Back to Journals » Clinical and Experimental Gastroenterology » Volume 9
An update on antibody-based immunotherapies for Clostridium difficile infection
Received 22 December 2015
Accepted for publication 18 April 2016
Published 1 August 2016 Volume 2016:9 Pages 209—224
DOI https://doi.org/10.2147/CEG.S84017
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Andreas M. Kaiser
Greg Hussack,1 Jamshid Tanha1–3
1Human Health Therapeutics Portfolio, National Research Council Canada, Ottawa, 2School of Environmental Sciences, University of Guelph, Guelph, 3Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada
Abstract: Clostridium difficile continues to be one of the most prevalent hospital-acquired bacterial infections in the developed world, despite the recent introduction of a novel and effective antibiotic agent (fidaxomicin). Alternative approaches under investigation to combat the anaerobic Gram-positive bacteria include fecal transplantation therapy, vaccines, and antibody-based immunotherapies. In this review, we catalog the recent advances in antibody-based approaches under development and in the clinic for the treatment of C. difficile infection. By and large, inhibitory antibodies that recognize the primary C. difficile virulence factors, toxin A and toxin B, are the most popular passive immunotherapies under investigation. We provide a detailed summary of the toxin epitopes recognized by various antitoxin antibodies and discuss general trends on toxin inhibition efficacy. In addition, antibodies to other C. difficile targets, such as surface-layer proteins, binary toxin, motility factors, and adherence and colonization factors, are introduced in this review.
Keywords: antibody, Clostridium difficile, immunotherapy, toxin
Introduction
Clostridium difficile is one of the most prevalent hospital-acquired bacterial infections in the developed world, with symptoms ranging from mild diarrhea to colitis and death.1,2 Reducing the rate and duration of C. difficile infection (CDI) are critical goals for health care providers due to the enormous cost associated with CDI. This is a considerable challenge, given that aging populations are particularly susceptible to CDI. While broad-spectrum antibiotics and the more recent narrow-spectrum antibiotic fidaxomicin have shown some efficacy toward containing CDI, novel therapeutics are desired.2–5 There are a number of treatments under development for CDI, including but not limited to vaccines, fecal transplantation therapy, antibiotics, probiotics, and antibody-based immunotherapy.4,6–8 With the focus of this review on chronicling the recent advances in monoclonal antibody (mAb)- and single-domain antibody (sdAb)-based immunotherapy, we direct readers to the excellent reviews highlighting other CDI therapies under development.9–12
Before discussing the present antibody-based therapeutics under development for CDI, it is important to understand the mechanisms of CDI, host colonization, and associated virulence factors. CDI often begins with a patient on broad-spectrum antibiotics being exposed to C. difficile spores. Other risk factors for potential CDI include age, gastrointestinal (GI) surgery, inflammatory bowel disease, and immunosuppression.2 In general, patients on antibiotics have modified GI microbiota populations, allowing for C. difficile spores that travel to the lower GI tract an opportunity to begin their colonization process and transformation into vegetative cells.2,12 At this point, it is thought that the main C. difficile virulence factors toxin A (TcdA) and toxin B (TcdB) (Figure 1A–F) are transcribed and secreted from the bacteria through a mechanism that requires the holin-like protein TcdE.13–17 Individuals who possess circulating antitoxin antibodies or those who mount a rapid and effective response are often only asymptomatic carriers or experience less severe CDI with a lower risk of recurrent CDI.2,18–20 On the other hand, individuals who fail to respond quickly to the toxins develop symptoms of CDI, which include diarrhea and colitis. Both TcdA and TcdB are glucosyltransferase-containing multi-domain proteins that enter host epithelial cells, undergo an acid-induced conformational change, and release their glucosyltransferase domain (GTD; Figure 1D) inside the cell to inactivate GTPases, such as Rho, Rac, and Cdc42.14,21 GTPase inactivation causes a cascade of downstream effects, culminating in a loss of epithelial barrier function, proinflammatory responses, and toxins reaching underlying germinal centers.22,23 Individuals who eventually restore their natural GI tract microbiota and/or who mount an effective antitoxin immune response clear the infection, while those who fail to do so are prone to rounds of relapsing CDI.2 Given the importance of these two toxins in manifesting the severe symptoms associated with CDI, antibody-based immunotherapies have largely focused on targeting the toxins.
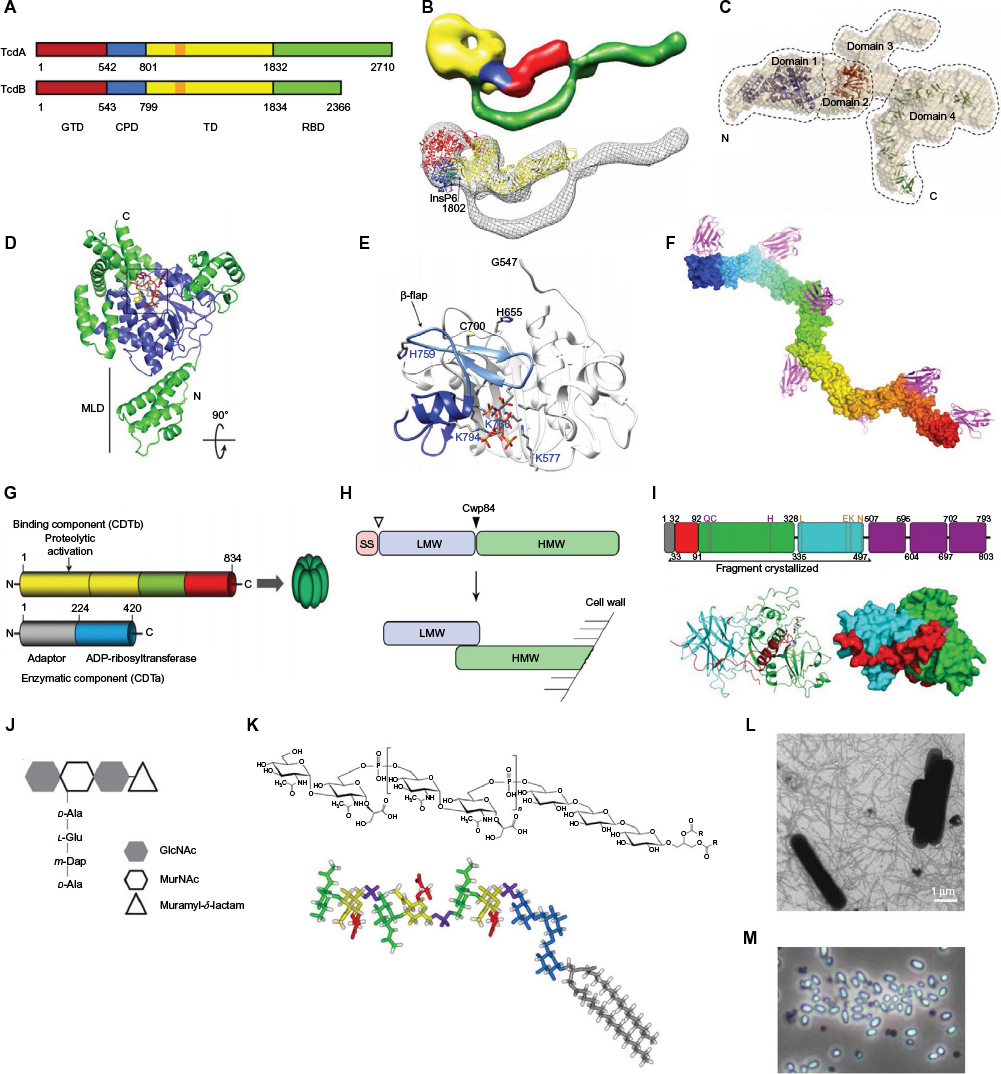 | Figure 1 Validated and potential C. difficile targets for antibody-based immunotherapy of CDI. Notes: (A–F) C. difficile toxin A (TcdA) and toxin B (TcdB). (A) A schematic of TcdA and TcdB.155 (B) A proposed global structure of TcdA.155,156 (C) A proposed global structure of TcdB.157 (D) A crystal structure of TcdA GTD.158 (E) A crystal structure of TcdA APD, including the CPD.156 (F) A model of TcdA RBD cocrystal structure in complex with A20.1 VHH.78 (G) A schematic of C. difficile binary toxin CDT.86 (H) A schematic of C. difficile SLPs104; arrows denote SS and Cwp84 cleavage sites. (I) A crystal structure of Cwp84.159,160 (J) A schematic of C. difficile spore peptidoglycan complex.161 (K) C. difficile LTA.113 (L) A photograph of C. difficile (630 strain) showing flagella.162 (M) A photograph of C. difficile (R20291 strain) spores (courtesy of Susan Logan, NRC, Canada). Abbreviations: C. difficile, Clostridium difficile; CDI, C. difficile infection; GTD, glucosyltransferase domain; APD, autoprocessing domain; CPD, cysteine protease–domain; RBD, receptor-binding domain; SLPs, surface-layer proteins; SS, signal sequence; LTA, lipoteichoic acid; TD, translocation domain; MLD, membrane localization domain; LMW, low-molecular weight SLP subunit; HMW, high-molecular weight SLP subunit. |
Conventional wisdom implies that the use of antibodies as therapeutic agents against bacterial infections is a logical choice, given the immune system, including antibodies, has evolved to combat bacterial infections. A stream of antibacterial antibodies in various stages of development, including many antibodies in various phases of clinical development, is in line with this idea and a testament to the optimism and confidence drug developers have in antibodies as effective antibacterial agents.24–26 The infections targeted by antibodies include, but are not limited to, CDI, hospital-acquired pneumonia, ventilator-associated pneumonia, and Shiga toxin-associated Escherichia coli-induced hemolytic uremic syndrome, caused by bacteria such as C. difficile, Staphylococcus aureus, Pseudomonas aeruginosa, and E. coli. In many cases such as in CDI, the antibody targets are toxins.
In this review, we chronicle the recent advances in antibody-based immunotherapy for treating CDI. Antitoxin approaches dominate the current immunotherapy pipeline with bezlotoxumab, which successfully passed a recent Phase III clinical trial, leading the way.24 We discuss other antitoxin antibody approaches in development and in particular take a detailed look at the toxin epitopes targeted by these antibodies. We review nontoxin-based C. difficile targets that show promise for potential antibody targeting (Figure 1G–M). Finally, we propose next-generation antibody formats for C. difficile targeting, including multiple specificities that go beyond targeting one type of C. difficile virulence factor. These novel formats may show promise as GI-targeting oral therapeutics, opening up a novel and potentially very efficacious delivery route to disrupt C. difficile before it can effectively establish infection.
Antibody-based immunotherapies
We previously documented various passive antibody-based immunotherapies under development for the treatment of CDI.6 Since then there have been numerous advances and additional formats of antibodies characterized, including key Phase III clinical trial data from the actoxumab/bezlotoxumab program. While there are other antibody-based approaches that have shown efficacy in treating CDI in animals, namely, polyclonal antibody preparations27–30 and intravenous (IV) immunoglobulin therapy,31 the focus of this review is on mAbs and sdAbs.
mAbs are a widely successful class of antibodies with at least 45 antibodies approved to treat a range of indications, including cancer, autoimmune disorders, cholesterol, and infectious disease (Figure 2A). Hybridoma technology allows for the isolation of murine mAbs, but these often require conversion into semihuman (chimeric) or humanized mAbs before use in human beings. More recent techniques using transgenic animals with fully human antibody repertoires or recombinant selection systems allow for the isolation of fully human mAbs. mAbs, specifically the immunoglobulin G1 (IgG1) isotype, offer long serum half-lives of up to 21 days32 and high target affinity and specificity, making them ideal antitoxin agents for use systemically.8 This long half-life suggests that a single infusion of IgG1 could offer protection of patients from CDI, or in the case of patients with CDI, this could reduce the chance of relapse.8,33 While the development of mAbs as antitoxin and antimicrobial agents is well documented, concerns over their potential costs and widespread adoption are warranted.24,34,35
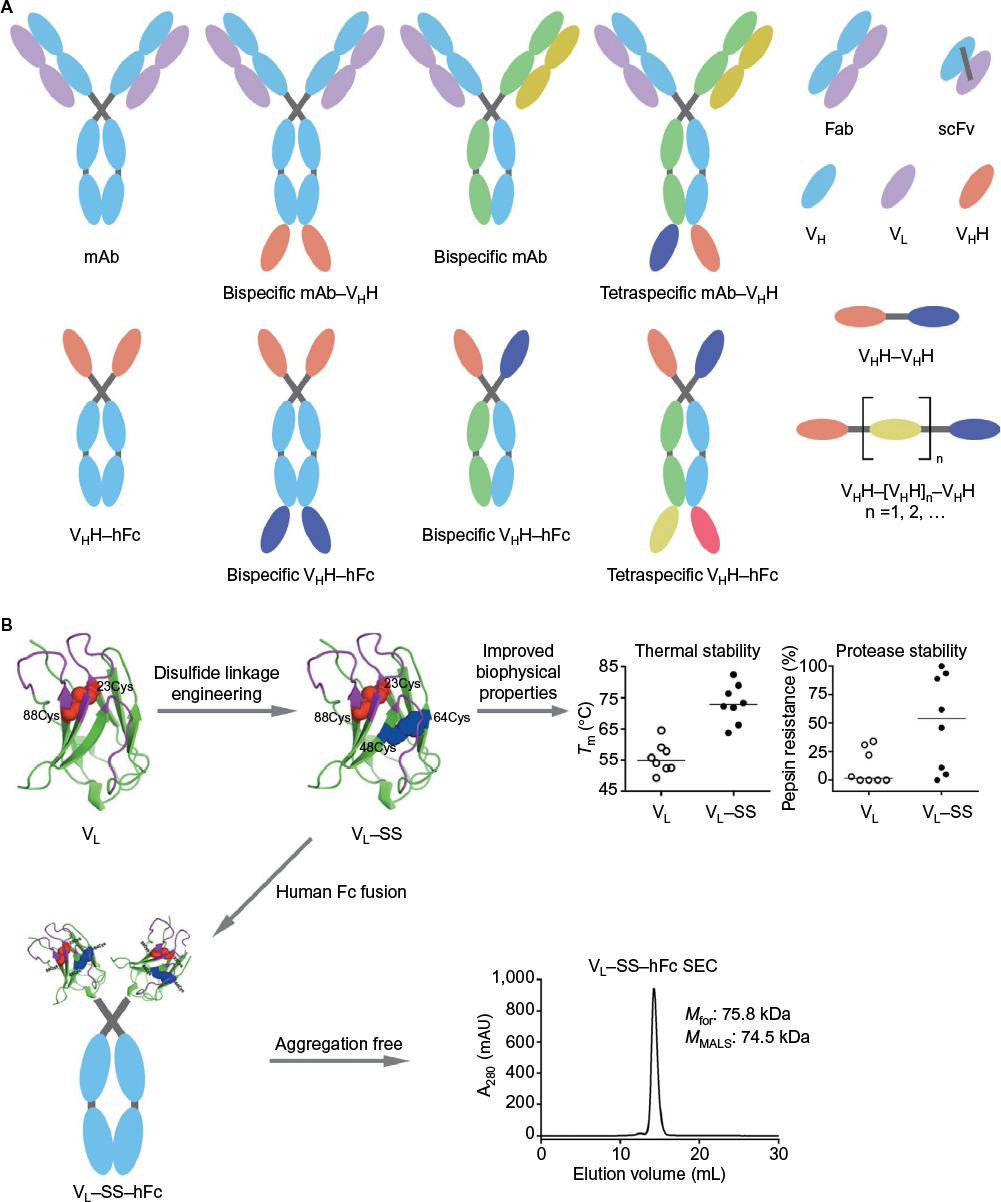 | Figure 2 Potential antibody formats for CDI immunotherapy. Notes: (A) Conventional mAbs and various multispecific targeting formats are now a reality. Antibody fragments such as Fab, scFv, VH, VL, and VHH allow for modular assembly of multispecific affinity reagents. Several examples, but by no means an exhaustive collection of possible antibody formats, are shown. For more antibody formats, refer to Spiess et al.120 (B) Engineering robust and efficacious sdAb (human VH, human VL, VHH) therapeutics. Dramatic improvements to sdAb thermal and proteolytic stability, for example, by disulfide linkage engineering of human VL domains, may allow for greater resistance to GI tract protease degradation and lead to more efficacious oral therapeutics targeting C. difficile. Furthermore, disulfide-engineered sdAbs (VL–SS) can be efficiently turned into highly stable, aggregation-free, efficacious systemic therapeutics by fusion to hFc (VL–SS–hFc). Abbreviations: CDI, C. difficile infection; mAbs, monoclonal antibodies; Fab, fragment antigen binding; sdAb, single-domain antibody; GI, gastrointestinal; C. difficile, Clostridium difficile; hFc, human Fc; Tm, melting temperature; mAU, milli absorbance unit; Mfor, formula molecular mass; MMALS, molecular mass determined by MALS; MALS, multiangle light scattering. |
sdAbs provide attractive therapeutic modalities against CDI. Defined as autonomous variable domains of antibodies with antigen-binding capabilities, sdAbs may be Ig VH domains, Ig VL domains, VHH domains derived from Camelidae heavy-chain IgGs, or VNAR domains derived from cartilaginous shark IgNAR antibodies (Figure 2).36–38 Some of their unique features compared to mAbs and other recombinant antibody fragments such as fragment antigen binding and single-chain variable fragment (scFv)39 include their single-domain nature; small size (13–15 kDa); high chemical, thermal, and proteolytic stability; high refoldability; high aggregation resistance; high-level expression in microorganisms; high modularity; tissue-penetrating properties; ability to access cryptic epitopes (eg, cavities in receptors, enzymes, toxins, and infectious agents); amenability to in vitro selection and engineering approaches for robust domains that are resistant to proteases (eg, GI protease); and acidic pH-induced and heat-induced aggregation, denaturation, and degradation.36,38,40–52 In addition, sdAbs can be fully human (VHs and VLs) or readily humanized (VHHs, VNARs) to reduce/eliminate their potential immunogenicity.36,38,53–56 Moreover, high-affinity sdAbs with equilibrium dissociation constants (KDs) in the low nanomolar to picomolar range are readily obtainable.36 The aforementioned features render sdAbs as efficacious therapeutics with low cost of goods.
Table 1 describes the various antitoxin antibodies isolated or characterized, and in particular, examines toxin target specificity, epitopes, and neutralizing potency and breadth. Table 1 is categorized by antitoxin antibody format, beginning with mAbs and then camelids (llama and alpaca) and human sdAbs.
 | Table 1 Summary of mAbs and sdAbs targeting C. difficile toxin A and toxin B Abbreviations: mAb, monoclonal antibody; sdAb, single-domain antibody; C. difficile, Clostridium difficile; Ab, antibody; RBD, receptor-binding domain; GTD, glucosyltransferase domain; N/d, not determined; IP, intraperitoneal; IV, intravenous; B. longum, Bifidobacterium longum; TD, translocation domain. |
Antitoxin A/B mAbs
The prime targets for therapeutic antibodies against CDI have been the toxin A/toxin B pair (Figure 1A–F). Owing to their remarkable specificity combined with the fact that they target toxins, antitoxin A and B antibodies are not expected to induce broad resistance among bacteria or disturb the healthy microbiota, as is the case with most conventional antibiotics, and presumably the reason for their ability to reduce the recurrence of CDI.
The most clinically advanced and characterized antibodies for the treatment of CDI are actoxumab and bezlotoxumab (Table 1). These fully human mAbs target TcdA and TcdB, respectively,57 and are the only antibodies for the treatment of C. difficile to have been tested in clinical trials.24,58 Actoxumab (previously named MK-3415, CDA1, MDX-066, 3D8) recognizes the C-terminal cell-surface receptor-binding domain (RBD) of TcdA and appears to bind each TcdA protein twice.59 The antibody was shown to potently neutralize TcdA in vitro.57 The mechanism of TcdA neutralization is through direct toxin neutralization and is thought not to involve effector functions.60 Bezlotoxumab (previously named MK-6072, CDB1, MDX-1388, 124-152) recognizes the C-terminal RBD of TcdB and appears to bind each TcdB protein twice.59 Orth et al61 showed that the bezlotoxumab binding site on TcdB overlaps with the putative TcdB carbohydrate-binding region. The antibody was shown to potently neutralize TcdB in vitro.57 In vivo, actoxumab, when combined with bezlotoxumab, was protective in multiple mouse and hamster models.57,60 In contrast, in a piglet model of CDI, prophylactic administration of bezlotoxumab alone or combined with actoxumab provided 100% protection from systemic and GI tract CDI.62 Piglets given actoxumab alone showed a similar lack of efficacy compared to placebo, with a mortality rate of 67%, suggesting that much of the protection was driven by the anti-TcdB mAb.62 Interestingly, in the same piglet study, administration of alpaca antitoxin A polyclonal antibodies also showed a similar lack of efficacy compared to placebo. Clinically, in Phase II trials, the combination of both antibodies significantly reduced the rate of CDI relapse compared to standard-of-care antibiotic therapy.58 In the recently completed Phase III trials, treatment of CDI with bezlotoxumab in conjunction with standard-of-care antibiotic therapy reduced CDI recurrence for 12 weeks compared to placebo.24 Treatment with both mAbs provided no added efficacy, and actoxumab alone did not prevent C. difficile recurrence.24 It is likely, based on the totality of the available data, that the contribution of antitoxin A antibodies to protection is related to the nature of the host species (human vs piglet vs rodent) rather than to the nature of the antibody itself.
The aforementioned mAbs do not reduce the severity of diarrhea during the initial CDI episode, time to resolution of diarrhea, or the duration of hospitalization for the initial episode,1,58 although Phase II and III clinical studies were not designed to assess these, leaving the door open for developing next-generation antibody therapeutics with improved and more complete efficacy. Ideally, such therapeutics should be produced at low cost, work with high efficacy, eliminate CDI recurrence, and reduce the severity of diarrhea, time to resolution of diarrhea, and duration of hospitalization. Additionally, the lack of efficacy of actoxumab in clinical trials does not necessarily demonstrate that targeting TcdB alone is sufficient to achieve full efficacy. Already a number of antitoxin A/B antibody combinations are in the early stages of development and claim to have improved preclinical efficacy in reducing CDI recurrence and severity of diarrhea relative to actoxumab/bezlotoxumab.59,63 Thus, the next-generation CDI therapeutics will most likely require combinations of antitoxin A and antitoxin B antibodies.
Recently, Anosova et al63 presented data on a human mAb to TcdA and a pair of human mAbs to TcdB, isolated from healthy human donors using a high-throughput B-cell cloning strategy. The TcdA-neutralizing mAb A2 recognizes the C-terminal RBD of TcdA, possibly a linear epitope represented by the TGWQTI motif. In vitro, A2 neutralized TcdA from toxinotypes 0, III, and V in both Vero cell cytotoxicity and T84 transepithelial electrical resistance (TEER) assays. To target TcdB, the group isolated two mAbs, B1 and B2, which targeted the N-terminal GTD. B1 possibly recognizes a linear epitope containing residues SGRNK in a four-helix bundle near the N-terminus of the GTD, and B2 is thought to bind a conformational epitope that is within the GTD but distinct from the B1 mAb. The pair of TcdB mAbs neutralized toxinotypes 0, III, V, VIII, and X in Vero cell assays and T84 TEER assays. Using the hamster model of CDI, simultaneous intraperitoneal (IP) administration of all three mAbs protected the animals from mortality, following challenge with clinical C. difficile strain 630. Treatment of hamsters with A2 alone was not protective, while treatment with B1 alone, B2 alone, or B1 + B2 resulted in survival rates of 70%, 40%, and 50%, respectively.
In an earlier study, Davies et al59 also showed strong protection against CDI in hamsters using a single anti-TcdA mAb and a pair of anti-TcdB mAbs. CA997 is a humanized mAb that binds the C-terminal RBD of TcdA and was estimated to bind at least 12 times per toxin. Using Caco-2 cells, CA997 neutralized TcdA from ribotypes 003, 027, and 078 and was more potent than actoxumab. In addition, CA997 inhibited TcdA-induced TEER loss in TEER assays, while actoxumab was poorly protective in the TEER assay. CA1125 and CA1151 are humanized TcdB mAbs that target nonoverlapping epitopes in the RBD and bind with valencies of one and two, respectively. In vitro, single mAbs were weakly neutralized in Caco-2 inhibition assays; however, when combined, potent TcdB neutralization was reported. In vivo, using the hamster CDI model, simultaneous IP administration of all three mAbs provided 100% protection at 11 days. At 28 days, the combo of three antibodies showed higher levels of protection than the combination of actoxumab and bezlotoxumab (82% vs 28% survival, respectively).
Elsewhere, PA-50 is a humanized anti-TcdA mAb that targets TcdA RBD at multiple sites.64 This epitope is broadly conserved throughout C. difficile 027 ribotype strains and does not overlap with the TcdA epitope recognized by actoxumab. PA-50 was capable of TcdA neutralization from a broad range of C. difficile ribotypes (001, 002, 003, 012, 014, 017, 027, 078) and was more potent than actoxumab in vitro. The authors suggested a mechanism of toxin neutralization involving cooperative inhibition, possibly due to multivalent interactions with TcdA. Marozsan et al64 also isolated a potent TcdB inhibiting antibody termed PA-41. PA-41 targets the N-terminal GTD of TcdB and is thought to recognize a single epitope that is conserved among 027 ribotypes. In vitro, PA-41 was capable of broad TcdB neutralization, inhibiting toxins from the same C. difficile ribotypes described for PA-50-based TcdA neutralization. The fact that PA-41 showed superior neutralizing efficacy compared to bezlotoxumab suggests that targeting the GTD is an effective and potent area to target on TcdB. Using the hamster CDI model, PA-50 in combination with PA-41 resulted in long-term survival of hamsters compared to 0% survival for animals treated with the standard antibiotic vancomycin or, surprisingly, with a combination of actoxumab and bezlotoxumab.
A number of earlier mouse mAbs were isolated and capable of toxin neutralization. PCG-465–69 is a TcdA targeting mouse IgG2a mAb that binds the RBD with a valency of five or six and recognizes epitopes in amino acids 2097–2141 and 2355–2398 (Figure 1A). In vitro, Lyerly et al65 reported that the antibody failed to inhibit TcdA in Chinese hamster ovary (CHO) K1 cell-based assays; however, Demarest et al67 reported modest in vitro TcdA neutralization with the antibody. In vivo, PCG-4 neutralized the effects of TcdA (diarrhea and death) when TcdA was administered intragastrically to hamsters and when TcdA was used in an intestinal loop model in rabbits. Demarest et al67 characterized several mouse mAbs capable of TcdA inhibition. mAbs 3358 (IgG2a) and 3359 (IgG1) bound nonoverlapping epitopes in the TcdA RBD at up to 14 and nine sites, respectively. The authors revealed that 3358 and PCG-4 (expressed as a recombinant mouse/human chimeric antibody) contained overlapping TcdA RBD epitopes. Both 3358 and 3359 neutralized TcdA in CHO K1 cell-based assays, and the combination of the two resulted in increased neutralizing potency compared to individual mAbs. Interestingly, 3358 appeared to increase the detectable level of RBD on the surface of CHO cells, while 3359 inhibited RBD binding to the CHO cell surface. Elsewhere, Zhang et al70 isolated a panel of TcdA-specific mouse mAbs (1G3, 1B5, 2D4, 2C7, 4A4, 5D8) that recognized linear RBD epitopes and were capable of TcdA inhibition in CHO and HT-29 cell-based assays. In mouse models with IP administration of TcdA, 50% of mice given mAb 4A4 survived, while 2C7 and 5D8 mAbs were not as strongly protective. When combined, 4A4/2C7 and 4A4/5D8 pairs were more protective than the 4A4 mAb alone. Another TcdA-specific mouse IgG2a mAb, A1H3, has been used in several studies71–73 but is poorly characterized with respect to TcdA epitope and neutralizing potency. A1H3 enhanced TcdA-mediated cellular effects in murine macrophages and human monocytes through FcγR1-mediated endocytosis. A1H3 also facilitated cell-surface recruitment of TcdA, likely contributing to enhanced cytotoxicity. Corthier et al74 characterized three TcdA-binding mouse mAbs (A9, 141-2, C11) produced from ascites. All bound to a region of the TcdA RBD covered by amino acids 1964–2682. Mice were protected from C. difficile challenge upon IV administration of the antibodies. Finally, one of the first mouse mAbs (IgG1) isolated, G-2, which recognized both TcdA and TcdB, was not capable of in vitro TcdA or TcdB neutralization.65
Antitoxin A/B sdAb fragments
In addition to antitoxin human(ized) and mouse mAbs under development, a number of sdAb fragments are being explored as alternative immunotherapeutic agents for treating CDI (Figure 2). The potential advantages of using sdAb fragments are described earlier (Antibody-based immunotherapies). One of the first sdAbs isolated against C. difficile toxins was from our laboratory.75–77 In this work, a number of llama VHHs were isolated from an immune phage display library constructed after immunization with C-terminal RBD fragments from each toxin.76 A number of TcdA-binding VHHs were found to potently inhibit TcdA in cell-based assays, and combinations of various VHHs significantly improved neutralizing potency. VHHs A4.2, A5.1, A20.1, A24.1, and A26.8 recognized conformational RBD epitopes, while A19.2 bound a linear epitope. The A20.1 epitope was found to not overlap with those for A4.2, A5.1, or A26.8, which appeared to be overlapping, and none of the VHHs prevented free trisaccharide from binding the RBD carbohydrate-binding pocket.76 Cocrystal structures of A20.1 and A26.8 in complex with a TcdA fragment confirmed this binding pattern, with A20.1 binding very close to the carbohydrate-binding pocket with a proposed valency of seven VHHs per RBD based on modeling (Figure 1F).78 A26.8 appeared to bind the extreme C-terminus of TcdA with a stoichiometry of 1:1.78 Recently, Shkoporov et al79 expressed A20.1 and A26.8 in Bifidobacterium longum and showed TcdA neutralization in vitro. The group went on to administer the probiotic bacteria to mice and confirmed the in vivo expression (secretion) of the VHHs in the gut of mice. Several VHHs and human VLs that bind the TcdB RBD have also been isolated and characterized,76–78 and despite some with very high affinity binding, none were capable of toxin inhibition in cell-based assays. This includes B5.2, B13.6, and B15.5 VHHs76; B39 VHH78; and B4, B5, B12 and B17 human VLs.77 B39 has been proposed to bind to TcdB at four sites.78 Interestingly, B5.2 showed cross-reactive binding to TcdA, but did not neutralize the toxin.
Yang et al80 later reported the isolation of TcdA- and TcdB-specific alpaca VHHs. The AH3 VHH bound the N-terminal GTD of TcdA, the AA6 VHH recognized the central translocation domain, and the E3 VHH bound the GTD of TcdB. The group constructed a novel, tandem-linked molecule consisting of a string of four VHHs (AH3–E3–E3–AA6), with the TcdA-targeting antibodies at the termini and the TcdB-targeting antibody, represented twice, in the middle. The tetramer broadly neutralized both toxins from several clinical C. difficile isolates in cell-based (Vero and CT26) assays, including ribotypes 001, 002, 012, 014, 015, 023, 027, 078, and 106. The tetramer protected mice against a lethal systemic challenge of a mixture of TcdA/TcdB, at a tetramer concentration as low as 3.2 μg/kg, and reversed CDI in mice infected with the 027 strain after a single 1 mg/kg injection.
More recently, Andersen et al81 reported the isolation of four llama VHHs against the TcdB RBD after immunization with whole TcdB toxin. The four VHHs (B2, E2, G3, and D8) bind to three unique TcdB RBD epitopes (B2, E2/G3, and D8). As monomeric VHHs, B2, G3, and D8 neutralized the cytopathic effects of TcdB on MA-104 cells. In combinations as doublet and triplet sets, the antibodies had no additive effect compared to the most protective singlet VHH G3. The group expressed the antibodies on the surface of probiotic bacteria, and B2, G3, and D8 retained neutralizing potency. Interestingly, additional VHHs that were nonneutralizing as monomers became neutralizing when surface displayed, presumably due to the large steric effects imparted by the probiotic bacteria. In a prophylactic, oral treatment hamster model of CDI, a combination of two strains of Lactobacilli (one displaying B2 and the other displaying G3) delayed the death of hamsters challenged with TcdA- TcdB+ C. difficile spores, compared to zero survivors in the controls (infection-only group and non-VHH-expressing Lactobacillus group). Half of the hamsters receiving the VHH-expressing Lactobacillus survived until the end of the experiment at day 5. A mixture of B2, G3, and D8 dosed orally was not protective, possibly due to its degradation and/or transitory nature in the GI tract. Interestingly, Andersen et al81 also immunized llamas with whole TcdA, but failed to isolate any neutralizing antibodies, which is exactly the opposite of Hussack et al76 who isolated TcdA neutralizers but not TcdB neutralizers.
Antibodies to other C. difficile targets
While the primary targets of numerous mAbs and sdAbs is C. difficile toxins A and B, many other C. difficile virulence factors such as binary toxin, surface-layer proteins (SLPs), and flagella are being used as possible targets for immunotherapy (Figure 1). One of the oldest immunotherapies investigated for CDI is IV immunoglobulin therapy, in which human polyclonal mixtures of antibodies are infused into CDI patients.31 The exact composition of these mixtures is unknown, although they likely contain antitoxin antibodies and potentially antibodies to C. difficile virulence factors/surface proteins. Analysis of sera from patients with CDI82 has led to a number of novel C. difficile targets being explored as potential vaccines.12,83,84 Successful vaccine targets could conceivably drive the development of antibodies specific to these targets and guide the next generation of immunotherapies for CDI.
There are emerging opinions that a “total cure” against CDI may have to include antibodies to other targets (Figure 1) in addition to those against toxins A and B.1 In line with this new emerging trend, a vaccine candidate includes TcdA, TcdB, and components of C. difficile binary toxin CDT.85–87 Similar to toxins A and B, CDT was shown to cause death in mice and hamsters, and the aforementioned combination vaccine approach protected mice when challenged with native toxins A, B, and CDT.85 Unger et al88 isolated llama sdAbs against CDTa and CDTb and found several sdAbs to neutralize cytotoxicity in vitro. As the antitoxin antibodies do not prevent the initial C. difficile colonization step, complementing them with antibodies that target spores and prevent or eliminate their carriage or dissemination appears attractive. Antibodies that target cell-surface components involved in the colonization and adherence to gut tissues of hosts, such as SLPs,83,87,89–95 flagella,83,87,96–98 and Cwp84,83,87,98,99 are other promising complementary therapeutics. A number of experiments, including some in vivo animal studies, strongly suggest that targeting SLPs by antibodies is a viable therapeutic approach against CDI,83,94,95,100–103 and interestingly, SLP-specific sdAbs have been shown to inhibit the motility of C. difficile in in vitro assays.104 Several studies support flagella as a therapeutic target,96,98,101 including one immunization study where flagellin (FliC)- and flagellin filament cap protein (FliD)-immunized mice showed a significant decrease in C. difficile colonization101 and another where orally administered purified FliD-specific IgY protected hamsters from CDI when challenged with C. difficile strain 630.98 Moreover, mice vaccinated rectally with a combination of FliD, a flagella preparation, Cwp84, and cell wall extract showed a significant reduction in C. difficile colonization,101 and rectal as well as oral vaccination with Cwp84 partially protected hamsters from lethality.105,106 PSII polysaccharides, abundantly expressed on the surface of all C. difficile, are also attractive targets for therapeutic antibodies.83,107–109 In support of this view, studies point to PSII as a beneficial vaccine,83,108,110–112 and interestingly, PSII–toxin conjugates appear to be a promising vaccine for targeting both cell-surface PSII and toxins A and B.111 Finally, a related promising antibody target is the surface-exposed lipoteichoic acid polymer, which has been shown to be conserved in the majority of C. difficile strains and easily accessible to antibodies.113,114
Future perspectives and conclusion
Going forward, much will be learned from the late-stage clinical data produced from actoxumab/bezlotoxumab and from vaccine trials underway, to determine if the biologics/vaccine route is an effective one for CDI therapy. Another major consideration is the optimal delivery route (eg, systemic or oral administration) and the target population of antibody-based or vaccine-based CDI therapies. For now, the main route for antibody-based therapies is systemic delivery; however, improved delivery systems to the GI tract coupled with stabilized antibody formats may open the possibility for oral delivery. The target population for antibody-based therapies will likely be at-risk groups, including the elderly, the immunocompromised, those admitted to hospitals for antibiotics, and those admitted to hospitals during a CDI outbreak. Improving our understanding of C. difficile biology, host–pathogen interactions, and the identification of novel virulence factors will provide a source of new targets for immunotherapy, with an end goal of not only reducing CDI recurrence but also rapidly clearing primary CDI and its effects or even preventing them entirely.
With respect to antibody-based immunotherapy, an attractive alternative to the combination therapy approach (eg, administering two antibodies each with distinct specificity) is to combine the specificity of individual component antibodies in a therapeutic cocktail into one bispecific or multispecific antibody construct.115–120 The approach, in particular the bispecific one, has been applied to therapeutic antibodies against cancer and inflammatory and infectious diseases. Bispecific antibodies are in general more efficacious than their parental individual antibodies but similar in efficacy to combined antibody pairs. The costs associated with resources for manufacturing, clinical studies, and regulatory reviews are substantially lower in the bispecific or multispecific antibody approach than the combination therapy approach since the number of antibodies to be developed and approved is reduced in the former scenario. However, issues such as manufacturability and the stability of bispecific and multispecific antibodies still need to be resolved. A good example of an antibacterial bispecific antibody is MEDI3902 (BiS4αPa), which is a dual-targeting antibody that is protective in a lethal pneumonia mouse model, whereas a monotherapeutic is not protective.121,122 Figure 2A proposes a number of bispecific and multispecific antibody formats as the next generation of CDI therapeutics.
The current trend for administrating antitoxin antibodies relies on systemic delivery by injection. Presumably, the systemically delivered antibodies should traverse the mucosal barrier into the GI tract to be effective, and a recent study indicates that they do so by a toxin-mediated, FcRn-independent paracellular transport mechanism through the compromised (leaky) gut wall barrier in infected animals.123 The need for crossing the barrier may be a bottleneck to efficacy by preventing the delivery of sufficient quantities of antibody drugs to the site of infection. This drawback does not exist with orally administered antibody therapeutics as they bypass the requirement for traversing the mucosal barrier and can directly reach the site of infection. Other advantages of oral therapy include patient compliance; simplicity of administration; reduced cost, partly due to less stringent manufacturing requirements than injectable antibodies; enhanced potency and specificity; needle-free nature; higher safety; immune-tolerant nature, which allows repeated ingestion of therapeutic antibodies without compromising safety and eliminates the need to humanize antibodies; minimized systemic drug exposure and its subsequent unwanted side effects; and low dosage requirements.124–126 Studies with bovine immune Ig preparations6,7,30,124,127 and IgA preparations6,7,124,128 have shown that an oral antibody-mediated approach against CDI is possible. However, significant challenges facing oral antibody therapeutics include the presence of the protein-degrading enzymes, such as pepsin, trypsin, chymotrypsin, carboxypeptidase, and elastase, in the stomach and intestine; the denaturing acidic character of the gastric fluid (pH 1–3.5); the transient presence of therapeutic antibodies in the GI tract; and the requirement for repeated dosing leading to increased costs.6,124,125 However, these challenges are not insurmountable. To begin, one may utilize certain classes of antibodies that are more resistant to proteases such as IgA,6,7,125 IgY,6,7,125,129 and sdAbs.6,37,38,41,130 Furthermore, advances in protein engineering allow for the creation of robust antibodies that are resistant to the GI tract proteases and acidic environment of the stomach. Their stability can further be enhanced by applying strategies such as decoy proteins, pH modulation, masking protease active sites, enzyme inhibition, and encapsulation.124,125 As for the transient presence of oral therapeutic antibodies, theoretically, the GI half-life of antibodies can be improved by fusing them to GI retaining molecules. Moreover, engineered probiotic bacteria (eg, lactic acid bacteria) displaying or secreting recombinant antibody fragments present a promising solution: residing within the microbiota of the intestine, they can “administer” antibodies to the lower GI tract for prolonged periods of time.81,126,131–138 Additional advantages of the approach include the generally-regarded-as-safe nature of the carrier bacteria, cost-effective production, long shelf life (eg, in lyophilized form), simple distribution logistics, ease of administration, and possibility of engineering probiotic bacteria that surface display recombinant antibodies with high avidity and multiple specificity for improved efficacy. The approach has shown promising results in in vivo protection studies with several GI viral and bacterial pathogens, including C. difficile, where the target antigen was TcdB.81,126,131–136,138–140 Another promising oral therapy approach takes advantage of the ability of the gut’s enteroendocrine cells to produce and release large quantities of proteins into the gut environment in an intermittent or continuous fashion and a natural, nontoxic, and biocompatible polymer that serves as a protective carrier of the drug-encoding nucleotides into the enteroendocrine cells (http://www.engeneinc.com/).141 Both of the latter oral therapeutic approaches bypass the adverse GI tract conditions (presence of proteases and low acidic pH) and the need for purified antibodies.
With regard to systemic applications, unlike mAbs, sdAbs have very fast half-lives as low as 5 minutes (in mice) due to their small size, being below the renal filtration molecular mass cut-off, and lack of Ig Fc region.142–150 Several approaches can be applied to sdAbs for half-life extension.142,151,152 In particular, two popular recombinant approaches can be efficiently applied to therapeutic sdAbs, thanks to the highly modular nature of sdAbs. These include their tandem fusion to a second sdAb that is specific to human serum albumin or fusion to human IgG Fc region, with the latter format emulating the overall structure of naturally occurring Camelidae heavy-chain antibodies (Figure 2).38,56,142–144,148–150,153,154 In these cases, the half-life extension is conferred directly (ie, sdAb–Fc fusion) or indirectly, through serum albumin (ie, sdAb–sdAb fusion) binding to FcRn and through an increase in size (ie, sdAb–Fc fusion). Modularity of sdAbs also allows efficient generation of bispecific or multispecific recombinant antibodies against C. difficile targets, including toxins A and B. The options include fusing different sdAbs in tandem to make dimers, trimers, or tetramers or fusing sdAbs to mAbs to construct sdAb–mAb hybrid fusions (Figure 2). In the case of dimers and trimers, one may include a human serum albumin-specific sdAb to confer sufficient serum half-life, but in the case of a tetramer, this augmentation may not be necessary as its size surpasses the renal filtration molecular mass cut-off giving it decent half-life for sufficient levels of in vivo efficacy. In fact, a bispecific antitoxin A/antitoxin B tetramer was shown to be protective of death in CDI-inflicted mice.80 This also supports the finding that Fc-mediated host effector functions are not needed for in vivo protection of antitoxin A/antitoxin B antibodies and direct toxin neutralization will serve the purpose.80 The increase in size and avidity in the fusion constructs may further enhance the neutralization potency of sdAbs.
The stability of sdAbs against the denaturing acidic environment and proteases of the GI tract, their superior and efficient folding, and the fact that they can be engineered into robust molecules make sdAbs attractive oral therapeutics against GI diseases such as CDI. One simple yet general stability engineering approach is disulfide engineering of sdAbs. The approach appears to be universally applicable to all VHs, VLs, and VHHs and results in antibodies with very high thermostability, resistance to low acidic (stomach) pH, and resistance to GI proteases (Figure 2B).36,41,49,130 As an example, disulfide engineering applied to antitoxin A VHHs (A4.2, A5.1, A20.1, and A26.8) rendered them highly thermostable and acidic pH and pepsin resistant without significantly compromising their neutralization capabilities.41,76 In addition, these robust domains can also be efficiently turned into aggregation-free sdAb–human Fc molecules (Figure 2B; example given for VLs). With respect to probiotic antibody-mediated oral therapy applications, sdAbs are the ideal therapeutic antibody format, both in secreted and cell-surface display modes, and several studies have shown the protection capability of VHH displaying Lactobacilli in animal models of infections.79,81,131,133,135–138,140 Owing to their stability and superior folding and expression properties, sdAbs are produced feasibly and efficiently by engineered host probiotic bacteria at higher levels than scFvs, in both secretion and cell-surface display formats, resulting in higher therapeutic efficacies.79,131,133 Interestingly, the aforementioned antitoxin A A20.1 and A26.8 VHHs were shown to be secreted in a functional form by the probiotic bacterium B. longum at a much higher efficiency than an scFv.79
In conclusion, antibodies are poised to become a critical tool for health care professionals to use in their fight against CDI; however, concerns over their cost and widespread adoption by physicians remain valid. Efforts to obtain more efficacious antitoxin antibodies and antibodies to novel C. difficile targets, as well as the search for alternative antibody delivery routes (eg, GI delivery), could provide low-cost options for treating primary CDI and preventing cases of relapse.
Disclosure
The authors report no conflicts of interest in this work.
References
Ghose C. Clostridium difficile infection in the twenty-first century. Emerg Microbes Infect. 2013;2(9):e62. | ||
Leffler DA, Lamont JT. Clostridium difficile infection. N Engl J Med. 2015;372(16):1539–1548. | ||
Rea MC, Dobson A, O’Sullivan O, et al. Effect of broad- and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4639–4644. | ||
Chaparro-Rojas F, Mullane KM. Emerging therapies for Clostridium difficile infection – focus on fidaxomicin. Infect Drug Resist. 2013;6:41–53. | ||
Mullane K. Fidaxomicin in Clostridium difficile infection: latest evidence and clinical guidance. Ther Adv Chronic Dis. 2014;5(2):69–84. | ||
Hussack G, Tanha J. Toxin-specific antibodies for the treatment of Clostridium difficile: current status and future perspectives. Toxins. 2010;2(5):998–1018. | ||
Simon MR, Chervin SM, Brown SC. Polyclonal antibody therapies for Clostridium difficile infection. Antibodies. 2014;3(4):272–288. | ||
Humphreys DP, Wilcox MH. Antibodies for treatment of Clostridium difficile infection. Clin Vaccine Immunol. 2014;21(7):913–923. | ||
Mathur H, Rea MC, Cotter PD, Ross RP, Hill C. The potential for emerging therapeutic options for Clostridium difficile infection. Gut Microbes. 2014;5(6):696–710. | ||
Ivarsson ME, Leroux JC, Castagner B. Investigational new treatments for Clostridium difficile infection. Drug Discov Today. 2015;20(5):602–608. | ||
Rupnik M, Wilcox MH, Gerding DN. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol. 2009;7(7):526–536. | ||
Mizrahi A, Collignon A, Péchiné S. Passive and active immunization strategies against Clostridium difficile infections: state of the art. Anaerobe. 2014;30:210–219. | ||
Jank T, Aktories K. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol. 2008;16(5):222–229. | ||
Jank T, Giesemann T, Aktories K. Rho-glucosylating Clostridium difficile toxins A and B: new insights into structure and function. Glycobiology. 2007;17(4):15R–22R. | ||
Lyras D, O’Connor JR, Howarth PM, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458(7242):1176–1179. | ||
Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467(7316):711–713. | ||
Govind R, Dupuy B. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog. 2012;8(6):e1002727. | ||
Leav BA, Blair B, Leney M, et al. Serum anti-toxin B antibody correlates with protection from recurrent Clostridium difficile infection (CDI). Vaccine. 2010;28(4):965–969. | ||
Katchar K, Taylor CP, Tummala S, Chen X, Sheikh J, Kelly CP. Association between IgG2 and IgG3 subclass responses to toxin A and recurrent Clostridium difficile-associated disease. Clin Gastroenterol Hepatol. 2007;5(6):707–713. | ||
Monaghan TM, Robins A, Knox A, Sewell HF, Mahida YR. Circulating antibody and memory B-cell responses to C. difficile toxins A and B in patients with C. difficile-associated diarrhoea, inflammatory bowel disease and cystic fibrosis. PLoS One. 2013;8(9):e74452. | ||
Aktories K. Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Microbiol. 2011;9(7):487–498. | ||
El Feghaly RE, Bangar H, Haslam DB. The molecular basis of Clostridium difficile disease and host response. Curr Opin Gastroenterol. 2015;31(1):24–29. | ||
Solomon K. The host immune response to Clostridium difficile infection. Ther Adv Infect Dis. 2013;1(1):19–35. | ||
Morrison C. Antibacterial antibodies gain traction. Nat Rev Drug Discov. 2015;14(11):737–738. | ||
Oleksiewicz MB, Nagy G, Nagy E. Anti-bacterial monoclonal antibodies: back to the future? Arch Biochem Biophys. 2012;526(2):124–131. | ||
Bebbington C, Yarranton G. Antibodies for the treatment of bacterial infections: current experience and future prospects. Curr Opin Biotechnol. 2008;19(6):613–619. | ||
Kink JA, Williams JA. Antibodies to recombinant Clostridium difficile toxins A and B are an effective treatment and prevent relapse of C. difficile-associated disease in a hamster model of infection. Infect Immun. 1998;66(5):2018–2025. | ||
Lyerly DM, Bostwick EF, Binion SB, Wilkins TD. Passive immunization of hamsters against disease caused by Clostridium difficile by use of bovine immunoglobulin G concentrate. Infect Immun. 1991;59(6):2215–2218. | ||
Roberts A, McGlashan J, Al-Abdulla I, et al. Development and evaluation of an ovine antibody-based platform for treatment of Clostridium difficile infection. Infect Immun. 2012;80(2):875–882. | ||
van Dissel JT, de Groot N, Hensgens CM, et al. Bovine antibody-enriched whey to aid in the prevention of a relapse of Clostridium difficile-associated diarrhoea: preclinical and preliminary clinical data. J Med Microbiol. 2005;54(pt 2):197–205. | ||
Abougergi MS, Kwon JH. Intravenous immunoglobulin for the treatment of Clostridium difficile infection: a review. Dig Dis Sci. 2011;56(1):19–26. | ||
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–558. | ||
Taylor CP, Tummala S, Molrine D, et al. Open-label, dose escalation phase I study in healthy volunteers to evaluate the safety and pharmacokinetics of a human monoclonal antibody to Clostridium difficile toxin A. Vaccine. 2008;26(27–28):3404–3409. | ||
Saylor C, Dadachova E, Casadevall A. Monoclonal antibody-based therapies for microbial diseases. Vaccine. 2009;27(suppl 6):G38–G46. | ||
Reichert JM, Dewitz MC. Anti-infective monoclonal antibodies: perils and promise of development. Nat Rev Drug Discov. 2006;5(3):191–195. | ||
Kim DY, Hussack G, Kandalaft H, Tanha J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim Biophys Acta. 2014;1844(11):1983–2001. | ||
Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–797. | ||
Zielonka S, Empting M, Grzeschik J, Könning D, Barelle CJ, Kolmar H. Structural insights and biomedical potential of IgNAR scaffolds from sharks. MAbs. 2015;7(1):15–25. | ||
Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23(9):1126–1136. | ||
Kim DS, Song HN, Nam HJ, et al. Directed evolution of human heavy chain variable domain (VH) using in vivo protein fitness filter. PLoS One. 2014;9(6):e98178. | ||
Hussack G, Hirama T, Ding W, Mackenzie R, Tanha J. Engineered single-domain antibodies with high protease resistance and thermal stability. PLoS One. 2011;6(11):e28218. | ||
Jespers L, Schon O, Famm K, Winter G. Aggregation-resistant domain antibodies selected on phage by heat denaturation. Nat Biotechnol. 2004;22(9):1161–1165. | ||
To R, Hirama T, Arbabi-Ghahroudi M, et al. Isolation of monomeric human VHs by a phage selection. J Biol Chem. 2005;280(50):41395–41403. | ||
Christ D, Famm K, Winter G. Repertoires of aggregation-resistant human antibody domains. Protein Eng Des Sel. 2007;20(8):413–416. | ||
Dudgeon K, Rouet R, Kokmeijer I, et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012;109(27):10879–10884. | ||
Rouet R, Lowe D, Christ D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014;588(2):269–277. | ||
Famm K, Hansen L, Christ D, Winter G. Thermodynamically stable aggregation-resistant antibody domains through directed evolution. J Mol Biol. 2008;376(4):926–931. | ||
Hussack G, Riazi A, Ryan S, et al. Protease-resistant single-domain antibodies inhibit Campylobacter jejuni motility. Protein Eng Des Sel. 2014;27(6):191–198. | ||
Kim DY, Kandalaft H, Ding W, et al. Disulfide linkage engineering for improving biophysical properties of human VH domains. Protein Eng Des Sel. 2012;25(10):581–589. | ||
Harmsen MM, van Solt CB, van Zijderveld-van Bemmel AM, Niewold TA, van Zijderveld FG. Selection and optimization of proteolytically stable llama single-domain antibody fragments for oral immunotherapy. Appl Microbiol Biotechnol. 2006;72(3):544–551. | ||
Desmyter A, Spinelli S, Roussel A, Cambillau C. Camelid nanobodies: killing two birds with one stone. Curr Opin Struct Biol. 2015;32:1–8. | ||
Griffiths K, Dolezal O, Parisi K, et al. Shark variable new antigen receptor (VNAR) single domain antibody fragments: stability and diagnostic applications. Antibodies. 2013;2(1):66–81. | ||
Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–3284. | ||
Ben Abderrazek R, Vincke C, Hmila I, et al. Development of Cys38 knock-out and humanized version of NbAahII10 nanobody with improved neutralization of AahII scorpion toxin. Protein Eng Des Sel. 2011;24(9):727–735. | ||
Kovalenko OV, Olland A, Piché-Nicholas N, et al. Atypical antigen recognition mode of a shark immunoglobulin new antigen receptor (IgNAR) variable domain characterized by humanization and structural analysis. J Biol Chem. 2013;288(24):17408–17419. | ||
Kovaleva M, Ferguson L, Steven J, Porter A, Barelle C. Shark variable new antigen receptor biologics – a novel technology platform for therapeutic drug development. Expert Opin Biol Ther. 2014;14(10):1527–1539. | ||
Babcock GJ, Broering TJ, Hernandez HJ, et al. Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect Immun. 2006;74(11):6339–6347. | ||
Lowy I, Molrine DC, Leav BA, et al. Treatment with monoclonal antibodies against Clostridium difficile toxins. N Engl J Med. 2010;362(3):197–205. | ||
Davies NL, Compson JE, Mackenzie B, et al. A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin Vaccine Immunol. 2013;20(3):377–390. | ||
Yang Z, Ramsey J, Hamza T, et al. Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect Immun. 2015;83(2):822–831. | ||
Orth P, Xiao L, Hernandez LD, et al. Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J Biol Chem. 2014;289(26):18008–18021. | ||
Steele J, Mukherjee J, Parry N, Tzipori S. Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J Infect Dis. 2013;207(2):323–330. | ||
Anosova NG, Cole LE, Li L, et al. A combination of three fully human toxin A- and toxin B-specific monoclonal antibodies protects against challenge with highly virulent epidemic strains of Clostridium difficile in the Hamster Model. Clin Vaccine Immunol. 2015;22(7):711–725. | ||
Marozsan AJ, Ma D, Nagashima KA, et al. Protection against Clostridium difficile infection with broadly neutralizing antitoxin monoclonal antibodies. J Infect Dis. 2012;206(5):706–713. | ||
Lyerly DM, Phelps CJ, Toth J, Wilkins TD. Characterization of toxins A and B of Clostridium difficile with monoclonal antibodies. Infect Immun. 1986;54(1):70–76. | ||
Frey SM, Wilkins TD. Localization of two epitopes recognized by monoclonal antibody PCG-4 on Clostridium difficile toxin A. Infect Immun. 1992;60(6):2488–2492. | ||
Demarest SJ, Hariharan M, Elia M, et al. Neutralization of Clostridium difficile toxin A using antibody combinations. MAbs. 2010;2(2):190–198. | ||
Modi N, Gulati N, Solomon K, et al. Differential binding and internalization of Clostridium difficile toxin A by human peripheral blood monocytes, neutrophils and lymphocytes. Scand J Immunol. 2011;74(3):264–271. | ||
Lyerly DM, Phelps CJ, Wilkins TD. Monoclonal and specific polyclonal antibodies for immunoassay of Clostridium difficile toxin A. J Clin Microbiol. 1985;21(1):12–14. | ||
Zhang C, Jin K, Xiao Y, et al. Potent monoclonal antibodies against Clostridium difficile toxin A elicited by DNA immunization. Hum Vaccin Immunother. 2013;9(10):2157–2164. | ||
He X, Sun X, Wang J, et al. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect Immun. 2009;77(6):2294–2303. | ||
He X, Wang J, Steele J, et al. An ultrasensitive rapid immunocytotoxicity assay for detecting Clostridium difficile toxins. J Microbiol Methods. 2009;78(1):97–100. | ||
Steele J, Chen K, Sun X, et al. Systemic dissemination of Clostridium difficile toxins A and B is associated with severe, fatal disease in animal models. J Infect Dis. 2012;205(3):384–391. | ||
Corthier G, Muller MC, Wilkins TD, Lyerly D, L’Haridon R. Protection against experimental pseudomembranous colitis in gnotobiotic mice by use of monoclonal antibodies against Clostridium difficile toxin A. Infect Immun. 1991;59(3):1192–1195. | ||
Hussack G, Arbabi-Ghahroudi M, Mackenzie CR, Tanha J. Isolation and characterization of Clostridium difficile toxin-specific single-domain antibodies. Methods Mol Biol. 2012;911:211–239. | ||
Hussack G, Arbabi-Ghahroudi M, van Faassen H, et al. Neutralization of Clostridium difficile toxin A with single-domain antibodies targeting the cell receptor binding domain. J Biol Chem. 2011;286(11):8961–8976. | ||
Hussack G, Keklikian A, Alsughayyir J, et al. A VL single-domain antibody library shows a high-propensity to yield non-aggregating binders. Protein Eng Des Sel. 2012;25(6):313–318. | ||
Murase T, Eugenio L, Schorr M, et al. Structural basis for antibody recognition in the receptor-binding domains of toxins A and B from Clostridium difficile. J Biol Chem. 2013;289(4):2331–2343. | ||
Shkoporov AN, Khokhlova EV, Savochkin KA, Kafarskaia LI, Efimov BA. Production of biologically active scFv and VHH antibody fragments in Bifidobacterium longum. FEMS Microbiol Lett. 2015;362(12):fnv083. | ||
Yang Z, Schmidt D, Liu W, et al. A novel multivalent, single-domain antibody targeting TcdA and TcdB prevents fulminant Clostridium difficile infection in mice. J Infect Dis. 2014;210(6):964–972. | ||
Andersen KK, Strokappe NM, Hultberg A, et al. Neutralization of Clostridium difficile toxin B mediated by engineered lactobacilli producing single domain antibodies. Infect Immun. 2015;84(2):395–406. | ||
Péchiné S, Janoir C, Collignon A. Variability of Clostridium difficile surface proteins and specific serum antibody response in patients with Clostridium difficile-associated disease. J Clin Microbiol. 2005;43(10):5018–5025. | ||
Leuzzi R, Adamo R, Scarselli M. Vaccines against Clostridium difficile. Hum Vaccin Immunother. 2014;10(6):1466–1477. | ||
Ghose C, Kelly CP. The prospect for vaccines to prevent Clostridium difficile infection. Infect Dis Clin North Am. 2015;29(1):145–162. | ||
Wang S, Rustandi RR, Lancaster C, et al. Toxicity assessment of Clostridium difficile toxins in rodent models and protection of vaccination. Vaccine. 2016;34(10):1319–1323. | ||
Gerding DN, Johnson S, Rupnik M, Aktories K. Clostridium difficile binary toxin CDT: mechanism, epidemiology, and potential clinical importance. Gut Microbes. 2014;5(1):15–27. | ||
Janoir C. Virulence factors of Clostridium difficile and their role during infection. Anaerobe. 2016;37:13–24. | ||
Unger M, Eichhoff AM, Schumacher L, et al. Selection of nanobodies that block the enzymatic and cytotoxic activities of the binary Clostridium difficile toxin CDT. Sci Rep. 2015;5:7850. | ||
Fagan RP, Fairweather NF. Biogenesis and functions of bacterial S-layers. Nat Rev Microbiol. 2014;12(3):211–222. | ||
Sleytr UB, Beveridge TJ. Bacterial S-layers. Trends Microbiol. 1999;7(6):253–260. | ||
Calabi E, Ward S, Wren B, et al. Molecular characterization of the surface layer proteins from Clostridium difficile. Mol Microbiol. 2001;40(5):1187–1199. | ||
Calabi E, Calabi F, Phillips AD, Fairweather NF. Binding of Clostridium difficile surface layer proteins to gastrointestinal tissues. Infect Immun. 2002;70(10):5770–5778. | ||
Drudy D, O’Donoghue DP, Baird A, Fenelon L, O’Farrelly C. Flow cytometric analysis of Clostridium difficile adherence to human intestinal epithelial cells. J Med Microbiol. 2001;50(6):526–534. | ||
Merrigan MM, Venugopal A, Roxas JL, et al. Surface-layer protein A (SlpA) is a major contributor to host-cell adherence of Clostridium difficile. PLoS One. 2013;8(11):e78404. | ||
Takumi K, Susami Y, Takeoka A, Oka T, Koga T. S layer protein of Clostridium tetani: purification and properties. Microbiol Immunol. 1991;35(7):569–575. | ||
Stevenson E, Minton NP, Kuehne SA. The role of flagella in Clostridium difficile pathogenicity. Trends Microbiol. 2015;23(5):275–282. | ||
Tasteyre A, Barc MC, Collignon A, Boureau H, Karjalainen T. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect Immun. 2001;69(12):7937–7940. | ||
Mulvey GL, Dingle TC, Fang L, Strecker J, Armstrong GD. Therapeutic potential of egg yolk antibodies for treating Clostridium difficile infection. J Med Microbiol. 2011;60(pt 8):1181–1187. | ||
Pantaléon V, Soavelomandroso AP, Bouttier S, et al. The Clostridium difficile protease Cwp84 modulates both biofilm formation and cell-surface properties. PLoS One. 2015;10(4):e0124971. | ||
O’Brien JB, McCabe MS, Athié-Morales V, McDonald GS, Ní Eidhin DB, Kelleher DP. Passive immunisation of hamsters against Clostridium difficile infection using antibodies to surface layer proteins. FEMS Microbiol Lett. 2005;246(2):199–205. | ||
Péchiné S, Janoir C, Boureau H, et al. Diminished intestinal colonization by Clostridium difficile and immune response in mice after mucosal immunization with surface proteins of Clostridium difficile. Vaccine. 2007;25(20):3946–3954. | ||
Ní Eidhin DB, O’Brien JB, McCabe MS, Athié-Morales V, Kelleher DP. Active immunization of hamsters against Clostridium difficile infection using surface-layer protein. FEMS Immunol Med Microbiol. 2008;52(2):207–218. | ||
Drudy D, Calabi E, Kyne L, et al. Human antibody response to surface layer proteins in Clostridium difficile infection. FEMS Immunol Med Microbiol. 2004;41(3):237–242. | ||
Kandalaft H, Hussack G, Aubry A, et al. Targeting surface-layer proteins with single-domain antibodies: a potential therapeutic approach against Clostridium difficile-associated disease. Appl Microbiol Biotechnol. 2015;99(20):8549–8562. | ||
Péchiné S, Denéve C, Le Monnier A, Hoys S, Janoir C, Collignon A. Immunization of hamsters against Clostridium difficile infection using the Cwp84 protease as an antigen. FEMS Immunol Med Microbiol. 2011;63(1):73–81. | ||
Sandolo C, Péchiné S, Le Monnier A, et al. Encapsulation of Cwp84 into pectin beads for oral vaccination against Clostridium difficile. Eur J Pharm Biopharm. 2011;79(3):566–573. | ||
Ganeshapillai J, Vinogradov E, Rousseau J, Weese JS, Monteiro MA. Clostridium difficile cell-surface polysaccharides composed of pentaglycosyl and hexaglycosyl phosphate repeating units. Carbohydr Res. 2008;343(4):703–710. | ||
Oberli MA, Hecht ML, Bindschädler P, Adibekian A, Adam T, Seeberger PH. A possible oligosaccharide-conjugate vaccine candidate for Clostridium difficile is antigenic and immunogenic. Chem Biol. 2011;18(5):580–588. | ||
Monteiro MA, Ma Z, Bertolo L, et al. Carbohydrate-based Clostridium difficile vaccines. Expert Rev Vaccines. 2013;12(4):421–431. | ||
Adamo R, Romano MR, Berti F, et al. Phosphorylation of the synthetic hexasaccharide repeating unit is essential for the induction of antibodies to Clostridium difficile PSII cell wall polysaccharide. ACS Chem Biol. 2012;7(8):1420–1428. | ||
Romano MR, Leuzzi R, Cappelletti E, et al. Recombinant Clostridium difficile toxin fragments as carrier protein for PSII surface polysaccharide preserve their neutralizing activity. Toxins. 2014;6(4):1385–1396. | ||
Dapa T, Unnikrishnan M. Biofilm formation by Clostridium difficile. Gut Microbes. 2013;4(5):397–402. | ||
Reid CW, Vinogradov E, Li J, Jarrell HC, Logan SM, Brisson JR. Structural characterization of surface glycans from Clostridium difficile. Carbohydr Res. 2012;354:65–73. | ||
Cox AD, St Michael F, Aubry A, et al. Investigating the candidacy of a lipoteichoic acid-based glycoconjugate as a vaccine to combat Clostridium difficile infection. Glycoconj J. 2013;30(9):843–855. | ||
Kontermann RE. Dual targeting strategies with bispecific antibodies. MAbs. 2012;4(2):182–197. | ||
Castoldi R, Jucknischke U, Pradel LP, et al. Molecular characterization of novel trispecific ErbB-cMet-IGF1R antibodies and their antigen-binding properties. Protein Eng Des Sel. 2012;25(10):551–559. | ||
Jachimowicz RD, Borchmann S, Rothe A. Multi-specific antibodies for cancer immunotherapy. BioDrugs. 2014;28(4):331–343. | ||
Weidle UH, Tiefenthaler G, Weiss EH, Georges G, Brinkmann U. The intriguing options of multispecific antibody formats for treatment of cancer. Cancer Genomics Proteomics. 2013;10(1):1–18. | ||
Dimasi N, Fleming R, Hay C, et al. Development of a trispecific antibody designed to simultaneously and efficiently target three different antigens on tumor cells. Mol Pharm. 2015;12(9):3490–3501. | ||
Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67(2 pt A):95–106. | ||
DiGiandomenico A, Keller AE, Gao C, et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci Transl Med. 2014;6(262):262ra155. | ||
Kingwell K. Infectious diseases: two-hit antibody tackles bacteria. Nat Rev Drug Discov. 2015;14(1):15. | ||
Zhang Z, Chen X, Hernandez LD, et al. Toxin-mediated paracellular transport of antitoxin antibodies facilitates protection against Clostridium difficile infection. Infect Immun. 2015;83(1):405–416. | ||
Jones RG, Martino A. Targeted localized use of therapeutic antibodies: a review of non-systemic, topical and oral applications. Crit Rev Biotechnol. 2016;36(3):506–520. | ||
Reilly RM, Domingo R, Sandhu J. Oral delivery of antibodies. Future pharmacokinetic trends. Clin Pharmacokinet. 1997;32(4):313–323. | ||
Wells JM, Mercenier A. Mucosal delivery of therapeutic and prophylactic molecules using lactic acid bacteria. Nat Rev Microbiol. 2008;6(5):349–362. | ||
Numan SC, Veldkamp P, Kuijper EJ, van den Berg RJ, van Dissel JT. Clostridium difficile-associated diarrhoea: bovine anti-Clostridium difficile whey protein to help aid the prevention of relapses. Gut. 2007;56(6):888–889. | ||
Tjellström B, Stenhammar L, Eriksson S, Magnusson KE. Oral immunoglobulin A supplement in treatment of Clostridium difficile enteritis. Lancet. 1993;341(8846):701–702. | ||
Rahman S, Van Nguyen S, Icatlo FC Jr, Umeda K, Kodama Y. Oral passive IgY-based immunotherapeutics: a novel solution for prevention and treatment of alimentary tract diseases. Hum Vaccin Immunother. 2013;9(5):1039–1048. | ||
Kim DY, To R, Kandalaft H, et al. Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. MAbs. 2014;6(1):219–235. | ||
Pant N, Hultberg A, Zhao Y, et al. Lactobacilli expressing variable domain of llama heavy-chain antibody fragments (lactobodies) confer protection against rotavirus-induced diarrhea. J Infect Dis. 2006;194(11):1580–1588. | ||
Krüger C, Hu Y, Pan Q, et al. In situ delivery of passive immunity by lactobacilli producing single-chain antibodies. Nat Biotechnol. 2002;20(7):702–706. | ||
Martín MC, Pant N, Ladero V, et al. Integrative expression system for delivery of antibody fragments by lactobacilli. Appl Environ Microbiol. 2011;77(6):2174–2179. | ||
Andersen KK, Marcotte H, Álvarez B, Boyaka PN, Hammarström L. In situ gastrointestinal protection against anthrax edema toxin by single-chain antibody fragment producing lactobacilli. BMC Biotechnol. 2011;11:126. | ||
de Marco A. Recombinant antibody production evolves into multiple options aimed at yielding reagents suitable for application-specific needs. Microb Cell Fact. 2015;14:125. | ||
Álvarez B, Krogh-Andersen K, Tellgren-Roth C, et al. An exopolysaccharide-deficient mutant of Lactobacillus rhamnosus GG efficiently displays a protective llama antibody fragment against rotavirus on its surface. Appl Environ Microbiol. 2015;81(17):5784–5793. | ||
Günaydin G, Álvarez B, Lin Y, Hammarström L, Marcotte H. Co-expression of anti-rotavirus proteins (llama VHH antibody fragments) in Lactobacillus: development and functionality of vectors containing two expression cassettes in tandem. PLoS One. 2014;9(4):e96409. | ||
Pant N, Marcotte H, Hermans P, et al. Lactobacilli producing bispecific llama-derived anti-rotavirus proteins in vivo for rotavirus-induced diarrhea. Future Microbiol. 2011;6(5):583–593. | ||
Beninati C, Oggioni MR, Boccanera M, et al. Therapy of mucosal candidiasis by expression of an anti-idiotype in human commensal bacteria. Nat Biotechnol. 2000;18(10):1060–1064. | ||
Maffey L, Vega CG, Parreno V, Garaicoechea L. Controlling Rotavirus-associated diarrhea: could single-domain antibody fragments make the difference? Rev Argent Microbiol. 2015;47(4):368–379. | ||
Cheung AT, Dayanandan B, Lewis JT, et al. Glucose-dependent insulin release from genetically engineered K cells. Science. 2000;290(5498):1959–1962. | ||
Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. | ||
Coppieters K, Dreier T, Silence K, et al. Formatted anti-tumor necrosis factor a VHH proteins derived from camelids show superior potency and targeting to inflamed joints in a murine model of collagen-induced arthritis. Arthritis Rheum. 2006;54(6):1856–1866. | ||
Terryn S, Francart A, Lamoral S, et al. Protective effect of different anti-rabies virus VHH constructs against rabies disease in mice. PLoS One. 2014;9(10):e109367. | ||
Gainkam LO, Huang L, Caveliers V, et al. Comparison of the biodistribution and tumor targeting of two 99mTc-labeled anti-EGFR nanobodies in mice, using pinhole SPECT/micro-CT. J Nucl Med. 2008;49(5):788–795. | ||
Vugmeyster Y, Entrican CA, Joyce AP, et al. Pharmacokinetic, biodistribution, and biophysical profiles of TNF nanobodies conjugated to linear or branched poly(ethylene glycol). Bioconjug Chem. 2012;23(7):1452–1462. | ||
Rashidian M, Wang L, Eden JG, et al. Enzyme-mediated modification of single-domain antibodies for imaging modalities with different characteristics. Angew Chem Int Ed Engl. 2016;55(2):528–533. | ||
Iqbal U, Trojahn U, Albaghdadi H, et al. Kinetic analysis of novel mono- and multivalent VHH-fragments and their application for molecular imaging of brain tumours. Br J Pharmacol. 2010;160(4):1016–1028. | ||
Tijink BM, Laeremans T, Budde M, et al. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. 2008;7(8):2288–2297. | ||
Müller MR, Saunders K, Grace C, et al. Improving the pharmacokinetic properties of biologics by fusion to an anti-HSA shark VNAR domain. MAbs. 2012;4(6):673–685. | ||
Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015;29(4):215–239. | ||
Sleep D, Cameron J, Evans LR. Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta. 2013;1830(12):5526–5534. | ||
Rotman M, Welling MM, van den Boogaard ML, et al. Fusion of hIgG1-Fc to 111In-anti-amyloid single domain antibody fragment VHH-pa2H prolongs blood residential time in APP/PS1 mice but does not increase brain uptake. Nucl Med Biol. 2015;42(8):695–702. | ||
Richard G, Meyers AJ, McLean MD, Arbabi-Ghahroudi M, MacKenzie R, Hall JC. In vivo neutralization of a-cobratoxin with high-affinity llama single-domain antibodies (VHHs) and a VHH-Fc antibody. PLoS One. 2013;8(7):e69495. | ||
Pruitt RN, Chambers MG, Ng KK, Ohi MD, Lacy DB. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A. 2010;107(30):13467–13472. | ||
Chumbler NM, Rutherford SA, Zhang Z, et al. Crystal structure of Clostridium difficile toxin A. Nat Microbiol. 2016;1(1):15002. | ||
Albesa-Jove D, Bertrand T, Carpenter EP, et al. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J Mol Biol. 2010;396(5):1260–1270. | ||
Pruitt RN, Chumbler NM, Rutherford SA, et al. Structural determinants of Clostridium difficile toxin A glucosyltransferase activity. J Biol Chem. 2012;287(11):8013–8020. | ||
Bradshaw WJ, Kirby JM, Thiyagarajan N, et al. The structure of the cysteine protease and lectin-like domains of Cwp84, a surface layer-associated protein from Clostridium difficile. Acta Crystallogr D Biol Crystallogr. 2014;70(pt 7):1983–1993. | ||
Bradshaw WJ, Roberts AK, Shone CC, Acharya KR. Cwp84, a Clostridium difficile cysteine protease, exhibits conformational flexibility in the absence of its propeptide. Acta Crystallogr F Struct Biol Commun. 2015;71(pt 3):295–303. | ||
Gutelius D, Hokeness K, Logan SM, Reid CW. Functional analysis of SleC from Clostridium difficile: an essential lytic transglycosylase involved in spore germination. Microbiology. 2014;160(pt 1):209–216. | ||
Twine SM, Reid CW, Aubry A, et al. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol. 2009;191(22):7050–7062. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.