Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
An optimized nanoparticle delivery system based on chitosan and chondroitin sulfate molecules reduces the toxicity of amphotericin B and is effective in treating tegumentary leishmaniasis
Authors Ribeiro T, Ribeiro J, Fuscaldi L, Santos M, Duarte M, Lage P, Martins V, Costa L, Fernandes S, Cardoso V, Castilho R, Soto M, Tavares CA, Faraco A ![]() , Coelho E, Chávez-Fumagalli MA
, Coelho E, Chávez-Fumagalli MA
Received 7 June 2014
Accepted for publication 26 July 2014
Published 19 November 2014 Volume 2014:9(1) Pages 5341—5353
DOI https://doi.org/10.2147/IJN.S68966
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Tatiana G Ribeiro,1 Juçara R Franca,1 Leonardo L Fuscaldi,1 Mara L Santos,2 Mariana C Duarte,3 Paula S Lage,3 Vivian T Martins,4 Lourena E Costa,3 Simone OA Fernandes,1,5 Valbert N Cardoso,1,5 Rachel O Castilho,1,6 Manuel Soto,7 Carlos AP Tavares,4 André AG Faraco,1,6 Eduardo AF Coelho,3,8,* Miguel A Chávez-Fumagalli3,*
1Programa de Pós-Graduação em Ciências Farmacêuticas, Faculdade de Farmácia, 2Departamento de Morfologia, Instituto de Ciências Biológicas, 3Programa de Pós-Graduação em Ciências da Saúde: Infectologia e Medicina Tropical, Faculdade de Medicina, 4Departamento de Bioquímica e Imunologia, Instituto de Ciências Biológicas, 5Departamento de Análises Clínicas e Toxicológicas, 6Departamento de Produtos Farmacêuticos, Faculdade de Farmácia, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil; 7Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Departamento de Biología Molecular, Universidad Autónoma de Madrid, Madrid, Spain; 8Departamento de Patologia Clínica, COLTEC, Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais, Brazil
*These authors contributed equally to this work
Abstract: Amphotericin B (AmpB) is active against leishmaniasis, but its use is hampered due to its high toxicity observed in patients. In this study, a nanoparticles-delivery system for AmpB (NQC-AmpB), containing chitosan and chondroitin sulfate molecules, was evaluated in BALB/c mice against Leishmania amazonensis. An in vivo biodistribution study, including biochemical and toxicological evaluations, was performed to evaluate the toxicity of AmpB. Nanoparticles were radiolabeled with technetium-99m and injected in mice. The products presented a similar biodistribution in the liver, spleen, and kidneys of the animals. Free AmpB induced alterations in the body weight of the mice, which, in the biochemical analysis, indicated hepatic and renal injury, as well as morphological damage to the kidneys of the animals. In general, no significant organic alteration was observed in the animals treated with NQC-AmpB. Mice were infected with L. amazonensis and treated with the nanoparticles or free AmpB; then, parasitological and immunological analyses were performed. The NQC-AmpB group, as compared to the control groups, presented significant reductions in the lesion size and in the parasite burden in all evaluated organs. These animals presented significantly higher levels of IFN-γ and IL-12, and low levels of IL-4 and IL-10, when compared to the control groups. The NQC-AmpB system was effective in reducing the infection in the animals, and proved to be effective in diminishing the toxicity evoked by AmpB, which was observed when it was administered alone. In conclusion, NQC-AmpB could be considered a viable possibility for future studies in the treatment of leishmaniasis.
Keywords: in vivo treatment, Leishmania amazonensis, nanoparticles, chitosan, chondroitin sulfate
Introduction
Leishmaniasis is a disease with a wide spectrum of clinical manifestations caused by protozoa belonging to the Leishmania genus.1 Currently, nearly 350 million people in 98 countries are at risk of contracting the infection,2 whereas between 700,000 and 1.2 million cases of cutaneous leishmaniasis, and about 500,000 cases of visceral leishmaniasis, are diagnosed annually worldwide.3
The first choice to treat leishmaniasis has been the employ of pentavalent antimony; however, the side effects, such as myalgias, arthralgias, pancreatitis, leukopenia, and cardiotoxicity, are problems reported by patients.4,5 Amphotericin B (AmpB), a polyene used in the treatment of the disease, is a highly hydrophobic antifungal drug. It is active against Leishmania, but its clinical use is hampered mainly due to its high toxicity, including nephrotoxicity, hemolysis, and liver damage, as well as nausea and fever.6,7
To improve the therapeutic index and reduce the toxicity of this drug, lipid-based formulations have been developed. In this context, the World Health Organization has recommended the use of liposomal AmpB.8 Despite the improvement in the therapeutic index for its formulations, its use still remains limited due to the high cost.9 Therefore, the development of other delivery systems by incorporating effective drugs to treat leishmaniasis, and aimed at reducing their side effects, as well as presenting an accessible cost, could be considered desirable.10
Delivery systems using nanospheres, liposomes, or microspheres could result in a higher concentration and a slower distribution of drugs in organs, such as the spleen, liver, and kidneys.11–16 In one prior study performed by our group, an optimized nanoparticles system composed of chitosan (Cs), chondroitin sulfate (ChS), and AmpB (namely, NQC-AmpB); was developed and evaluated to determine its in vitro antileishmanial activity against extracellular promastigotes and intracellular amastigotes of L. amazonensis and L. infantum.17 In this study, the Cs nanoparticles (NQ), Cs–ChS nanoparticles (NQC), and NQC-AmpB nanoparticles all presented an in vitro antileishmanial activity, associated with a low toxicity in murine macrophages, as well as a null hemolytic activity in type O+ human red blood cells, whereas the use of AmpB in a free form evoked a high toxicity in the mammal cells. The study concluded that the engineered NQC-AmpB nanoparticles presented the best results against Leishmania, and led to the possibility of this nanosystem being evaluated in in vivo models against leishmaniasis.17
In this context, the present study aims to investigate NQC-AmpB nanoparticles by evaluating their in vivo biodistribution profile in mice, as well as toxicological and biochemical parameters in the treated animals, to verify the effectiveness of this system in treating mice infected with L. amazonensis, in an attempt to find better chemotherapeutic alternatives against leishmaniasis.
Materials and methods
Ethics statement
Experiments were performed in compliance with the National Guidelines of the Institutional Animal Care and Use Committee for the Ethical Handling of Research Animals (CEUA) from the Federal University of Minas Gerais (UFMG) (Law number 11.794, 2008), which approved this study under protocol number 182/2012.
Mice
Female BALB/c and Swiss mice (8 weeks age) were obtained from the breeding facilities of the Department of Biochemistry and Immunology, Institute of Biological Sciences, UFMG, and were maintained under specific pathogen-free conditions. All animals used in the present study were euthanized at the end of the experiments, with the toxicological, biochemical, parasitological, and/or immunological analyses being used as the murine endpoints in the experiments.
Preparation of NQ, NQC, and NQC-AmpB nanoparticles
All nanoparticles used in this study were prepared and characterized by polyelectrolyte complexes technique, as described.17
In vivo biodistribution studies
The in vivo biodistribution studies were conducted using nanoparticles radiolabeled with technetium-99m (99mTc). The prepared nanoparticles were radiolabeled with 99mTc, according to the following procedure. Briefly, 1 mL of aqueous solution containing the nanoparticles (1 mg/mL), 10 μL of a SnCl22H2O solution (diluted in 0.25 N HCl 1 mg/mL), and 10 μL of a NaHB4 solution (diluted in 0.25 N NaOH 5 mg/mL) were added to a vial, with the pH adjusted to 7.0. After, the vial was sealed, and a vacuum was made. Next, an aliquot containing 37 MBq of Na99mTcO4 was added to the mixture. The solution was stirred at 60°C for 30 min and filtered using a sterile syringe filter with a 0.45 μm pore size hydrophilic PVDF membrane (Thomas Scientific, Swedesboro, NJ, USA). Radiochemical purity of 99mTc nanoparticles was determined by means of two chromatographic systems: ascending chromatography on a silica gel strip (Merck KGaA, Darmstadt, Germany) using methyl ethyl ketone to determine the amount of pure technetium (99mTcO4−); and by descending chromatography on a strip of Whatman paper No. 1 that had been previously saturated with 1% serum bovine albumin solution, using saline to determine the amount of hydrolyzed technetium (99mTcO2). Radioactivity was measured using an automatic gamma counter (Wallac 1470 Wizard Gamma Counter, Perkin Elmer, Turku, Finland).18–20
For scintigraphic images, Swiss mice (n=8 per group) were assigned to one of the following groups: NQ, NQC, and NQC-AmpB nanoparticles. Aliquots containing 3.7 MBq of the respective 99mTc nanoparticles were administered intravenously into the animals. At 0.5, 2, 4, 8, and 24 hours after administration, mice were anesthetized and placed in a supine position under a gamma camera (Mediso Medical Imaging Systems, Budapest, Hungary) using a low-energy high-resolution collimator. Images were acquired using a 256×256×16 matrix size, with a 20% energy window set at 140 keV for a period of 600 seconds. For the in vivo biodistribution analyses, Swiss mice (n=8 per group) were placed in those groups that received NQ, NQC, or NQC-AmpB nanoparticles. For this, 3.7 MBq of the respective 99mTc nanoparticles were administered intravenously into the animals, which were anesthetized and euthanized at 0.5, 2, 4, 8, and 24 hours, after this administration. Blood samples, heart, lungs, spleen, liver, and kidneys were harvested for analysis. Each organ or tissue was weighed, and its associated radioactivity was determined in an automatic gamma counter. A standard dose, containing the same amount of radioactivity injected into the animals, was analyzed simultaneously and defined as 100% of radioactivity. The results were expressed as the percentage of the injected dose per gram of tissue (% ID/g) according to the following equation:
| (1) |
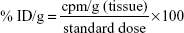
In vivo toxicity studies
Swiss mice (n=8 per group) were treated daily, from day 0 to 10 days after the first administration of the dose, by intravenous injection, as described below:
- Control group: mice received 100 μL of a phosphate-buffered saline (PBS) 1× solution.
- AmpB group: mice received 100 μL of a solution containing 1 mg/kg body weight of AmpB.
- NQ group: mice received 100 μL of Cs nanoparticles, with the same amount of Cs in the NQC-AmpB nanoparticles.
- NQC group: mice received 100 μL of Cs–ChS nanoparticles, with an equivalent distribution of these molecules in the nanoparticles.
- NQC-AmpB group: mice received 100 μL of AmpB-loaded Cs–ChS nanoparticles containing 1 mg/kg of body weight of AmpB.
During 12 days, variations in the body weight of the mice were monitored, as was the time at which toxicity appeared or death occurred. In addition, 2 days after the end of the treatment, blood samples were collected for biochemical and hematological analysis. The cardiac function was analyzed by the dosage of creatine kinase-myocardial band isoenzyme (CK/MB); and the hepatic function was analyzed by dosages of gamma-glutamyl transferase (GGT), aspartate aminotransferase (AST), and alanine aminotransferase (ALT). Nephrotoxicity was evaluated in the blood samples by examining the levels of blood urea nitrogen (BUN) and serum creatinine. All biochemical assays were performed using commercial kits (Labtest Diagnostica, Minas Gerais, Brazil) and an auto-analyzer apparatus (Thermo Plate TP analyzer), according to manufacturer instructions.
For the histopathological analyses, heart, liver, and spleen were removed from the animals, 2 days after the end of the treatments. The organs were washed in a NaCl 0.9% solution, and set in 10% buffered formalin. All tissues were embedded in paraffin blocks, sectioned in 5 μm thickness using a microtome (Leica RM2245; Leica Microsystems, Wetzlar, Germany), and placed onto glass slides. After hematoxylin–eosin staining, slides were observed and photos were taken using an optical microscope.
In vivo treatment
Experiments were carried out using L. amazonensis (IFLA/BR/1967/PH-8). Parasites were grown at 24°C in Schneider’s medium (Sigma-Aldrich Co., St Louis, MO, USA), supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich Co.), 20 mM L-glutamine, 200 U/mL penicillin, and 100 μg/mL streptomycin, at pH 7.4. The soluble Leishmania antigen (SLA) extract was prepared from 1×1010 stationary-phase promastigotes, as previously described.21
BALB/c mice (n=8 per group) were infected through subcutaneous injection with 5×106 stationary-phase promastigotes of L. amazonensis, and the development of lesions was monitored using an electronic caliper (799–6/150 model; Starrett®, Brazil). After the development of ulcerated lesions (at approximately 87–94 days postinfection), the animals were divided into groups according to lesion size to ensure similar average lesion sizes among the treated groups. Animals were then treated daily for 10 days, receiving intravenous injections containing one of the following regimens:
- Control group: mice received 100 μL of a PBS 1× solution.
- AmpB group: mice received 100 μL of 1 mg/kg body weight of AmpB.
- NQ group: mice received 100 μL of Cs nanoparticles, at equivalent amounts of Cs in relation to the NQC-AmpB nanoparticles.
- NQC group: mice received 100 μL of empty Cs–ChS nanoparticles, at equal amounts of Cs–ChS in relation to the NQC-AmpB nanoparticles.
- NQC-AmpB group: mice received 100 μL of AmpB-loaded Cs–ChS nanoparticles in the dosage of 1 mg/kg body weight of AmpB.
During and after treatment, lesion size was followed weekly to measure the diameter of the lesions.22 Further evaluations, through the careful observation of the lesions – including observations of the appearance of relapses and nodules, as well as metastasis in other regions of the body of the animals – were also performed. The infected mice were observed for an additional 30-day period after the interruption of treatment.
Animals were euthanized at 30 days after the end of treatment. Efficacy was evaluated by measuring the lesion size and the parasite load at the site of infection, as well as in the spleen, liver, and draining lymph nodes (dLN), using a limiting dilution technique.23 Briefly, organs were weighed and homogenized, using a glass tissue grinder in sterile PBS 1×. Tissue debris was removed by centrifugation at 150× g, and cells were concentrated by centrifugation at 2,000× g. The pellet was resuspended in 1 mL of Schneider’s medium supplemented with 20% fetal bovine serum. Two hundred and twenty microliters of the resuspension was plated onto 96-well flat-bottom microtiter plates (Nunc, Nunclon, Roskilde, Denmark), and diluted in log-fold serial dilutions in supplemented Schneider’s culture medium with a 10−1 to 10−12 dilution. Each sample was plated in triplicate and read 7 days after the beginning of the culture, at 24°C. Pipette tips were discarded after each dilution to avoid carrying adhered parasites from one well to another. Results are expressed as the negative log of the titer (ie, the dilution corresponding to the last positive well) adjusted per microgram of tissue.
Splenocyte cultures and cytokine assays were performed at 30 days after the end of the treatment, as previously described.21 Briefly, single-cell preparations from spleen tissue were plated in duplicate in 24-well plates (Nunc) at 5×106 cells per mL. Cells were incubated in Dulbecco’s Modified Eagle’s Medium (nonstimulated, background control), or separately stimulated with SLA (50 μg·mL−1) or with the respective nanoparticles (10 μg·mL−1), at 37°C in 5% CO2 for 48 h. IFN-γ, IL-4, IL-10, and IL-12 levels were assessed in the supernatants by a sandwich enzyme-linked immunosorbent assay (ELISA) provided in commercial kits (BD OptEIA TM set mouse IFN-γ, IL-4, IL-10, and IL-12; Pharmingen, San Diego, CA, USA), following the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism™ (version 6.0 for Windows). Results are expressed as mean ± standard deviation. The normality and homogeneity of the variance analysis of the data was performed using the Kolmogorov–Smirnov and Bartlett’s tests, respectively. In the biodistribution assays, two-way analysis of variance (ANOVA) was used to compare differences between the groups. To analyze the variation in body weight, the value before treatment was used as the covariate,24 and one-way ANOVA, followed by Bonferroni’s post-test, was used to compare differences between the groups. To analyze biochemical parameters, the computed normalized absorbance ratios were used, by correcting the mean optical density value for each sample from NQ, NQC, NQC-AmpB, and free AmpB groups, and dividing it by the mean optical density value from the control group. As a consequence, the normalized absorbance ratios represent multiples of reactivity in relation to the control group.25 To analyze the lesion size, parasite burden, and immunological response between the groups, the two-way ANOVA and the area under the curve, followed by Bonferroni’s post-test, were used. Differences were considered statistically significant when P<0.05. Data shown are representative of two independent experiments, which presented similar results.
Results
In vivo biodistribution studies
The NQ, NQC, and NQC-AmpB nanoparticles radiolabeled with 99mTc presented radiochemical purities of 89.4%±3.2%, 86.3%±2.0%, and 84.0%±2.0%, respectively, thus allowing for further in vivo assays. The NQC and NQC-AmpB nanoparticles presented similar biodistribution profiles in the spleen and liver of the treated animals, during the period of time that they were evaluated; however, at 24 h after administration, the NQC presented a higher presence in the spleen, when compared to the NQC-AmpB nanoparticles (Figure 1A). This evaluation could be also observed in the scintigraphic images, demonstrating a decreasing of the biodistribution profile of the nanoparticles in the abdominal region of the animals, after 24 h of administration (Figure 1B).
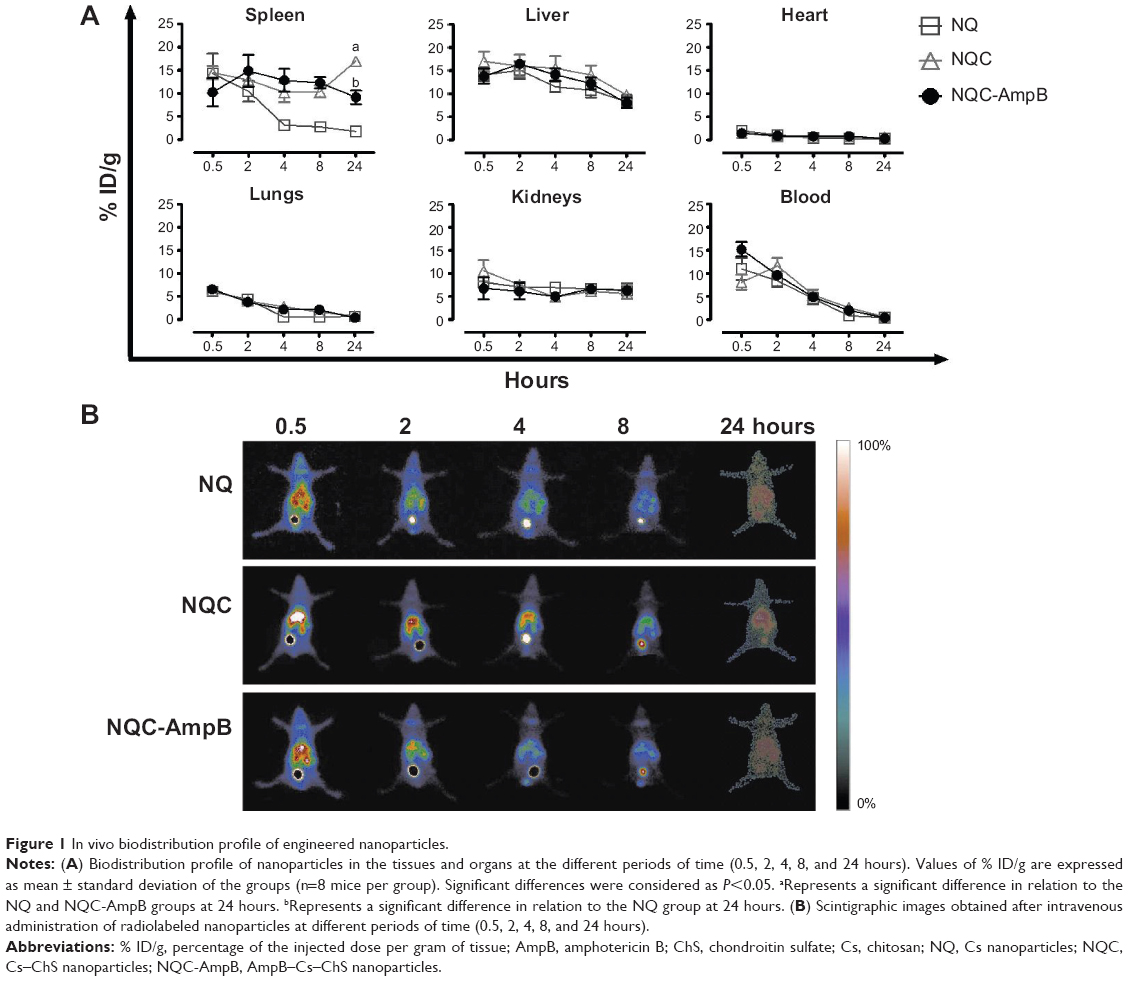 | Figure 1 In vivo biodistribution profile of engineered nanoparticles. |
In vivo toxicity studies
Some clinical symptoms, such as ataxia and weakness, were observed in the free AmpB-treated mice, while in the animals treated with NQ, NQC, or NQC-AmpB nanoparticles, no clinical alteration was observed. No death was reported in any group during the experiments. The body weight of the mice was monitored from 0 to 12 days during the treatment, and Figure 2 shows the results. The control group (nontreated and noninfected) presented a positive variation in weight, reaching a maximum of up to 6% relative to the weight of the animals, as compared to the beginning of the treatment. The NQ and NQC groups presented a light negative variation in weight of up to 4%. The NQC-AmpB group presented a negative variation in weight, reaching a maximum of 5%. However, for the free AmpB group, the loss of body weight presented a negative variation of 15%, possibly reflecting the high toxicity of this drug observed in the animals (Figure 2).
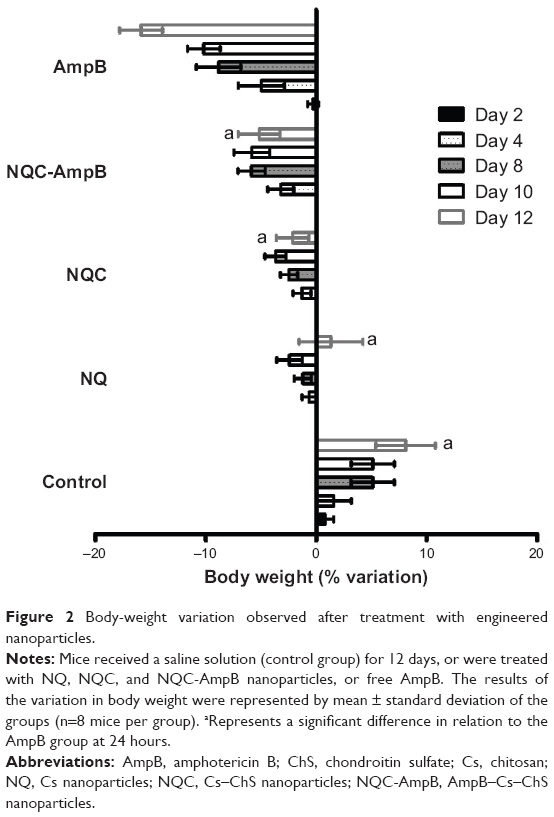 | Figure 2 Body-weight variation observed after treatment with engineered nanoparticles. |
The results of dosage of markers of cardiac, hepatic, and renal damage are shown in Figure 3A. The administration of free AmpB did not affect the CK/MB levels, indicating no cardiac damage, but the levels of GGT, AST, ALT, BUN, and serum creatinine increased in these animals. In the animals treated with NQ, NQC, and NQC-AmpB nanoparticles, no alteration was observed. Thus, this finding indicates that the NQC-AmpB system may well be able to diminish the hepatic and renal toxicity caused by free AmpB. Moreover, no hematological alteration was observed in any of the evaluated groups (data not shown).
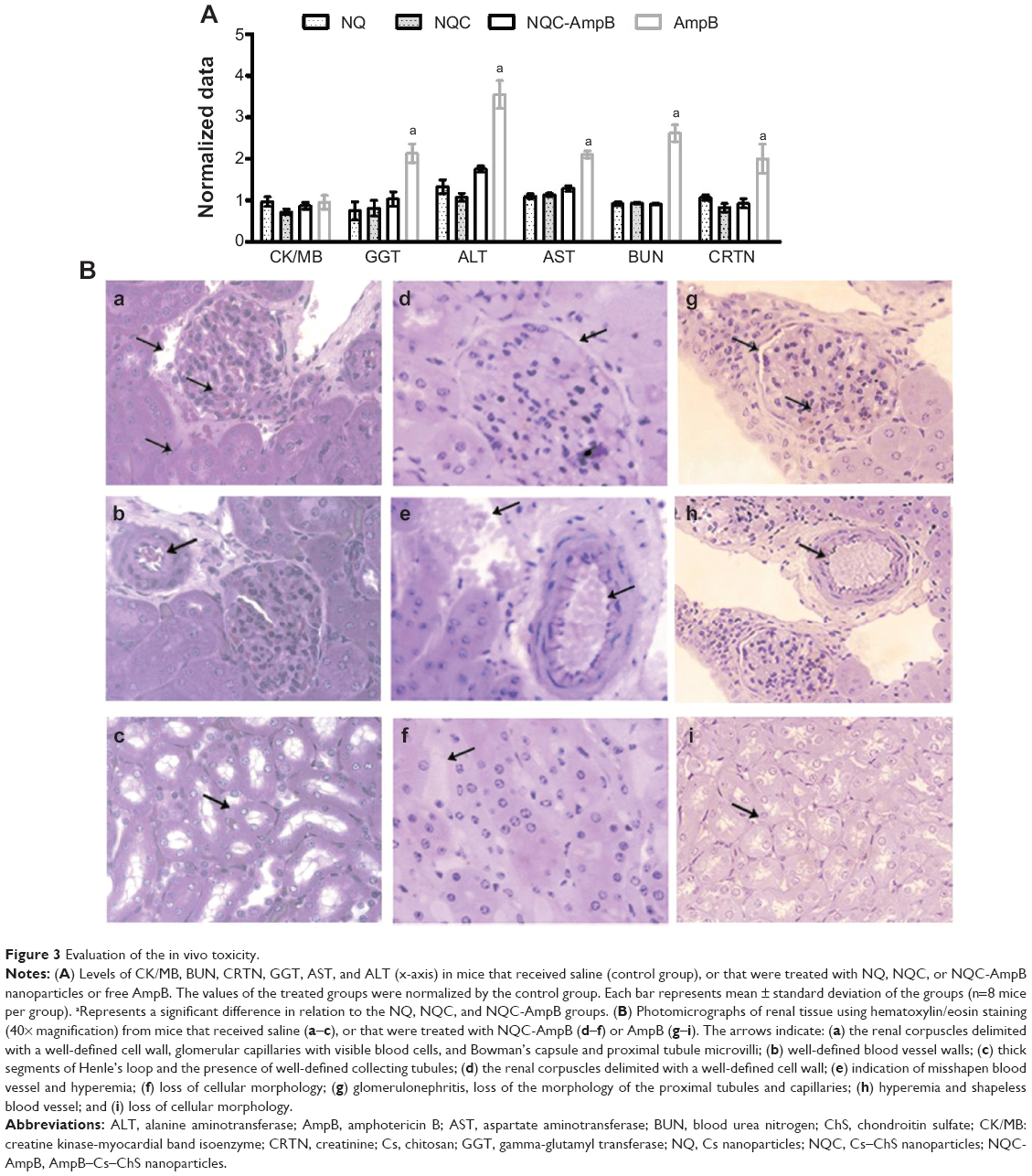 | Figure 3 Evaluation of the in vivo toxicity. |
The histological photomicrographs of animals treated with free AmpB exhibited mild to moderate renal tubular nephrosis, with glomerulonephritis; loss of the morphology of the proximal tubules and capillaries; hyperemic and shapeless blood vessels; and loss of the morphology of the animals’ tissue cells (Figure 3B). Interstitial and intraluminal calcified foci appeared as a result of the mineralization of injured support and epithelial cells. A mild damage in the kidneys of two of the eight mice treated with NQC-AmpB was observed, and no morphological alteration was found in the animals treated with NQ or NQC nanoparticles.
In vivo efficacy against L. amazonensis infection
The lesion development in the infected animals was monitored for 115 days. The saline (control), NQ, NQC, NQC-AmpB, and free AmpB were administered for 10 days. In the results, a significant reduction in the average lesion size could be observed in the animals treated with NQ, NQC, and NQC-AmpB nanoparticles, as well as in the free AmpB group, when compared to the saline group (Figure 4A). No significant difference could be found between the NQ and NQC groups. The NQC-AmpB group presented a smaller lesion size, which was sustained up to 30 days after the treatment in relation to the AmpB group and others, when the area under the curve was calculated (Figure 4B). To evaluate the parasite burden in the animals, the infected footpad, liver, spleen, and dLN were collected and cultured. All groups presented reductions in the parasite load in the evaluated organs, when compared to the control group (Figure 4C); however, animals treated with NQC-AmpB, when compared to the others, displayed better results in reducing the parasite load, thus demonstrating that this preparation was more effective in treating the infected animals.
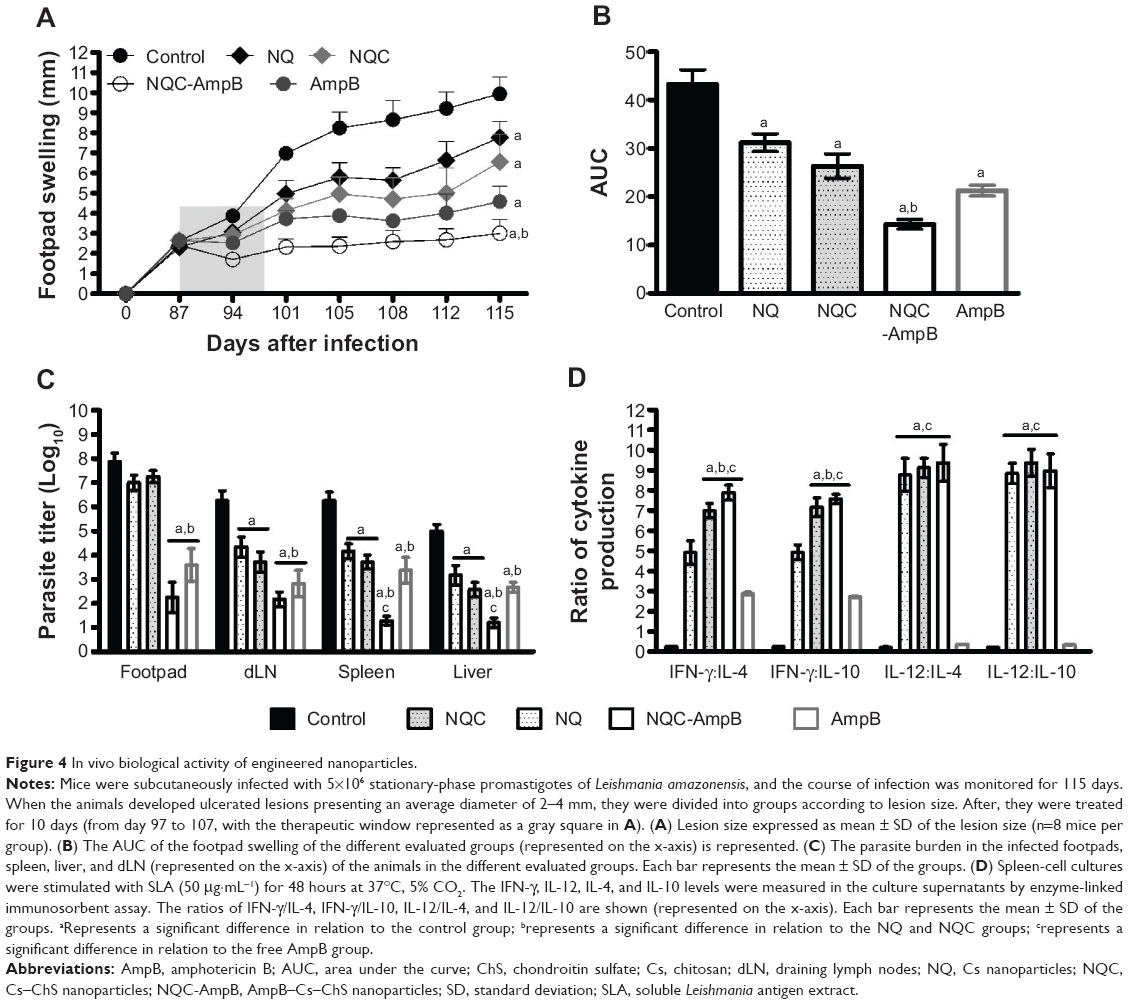 | Figure 4 In vivo biological activity of engineered nanoparticles. |
Immune-response profile
To evaluate if the treatment regimens were able to alter the immunological response associated with the resistance and/or susceptibility of BALB/c mice infected with L. amazonensis, the IFN-γ, IL-12, IL-4, and IL-10 cytokines were evaluated in the spleen cells, at 30 days after treatment. As shown in Figure 4D (see also Figure S1), spleen cells from mice treated with NQ, NQC, and NQC-AmpB nanoparticles produced significantly higher levels of SLA-specific IFN-γ and IL-12 than did those produced by spleen cells of animals treated with free AmpB or the control group, suggesting a polarized T helper (Th) 1 response in these animals. The ratio between IFN-γ/IL-4, IFN-γ/IL-10, IL-12/IL-4, and IL-12/IL-10 was also calculated, and the results showed that nanoparticles primed the infected animals to produce high levels of IFN-γ and IL-12, corroborating with the significant reduction observed in the parasite burden (Figure 4D). Nevertheless, spleen cells of animals treated with free AmpB, as compared to the NQC-AmpB group, also presented significantly higher IFN-γ/IL-4 and IFN-γ/IL-10 ratios, but lower IL-12/IL-4 and IL-12/IL-10 ratios.
Discussion
Current treatments for leishmaniasis have been considered to have low satisfaction, mainly due to the high toxicity of the products. For instance, some attempts have been made over the years to reduce the side effects evoked by AmpB.26 Lipid-based formulations, such as AmBisome®, AmhocilH®, and Abelcet®, have been developed and, despite improvements in the therapeutic index of these drugs, their uses still remain limited, mainly due to the high cost.9 In this context, this study aims to use an in vivo model to validate a preparation based on engineered nanoparticles carrying AmpB, using BALB/c mice challenged with L. amazonensis.
In a previous study, Ribeiro et al developed an AmpB delivery system based on Cs–ChS nanoparticles loaded with this drug (NQC-AmpB).17 To the best of our knowledge, this is the first time that the formulation of a Cs–ChS-based system for AmpB delivery has been developed to treat leishmaniasis. In this context, the in vivo biodistribution profile and the efficacy of NQC-AmpB nanoparticles to treat BALB/c mice that were chronically infected with L. amazonensis, as well as the biochemical, hematological, immunological, and morphological alterations induced by this preparation, as compared to the use of free AmpB, were also investigated.
Some prior studies developed by our group have focused on experiments to evaluate in vitro biodistribution profiles using products radiolabeled with 99mTc.27,28 In the present work, these experiments were also applied, and in vivo biodistribution studies and scintigraphic images showed a high radioactivity uptake of NQC-AmpB in the liver and spleen of the animals, up to 24 hours after their administration. The moderate levels of radioactivity observed in the kidneys could be explained by the leakage of 99mTc from the nanoparticles, most likely as a result of the degradation process of them, as well as due to the slight uptake of the delivery system in the heart, probably due to the positive charge of the nanoparticles.20 It has been postulated that particulate drug carriers can substantially influence not only pharmacokinetics, but also the biodistribution of drugs.29 In this context, the particulate nature of vehicles may well facilitate the passive guidance of entrapped molecules to the inside of the macrophages.30,31
Recently, He et al described that the control of particle size and surface charge has a direct effect on biodistribution, and is a significant issue in the rational design of drug nanocarriers.32 In the present study, all of the obtained nanoparticles presented a positive charge and sizes of approximately 79–136 nm.17 Furthermore, positively charged nanoparticles accumulate in mononuclear phagocytes at about two-fold more than negatively charged carriers.33,34 Danesh-Bahreini et al described that the positively charged nanoparticles, as compared to negative or neutral charged nanoparticles, are more quickly taken up by macrophages through phagocytosis.35
The mechanism of action proposed for the toxic effect of AmpB is derived from its interaction with sterols in bilayer membranes, such as cell walls, causing pore formation in the membrane, in turn leading either to its destruction or to the inhibition of membrane repair. In this context, AmpB could also form pores in cholesterol-containing membranes, explaining the high toxicity observed in the host cells.35 In a controlled drug-delivery system, an active product is incorporated into a polymeric network structure in such a way that the drug is slowly released and in a predefined manner.36,37 Depending on the drug delivery and the application route, the release time may be a few hours to several years.38 In this context, it can be suggested that the reduced toxicity of AmpB in the NQC-AmpB preparation observed in this study could be attributed to the slower release of the drug, when incorporated in the nanoparticles system, inside the macrophages, in turn favoring its leishmanicidal activity and the low toxicity.
The biodistribution profile observed in the present study demonstrates higher accumulation of the NQC-AmpB nanoparticles in the spleen and liver of the animals, which corroborates with findings evaluating other nanostructured systems, such as polymeric micelles39 and gold nanoparticles.40 Akiyama et al reported that ChS is able to induce the Th1-type cytokine (such as IFN-γ, IL-2, and IL-12) secretion and suppress the Th2-type cytokine (such as IL-5 and IL-10) secretion in ovalbumin-sensitized splenocytes of mice. The authors showed that O-sulfo groups in the ChS molecule are important for the Th1-promoted activity of murine splenocytes, in terms of the cytokine production and Th1/Th2 balance.41 ChS has been found in many tissues42 and cells,43–45 and has been reported to interact with various biologically important molecules and regulate their functions. It has been reported that the Th1-promoted and Th2-inhibitory activity of ChS could be associated with its binding to adhesion molecules, such as L- and P-selectins, CD44, and chemokines. In the same study, authors reported that the immunostimulatory activity of ChS could be associated with its binding to L-selectins expressed on T-cells’ surface.41 As the spleen is considered a systemic organ of transit and homing of T-cells in mammal hosts, one could speculate that the higher presence of the NQC and NQC-AmpB nanoparticles in this organ, in detriment to observed levels of NQ, could be due to the presence of a moderate to high number of T-cells in this organ, which could have their L-selectins adhered to ChS present in the nanoparticles. This fact could explain, at least in part, the higher presence of NQC and NQC-AmpB in the spleen of the treated animals.
In the present study, the liver could be also considered a site of accumulation of the nanoparticles in the mice; in this context, a toxicological study was performed on this organ. Clinically significant increases in hepatocyte-associated serum enzymes were observed in the animals treated with free AmpB, with a significant difference observed in the levels of GGT, AST, and ALT between the groups that received NQC-AmpB and free AmpB, which may well indicate a decrease in the hepatic toxicity when AmpB is administered in the NQC system, although used at the same dose.
The nephrotoxicity is considered perhaps the most described adverse drug event for AmpB.46 The pathophysiology and pharmacology of this activity have been well-documented, and the proposed mechanism has been described as being multifactorial.47,48 The BUN and serum creatinine showed significant increases in the animals treated with AmpB. The microscopic observation of kidneys of animals treated with AmpB is in accordance with previous studies.47–49 By contrast, in the animals treated with NQ, NQC, and NQC-AmpB nanoparticles, no significant alteration could be observed. It can therefore be concluded that the decrease in toxicity of AmpB in the NQC-AmpB preparation, in relation to free AmpB, may well have occurred because AmpB encapsulated in the nanoparticles does not interact well with the epithelial cell membranes within the kidney tubule, which would minimize the nephrotoxicity. More recently, other groups have used the same strategy to obtain a less toxic product than free AmpB.50–54
In the present work, the clinical symptoms, as well as hematological and biochemical alterations, were evaluated in the animals after the intravenous administration of saline, NQ, NQC, or NQC-AmpB nanoparticles, or free AmpB. As found in prior reports,55,56 clinical symptoms such as ataxia, weakness, and loss of body weight were associated with the treatment using free AmpB. These animals presented a significant loss of body weight when compared to the animals that received NQC-AmpB, indicating that the controlled release of this drug could well be responsible for the lower incidence of toxic side effects.17 Mice that received NQ and NQC treatment showed a loss of body weight, which could be associated with the recent role of Cs, which is involved in the modulation of adipokines.57
L. amazonensis is a member of the L. mexicana complex and is the etiological agent for a broad spectrum of leishmaniasis in South American countries.58 Among the causative species of cutaneous leishmaniasis in Brazil, recent data indicate that about 8% are attributed to L. amazonensis.59 The present study evaluated the efficacy of NQ, NQC, and NQC-AmpB nanoparticles in treating chronically L. amazonensis-infected BALB/c mice, as compared to treatment with free AmpB. Both the NQC-AmpB and AmpB treatments were able to promote a significant reduction in both the lesion size and parasite load of the infected animals. The lesion size of the NQ and NQC groups was lower than the control group, but higher than that found in the NQC-AmpB and free AmpB groups, although the parasitism levels in the treated mice with NQ and NQC were similar to that obtained in the AmpB group, showing the antileishmanial activity of Cs and its synergic effect with ChS.
The immune response of the treated and infected animals was also evaluated. Mice treated with nanoparticles presented higher levels of IFN-γ and IL-12, and lower levels of IL-4 and IL-10. By contrast, control animals displayed high levels of IL-4 and IL-10 in their splenic cultures. These results are in concordance with Asthana et al, who showed that low levels of IL-4 and IL-10, associated with a high production of IFN-γ and IL-12 by splenic cultures of infected hamsters, are related to the efficacy of the treatment using a template-based nanoemulsion loaded with AmpB.33 Altogether, these results indicate that treatment with NQC-AmpB nanoparticles was able to induce a Leishmania-specific Th1 immune response in the treated mice.
Traditionally, the systemic treatment of leishmaniasis has been with pentavalent antimony compounds (usually performed using 20 mg per kg per day, given by intravenous or intramuscular routes), over a 20-day period. This regimen is able to cure about 85%–90% of the patients; however, the treatment is associated with side toxic effects, such as electrocardiographic changes, ventricular tachyarrhythmias, and laboratory abnormalities, like elevated liver and pancreatic enzymes, and bone marrow suppression.60,61 The development of lipid formulations for AmpB has greatly reduced the toxicity of this drug in the treatment of disease. In this context, lipid-based AmpB products can be considered as alternatives to treatments of leishmaniasis.17 In general, this formulation is less nephrotoxic than free AmpB, since it is taken up selectively by macrophages. Some adverse effects including mild urticarial rash and renal impairment are resolved after therapy.63 The liposomal formulation AmBisome®, the AmpB colloidal dispersion (Amphocil™), and the AmpB lipid complex (Abelcet®) have been used in the treatment.62–64 However, the main restriction against the widespread use of the current approved lipid-AmpB formulations is their high cost. So, the search remains to obtain a low-cost formulation, which should have also an effective activity against Leishmania.64
The NQC-AmpB nanoparticles described in this study present characteristics that could be attractive to the treatment of leishmaniasis. Firstly, the dosage employed of the drug in this system is considered low (1 mg per kg per day), which could be impacting in the null side effects observed in the treated mice. Although the mouse model cannot be extrapolated to the model, it is a good perspective that NQC-AmpB could also be less toxic in the patients. Secondly, although NQC-AmpB does not at present have an estimated value of sale, due to the fact that this product is composed of two known commercial products – ChS and Cs – one could speculate that the NQC-AmpB nanoparticles may well have a lesser cost in comparison to the other available commercial lipid-AmpB formulations.17
Therapeutic and vaccine studies concerning leishmaniasis call for the critical evaluation of the parasitological and immunological parameters of the most common animal models today. It is extremely important to optimize the conditions of the artificial infection to the point where it can be firmly argued that the conditions best represent those of the natural infection caused by the sand fly.65 The present study showed that BALB/c mice, when infected with a highly infective inoculum, developed an exponential parasite burden in organs such as dLN, footpad, spleen, and liver, which suggests a chronic infection in the animals, and makes it very difficult to control the replication of parasites.66 In this context, one could speculate that even using a very effective treatment regimen, the infected animals would not be able to clear all parasites in the different organs. In this light, the NQC-AmpB nanoparticles could be considered as effective therapeutic agents; once the results obtained in the animals treated with this product were compared with the data shown in the free-AmpB-treated group, although not all parasites had been eliminated. However, additional studies are certainly necessary to improve the therapeutic index of NQC-AmpB, either by increasing the number of doses of the product, or the duration of the treatment; in order to clear the largest possible number of parasites in the infected animals. On the other hand, the evaluation of the treatment in other mammal models could also be of interest, in order to estimate the efficacy of the nanoparticles, in mammals that are not as susceptible as BALB/c mice.
Conclusion
In conclusion, based on the results involving the in vivo biodistribution, and with the evaluation of the biochemical, toxicological, parasitological, and immunological parameters associated with the treatment of mice infected with L. amazonensis, the NQC-AmpB nanoparticles could be applied as an alternative AmpB delivery system, maintaining the high activity of this drug against Leishmania, but reducing by significant levels its toxicity, compared to when it is administered in a free form. Therefore, this new formulation presents a high potential for use in future clinical studies aimed at treating leishmaniasis.
Acknowledgments
This work was supported by grants from Pró-Reitoria de Pesquisa from UFMG (Edital 01/2014), Instituto Nacional de Ciência e Tecnologia em Nano-biofarmacêutica (INCT-Nanobiofar), FAPEMIG (CBB-APQ-00496-11 and CBB-APQ-00819-12), and CNPq (APQ-472090/2011-9 and APQ-482976/2012-8). MACF is a grant recipient of FAPEMIG/CAPES. EAFC, VNC, and AAGF are grant recipients of CNPq. Eduardo AF Coelho and André AG Faraco are co-senior authors of this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis. 2004;27(5):305–318. | ||
World Health Organization (WHO). Control of the leishmaniases: report of a meeting of the 399 WHO Expert Committee on the Control of Leishmaniases. 2010. Available from: http://whqlibdoc.who.int/trs/WHO_TRS_949_eng.pdf. Accessed October 9, 2014. | ||
Alvar J, Vélez ID, Bern C, et al. Leishmaniasis worldwide and global estimates of its incidence. PloS One. 2012;7(5):e35671. | ||
Grevelink SA, Lerner EA. Leishmaniasis. J Am Acad Dermatol. 1996; 34(2 Pt 1):257–272. | ||
Croft SL, Coombs GH. Leishmaniasis – current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003;19(11):502–508. | ||
Annaloro C, Olivares C, Usardi P, et al. Retrospective evaluation of amphotericin B deoxycholate toxicity in a single centre series of haematopoietic stem cell transplantation recipients. J Antimicrob Chemother. 2009;63(3):625–626. | ||
Denning DW. Therapeutic outcome of invasive aspergillosis. Clin Infect Dis. 1996;23(3):608–615. | ||
Bern C, Adler-Moore J, Berenguer J, et al. Liposomal amphotericin B for the treatment of visceral leishmaniasis. Clin Infect Dis. 2006;43(7): 917–924. | ||
Egger SS, Meier S, Leu C, et al. Drug interactions and adverse events associated with antimycotic drugs used for invasive aspergillosis in hematopoietic SCT. Bone Marrow Transplant. 2010;45(7): 1197–1203. | ||
Italia JL, Sharp A, Carter KC, Warn P, Kumar MN. Peroral amphotericin B polymer nanoparticles lead to comparable or superior in vivo antifungal activity to that of intravenous Ambisome® or Fungizone™. PLoS One. 2011;6(10):25744. | ||
Gershkovich P, Wasan EK, Lin M, et al. Pharmacokinetics and biodistribution of amphotericin B in rats following oral administration in a novel lipid-based formulation. J Antimicrob Chemother. 2009;64(1): 101–108. | ||
Bekersky I, Fielding RM, Dressler DE, Lee JW, Buell DN, Walsh TJ. Pharmacokinetics, excretion, and mass balance of liposomal amphotericin B (AmBisome) and amphotericin B deoxycholate in humans. Antimicrob Agents Chemother. 2002;46(3):828–833. | ||
Fielding RM, Smith PC, Wang LH, Porter J, Guo LS. Comparative pharmacokinetics of amphotericin B after administration of a novel colloidal delivery system, ABCD, and a conventional formulation to rats. Antimicrob Agents Chemother. 1991;35(6):1208–1213. | ||
Townsed RW, Zutshi A, Bekersky I. Biodistribution of 4-[(14)C]cholesterol-Ambisome following a single intravenous administration to rats. Drug Metab Dispos. 2001;29(5):681–685. | ||
Saxena S, Ghosh PC. Biodistribution of amphotericin B when delivered through cholesterol hemisuccinate vesicles in normal and A. fumigatus infected mice. Phar Res. 2000;17(10):1236–1242. | ||
Agrawal AK, Agrawal A, Pal A, Guru PY, Gupta CM. Superior chemotherapeutic efficacy of amphotericin B in tuftsin-bearing liposomes against Leishmania donovani infection in hamsters. J Drug Target. 2002;10:41–45. | ||
Ribeiro TG, Chávez-Fumagalli MA, Valadares DG, et al. Novel targeting using nanoparticles: an approach to the development of an effective anti-leishmanial drug-delivery system. Int J Nanomedicine. 2014;14: 877–890. | ||
Soares DC, Ferreira TH, Ferreira Cde A, Cardoso VN, de Sousa EM. Boron nitride nanotubes radiolabeled with 99mTc: preparation, physicochemical characterization, biodistribution study, and scintigraphic imaging in Swiss mice. Int J Pharm. 2012;423(2):489–495. | ||
Kean T, Thanou M. Biodegradation, biodistribution and toxicity of chitosan. Adv Drug Deliv Rev. 2010;62(1):3–11. | ||
Banerjee T, Singh AK, Sharma RK, Maitra AN. Labeling efficiency and biodistribution of Technetium-99m labeled nanoparticles: interference by colloidal tin oxide particles. Int J Pharm. 2005;289(1–2):189–195. | ||
Coelho EA, Tavares CA, Carvalho FA, et al. Immune responses induced by the Leishmania (Leishmania) donovani A2 antigen, but not by the LACK antigen, are protective against experimental Leishmania (Leishmania) amazonensis infection. Infect Immun. 2003;71(7):3988–3994. | ||
Nahrevanian H, Farahmand M, Aghighi Z, Assmar M, Amirkhani A. Pharmacological evaluation of anti-leishmanial activity by in vivo nitric oxide modulation in Balb/c mice infected with Leishmania major MRHO/IR/75/ER: An Iranian strain of cutaneous leishmaniasis. Exp Parasitol. 2007;116(3):233–240. | ||
Buffet PA, Sulahian A, Garin YJ, Nassar N, Derouin F. Culture microtitration: a sensitive method for quantifying Leishmania infantum in tissues of infected mice. Antimicrob Agents Chemother. 1995;39(9):2167–2168. | ||
Leite EA, Giuberti Cdos S, Wainstein AJ, et al. Acute toxicity of long-circulating and pH-sensitive liposomes containing cisplatin in mice after intraperitoneal administration. Life Sci. 2009;84(19–20):641–649. | ||
Ramanakumar AV, Thomann P, Candeias JM, Ferreira S, Villa LL, Franco EL. Use of the normalized absorbance ratio as an internal standardization approach to minimize measurement error in enzyme-linked immunosorbent assays for diagnosis of human papillomavirus infection. J Clin Microbiol. 2010;48(3):791–796. | ||
Tasdemir D, Kaiser M, Brun R, et al. Antitrypanosomal and leishmanicidal activities of flavonoids and their analogues: in vitro, in vivo, structure-activity relationship, and quantitative structure-activity relationship studies. Antimicrob Agents Chemother. 2006;50(4):1352–1364. | ||
de Barros AL, Mota Ld, Soares DC, et al. Long-circulating, pH-sensitive liposomes versus long-circulating, non-pH-sensitive liposomes as a delivery system for tumor identification. J Biomed Nanotechnol. 2013;9(9):1636–1643. | ||
Araújo JG, Mota Ld, Leite EA, et al. Biodistribution and antitumoral effect of long-circulating and pH-sensitive liposomal cisplatin administered in Ehrlich tumor-bearing mice. Exp Biol Med (Maywood). 2011;236(7):808–815. | ||
Martinez-Pomares L, Hanitsch LG, Stillion R, Keshav S, Gordon S. Expression of mannose receptor and ligands for its cysteine-rich domain in venous sinuses of human spleen. Lab Invest. 2005;85:1238–1249. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1): 6387–6392. | ||
Owais M, Gupta CM. Targeted drug delivery to macrophages in parasitic infections. Curr Drug Deliv. 2005;2(4):311–318. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Asthana S, Jaiswal AK, Gupta PK, Pawar VK, Dube A, Chourasia MK. Immunoadjuvant chemotherapy of visceral leishmaniasis in hamsters using amphotericin B-encapsulated nanoemulsion template-based chitosan nanocapsules. Antimicrob Agents Chemother. 2013;57(4): 1714–1722. | ||
Batrakova EV, Gendelman HE, Kabanov AV. Cell-mediated drugs delivery. Expert Opin Drug Deliv. 2011;8(4):415–433. | ||
Danesh-Bahreini MA, Shokri J, Samiei A, Kamali-Sarvestani E, Barzegar-Jalali M, Mohammadi-Samani S. Nanovaccine for leishmaniasis: preparation of chitosan nanoparticles containing Leishmania superoxide dismutase and evaluation of its immunogenicity in BALB/c mice. Int J Nanomedicine. 2011;6:835–842. | ||
Darole PS, Hegde DD, Nair HA. Formulation and evaluation of microemulsion based delivery system for amphotericin B. AAPS Pharm Sci Tech. 2008;9(1):122–128. | ||
Colombo P, Santi P, Bettini R, Brazel CS, Peppas CA. Drug Release from Swelling-Controlled Systems. In: Wise DL, Brannon-Peppas L, Klibanov AM, et al, editors. Handbook of Pharmaceutical Controlled Release Technology. New York, NY: Marcel Dekker, Inc.; 2000:183–210. | ||
Jogani V, Jinturkar K, Vyas T, Misra A. Recent patents review on intranasal administration for CNS drug delivery. Recent Pat Drug Deliv Formul. 2008;2(1):25–40. | ||
Koide H, Asai T, Hatanaka K, et al. Particle size-dependent triggering of accelerated blood clearance phenomenon. Int J Pharm. 2008;362(1–2):197–200. | ||
De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJ, Geertsma RE. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials. 2008;29(12):1912–1919. | ||
Akiyama H, Sakai S, Linhardt RJ, Goda Y, Toida T, Maitani T. Chondroitin sulphate structure affects its immunological activities on murine splenocytes sensitized with ovalbumin. Biochem J. 2004;382:269–278. | ||
Suzuki S, Satio H, Yamagata T, et al. Formation of three types of disulphated disaccharides from chondroitin sulphates by chondroitinase digestion. J Biol Chem. 1968;243:1543–1550. | ||
Ohhashi Y, Hasumi F, Mori Y. Comparative study on glycosaminoglycans synthesized in peripheral and peritoneal polymorphonuclear leucocytes from guinea pigs. Biochem J. 1984;217:199–207. | ||
Stevens RL, Fox CC, Lichtenstein LM, Austen KF. Identification of chondroitin sulphate E proteoglycans and heparin proteoglycans in the secretory granules of human lung mast cells. Proc Natl Acad Sci U S A. 1988;85:2284–2287. | ||
Petersen RL, Brandt E, Lindahl U, Spillmann D. Characterization of a neutrophil cell surface glycosaminoglycan that mediates binding of platelet factor 4. J Biol Chem. 1999;274:12376–12382. | ||
Deray G. Amphotericin B nephrotoxicity. J Antimicrob Chemother. 2002;49 Suppl 1:37–41. | ||
Yano T, Itoh Y, Kawamura E, et al. Amphotericin B-induced renal tubular cell injury is mediated by Na influx through ion-permeable pores and subsequent activation of mitogen-activated protein kinases and elevation of intracellular Ca2+ concentration. Antimicrob Agents Chemother. 2009;53(4):1420–1426. | ||
Patel GP, Crank CW, Leikin JB. An evaluation of hepatotoxicity and nephrotoxicity of liposomal amphotericin B (L-AMB). J Med Toxicol. 2011;7(1):12–15. | ||
Fielding RM, Singer AW, Wang LH, Babbar S, Guo LS. Relationship of pharmacokinetics and drug distribution in tissue to increased safety of amphotericin B colloidal dispersion in dogs. Antimicrob Agents Chemother. 1992;36(2):299–307. | ||
Wertlake PT, Butler WT, Hill GJ 2nd, Utz JP. Nephrotoxic tubular damage and calcium deposition following amphotericin B therapy. Am J Pathol. 1963;43(3):449–457. | ||
Mishra J, Dey A, Singh N, Somvanshi R, Singh S. Evaluation of toxicity and therapeutic efficacy of a new liposomal formulation of amphotericin B in a mouse model. Indian J Med Res. 2013;137(4):767–776. | ||
Prajapati VK, Awasthi K, Gautam S, et al. Targeted killing of Leishmania donovani in vivo and in vitro with amphotericin B attached to functionalized carbon nanotubes. J Antimicrob Chemother. 2011;66(4):874–879. | ||
Nahar M, Jain NK. Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm Res. 2009;26(12):2588–2598. | ||
Tiyaboonchai W, Limpeanchob N. Formulation and characterization of amphotericin B-chitosan-dextran sulfate nanoparticles. Int J Pharm. 2007;329(1–2):142–149. | ||
Szoka FC Jr, Milholland D, Barza M. Effect of lipid composition and liposome size on toxicity and in vitro fungicidal activity of liposome-intercalated amphotericin B. Antimicrob Agents Chemother. 1987;31(3):421–429. | ||
Boswell GW, Bekersky I, Buell D, Hiles R, Walsh TJ. Toxicological profile and pharmacokinetics of a unilamellar liposomal vesicle formulation of amphotericin B in rats. Antimicrob Agents Chemother. 1998;42(2):263–268. | ||
Walsh AN, Sweeney T, Bahar B, O’Doherty JV. Multi-functional roles of chitosan as a potential protective agent against obesity. PLoS One. 2013;8(1):e53828. | ||
Camara Coelho LI, Paes M, Guerra JA, et al. Characterization of Leishmania spp. causing cutaneous leishmaniasis in Manaus, Amazonas, Brazil. Parasitol Res. 2011;108(3):671–677. | ||
de Carvalho RF, Ribeiro IF, Miranda-Vilela AL, et al. Leishmanicidal activity of amphotericin B encapsulated in PLGA-DMSA nanoparticles to treat cutaneous leishmaniasis in C57BL/6 mice. Exp Parasitol. 2013;135(2):217–222. | ||
Herwaldt BL, Berman JD. Recommendations for treating leishmaniasis with sodium stibogluconate (Pentostam) and review of pertinent clinical studies. Am J Trop Med Hyg. 1992;46:296–306. | ||
Solomon M, Pavlotsky F, Leshem E, Ephros M, Trau H, Schwartz E. Liposomal amphotericin B treatment of cutaneous leishmaniasis due to Leishmania tropica. J Eur Acad Dermatol Venereol. 2011;25:973–977. | ||
Mohamed-Ahmed AH, Brocchini S, Croft SL. Recent advances in development of amphotericin B formulations for the treatment of visceral leishmaniasis. Curr Opin Infect Dis. 2012;25:695–702. | ||
Dietze R1, Milan EP, Berman JD, et al. Treatment of Brazilian kala-azar with a short course of amphocil (amphotericin B cholesterol dispersion). Clin Infect Dis. 1993;17:981–986. | ||
Sundar S, Murray HW. Cure of antimony-unresponsive Indian visceral leishmaniasis with amphotericin B lipid complex. J Infect Dis. 1996;173:762–765. | ||
Carrión J, Nieto A, Iborra S, et al. Immunohistological features of visceral leishmaniasis in BALB/c mice. Parasite Immunol. 2006;28:173–183. | ||
Oliveira DM, Costa MA, Chavez-Fumagalli MA, et al. Evaluation of parasitological and immunological parameters of Leishmania chagasi infection in BALB/c mice using different doses and routes of inoculation of parasites. Parasitol Res. 2012;110:1277–1285. |
Supplementary material
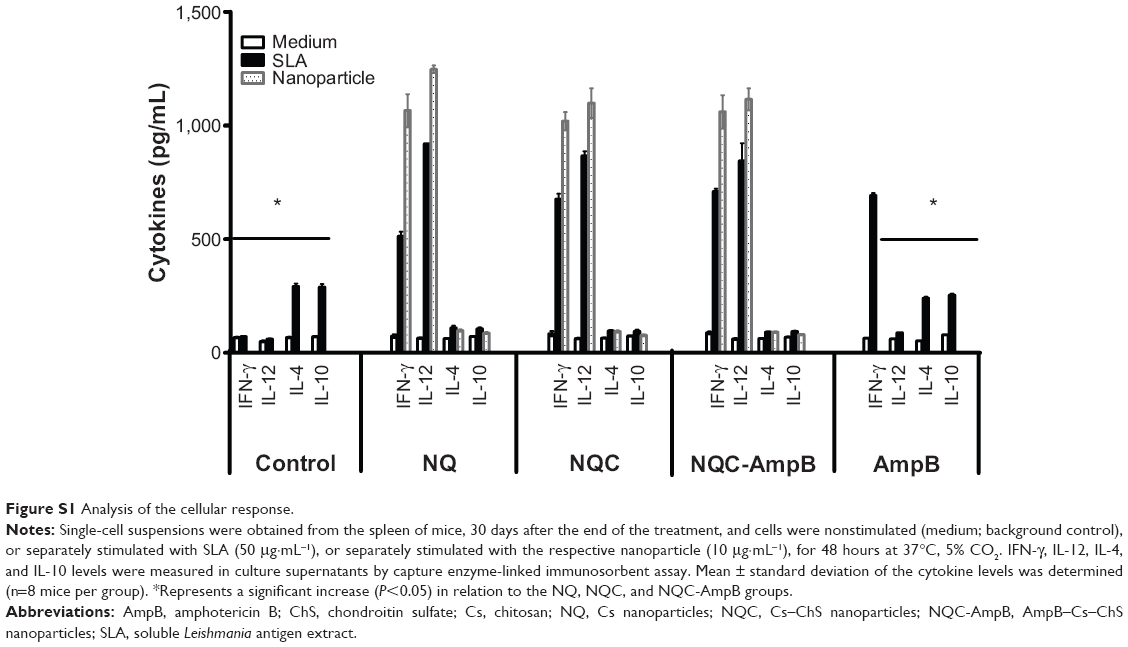 | Figure S1 Analysis of the cellular response. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.