. The better tumor-targeted ability of ACGs/DiR “solution” (~20 nm) compared to ACGs/DiR-NSps (~157 nm) suggested that the smaller size of ACGs nanoparticles (eg, 20–60 nm) warrants further study for ideal therapeutic efficacy and reduced side effects. Related work is ongoing.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (number U1401223) and the Youth Research Fund of Peking Union Medical College (number 3332015050).
Disclosure
The authors report no conflicts of interest in this work.
References
Liaw CC, Chang FR, Wu MJ, Wu YC. A novel constituent from Rollinia mucosa, rollicosin, and a new approach to develop annonaceous acetogenins as potential antitumor agents. J Nat Prod. 2003;66(2):279–281. | ||
Nakanishi Y, Chang FR, Liaw CC, Wu YC, Bastow KF, Lee KH. Acetogenins as selective inhibitors of the human ovarian 1A9 tumor cell line. J Med Chem. 2003;46(15):3185–3188. | ||
Zafra-Polo MC, Gonzalez MC, Estornell E, Sahpaz S, Cortes D. Acetogenins from Annonaceae, inhibitors of mitochondrial complex I. Phytochemistry. 1996;42(2):253–271. | ||
Alali FQ, Liu XX, McLaughlin JL. Annonaceous acetogenins: recent progress. J Nat Prod. 1999;62(3):504–540. | ||
Liaw CC, Wu TY, Chang FR, Wu YC. Historic perspectives on annonaceous acetogenins from the chemical bench to preclinical trials. Planta Med. 2010;76(13):1390–1404. | ||
McLaughlin JL. Paw paw and cancer: annonaceous acetogenins from discovery to commercial products. J Nat Prod. 2008;71(7):1311–1321. | ||
Hopp DC, Alali FQ, Gu ZM, McLaughlin JL. Three new bioactive bis-adjacent THF-ring acetogenins from the bark of Annona squamosa. Bioorg Med Chem. 1998;6(5):569–575. | ||
Ahammadsahib KI, Hollingworth RM, McGovren JP, Hui YH, McLaughlin JL. Mode of action of bullatacin: a potent antitumor and pesticidal annonaceous acetogenin. Life Sci. 1993;53(14):1113–1120. | ||
Chen Y, Chen JW, Li X. Cytotoxic bistetrahydrofuran annonaceous acetogenins from the seeds of Annona squamosa. J Nat Prod. 2011;74(11):2477–2481. | ||
Chiu HF, Chih TT, Hsian YM, Tseng CH, Wu MJ, Wu YC. Bullatacin, a potent antitumor annonaceous acetogenin, induces apoptosis through a reduction of intracellular cAMP and cGMP levels in human hepatoma 2.2.15 cells. Biochem Pharmacol. 2003;65(3):319–327. | ||
Miyoshi H, Ohshima M, Shimada H, Akagi T, Iwamura H, McLaughlin JL. Essential structural factors of annonaceous acetogenins as potent inhibitors of mitochondrial complex I. Biochim Biophys Acta. 1998;1365(3):443–452. | ||
Tormo JR, Estornell E. New evidence for the multiplicity of ubiquinone- and inhibitor-binding sites in the mitochondrial complex I. Arch Biochem Biophys. 2000;381(2):241–246. | ||
Chen Y, Chen JW, Zhai JH, Wang Y, Wang SL, Li X. Antitumor activity and toxicity relationship of annonaceous acetogenins. Food Chem Toxicol. 2013;58:394–400. | ||
Jiang P, Li WM, Gao NN, et al. Acute toxicity of total annonaceous acetogenins in mice. J Toxicol. 2012;26:473–474. | ||
Chen Y, Yu DQ. [Classification and NMR characteristics of the gamma-lactone and THF rings of antitumor bioactive annonaceous acetogenins]. Yao xue xue bao [Acta Pharm Sin]. 1998;33(7):553–560. | ||
Sun L, Yu JG, Li DY, Li J, Yang XD, Yang SL. [Determination of annonaceous acetogenins in Annonaceae plants by HPLC]. Yao xue xue bao [Acta Pharm Sin]. 2001;36(9):683–685. | ||
Pan XP, Yu DQ. [Studies on new cytotoxic annonaceous acetogenins from Uvaria grandiflora and absolute configurations]. Yao xue xue bao [Acta Pharm Sin]. 1997;32(4):286–293. | ||
Xu SS, Li X, Chen JW, Chen Y, Xie H. Preparation technology and determination of annonaceous acetogenins dripping pill. Chin J Exp Tradit Med Formulae. 2012;18(1):16–19. | ||
Dang YJ, Feng HZ, Zhang L, Hu CH, Zhu CY. In situ absorption in rat intestinal tract of solid dispersion of annonaceous acetogenins. Gastroenterol Res Pract. 2012;2012:9. [Article ID 879676]. | ||
Pan XP, Wu P, Lin LD. Determination of the total content of Annonaceous acetogenins in Anuoning emulsion injection by colorimetry. Chin Tradit Herbal Drugs. 2007;38:1658–1659. | ||
Keck CM, Muller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62(1):3–16. | ||
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
Patel VR, Agrawal YK. Nanosuspension: an approach to enhance solubility of drugs. J Adv Pharm Technol Res. 2011;2(2):81–87. | ||
Han M, Yu X, Guo Y, Wang Y, Kuang H, Wang X. Honokiol nanosuspensions: preparation, increased oral bioavailability and dramatically enhanced biodistribution in the cardio-cerebro-vascular system. Colloids Surf B Biointerfaces. 2014;116:114–120. | ||
Smyth T, Kullberg M, Malik N, Smith-Jones P, Graner MW, Anchordoquy TJ. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J Control Release. 2015;199:145–155. | ||
Muller RH, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19. | ||
Zhu Z. Effects of amphiphilic diblock copolymer on drug nanoparticle formation and stability. Biomaterials. 2013;34(38):10238–10248. | ||
Pagar KP, Vavia PR. Naltrexone-loaded poly[La-(Glc-Leu)] polymeric microspheres for the treatment of alcohol dependence: in vitro characterization and in vivo biocompatibility assessment. Pharm Dev Technol. 2014;19(4):385–394. | ||
Gandhi A, Jana S, Sen KK. In-vitro release of acyclovir loaded Eudragit RLPO((R)) nanoparticles for sustained drug delivery. Int J Biol Macromol. 2014;67:478–482. | ||
Yang L, Jiang J, Hong J, et al. High drug payload 10-hydroxycamptothecin nanosuspensions stabilized by cholesterol-PEG: in vitro and in vivo investigation. J Biomed Nanotechnol. 2015;11(4):711–721. | ||
Zeng L, Weber AP. Aerosol synthesis of nanoporous silica particles with controlled pore size distribution. J Aerosol Sci. 2014;76:1–12. | ||
Gou Q, Liu L, Wang C, et al. Polymeric nanoassemblies entrapping curcumin overcome multidrug resistance in ovarian cancer. Colloids Surf B Biointerfaces. 2015;126:26–34. | ||
Yang H, Zhang N, Li X, Chen J, Cai B. Structure-activity relationships of diverse annonaceous acetogenins against human tumor cells. Bioorg Med Chem Lett. 2009;19(8):2199–2202. | ||
Chen Y, Chen JW, Wang Y, Xu SS, Li X. Six cytotoxic annonaceous acetogenins from Annona squamosa seeds. Food Chem. 2012;135(3):960–966. | ||
Chen Y, Chen JW, Xu SS, et al. Antitumor activity of annonaceous acetogenins in HepS and S180 xenografts bearing mice. Bioorg Med Chem Lett. 2012;22(8):2717–2719. | ||
Chiannilkulchai N, Ammoury N, Caillou B, Devissaguet JP, Couvreur P. Hepatic tissue distribution of doxorubicin-loaded nanoparticles after i.v. administration in reticulosarcoma M 5076 metastasis-bearing mice. Cancer Chemother Pharmacol. 1990;26(2):122–126. | ||
Soma CE, Dubernet C, Barratt G, Benita S, Couvreur P. Investigation of the role of macrophages on the cytotoxicity of doxorubicin and doxorubicin-loaded nanoparticles on M5076 cells in vitro. J Control Release. 2000;68(2):283–289. | ||
Jain KK. Nanotechnology-based drug delivery for cancer. Technol Cancer Res Treat. 2005;4(4):407–416. | ||
Cabral H, Matsumoto Y, Mizuno K, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. | ||
Tang L, Yang X, Yin Q, et al. Investigating the optimal size of anticancer nanomedicine. Proc Natl Acad Sci U S A. 2014;111(43):15344–15349. | ||
Minagawa T, Sakanaka K, Inaba S, et al. Blood-brain-barrier transport of lipid microspheres containing clinprost, a prostaglandin I2 analogue. J Pharm Pharmacol. 1996;48(10):1016–1022. |
Supplementary materials
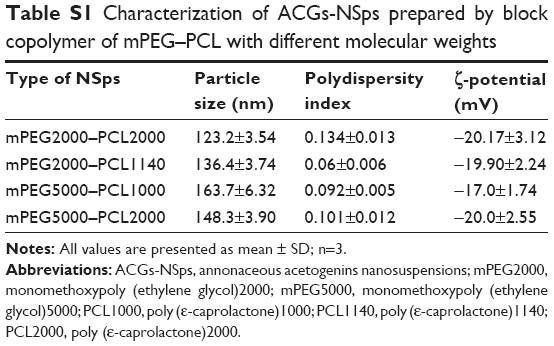 | Table S1 Characterization of ACGs-NSps prepared by block copolymer of mPEG–PCL with different molecular weights |
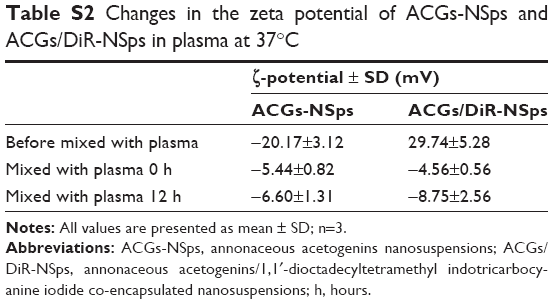 | Table S2 Changes in the zeta potential of ACGs-NSps and ACGs/DiR-NSps in plasma at 37°C |
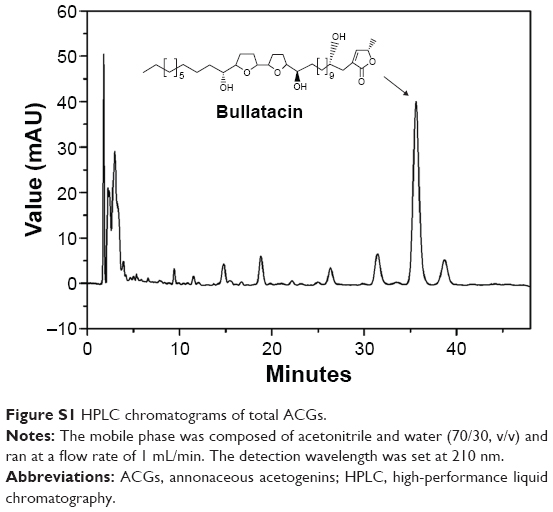 | Figure S1 HPLC chromatograms of total ACGs. |
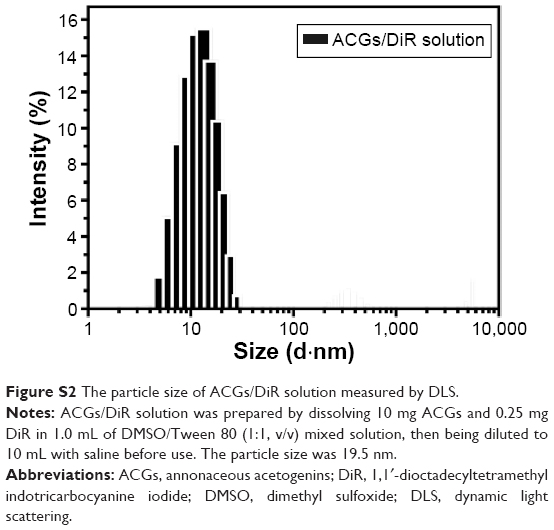 | Figure S2 The particle size of ACGs/DiR solution measured by DLS. |
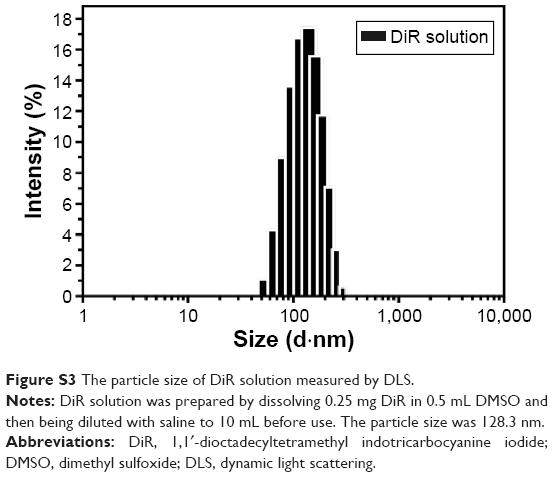 | Figure S3 The particle size of DiR solution measured by DLS. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.