Back to Journals » Drug Design, Development and Therapy » Volume 20
An LC-MS/MS Method for Simultaneous Determination of Almonertinib and Atorvastatin: Evaluating Their Drug-Drug Interactions in Rat
Authors Li H, Wu H, Gao S, Cui L, Xu J, Sun J ![]() , Liu Z, Wang Z
, Liu Z, Wang Z ![]() , Tao X
, Tao X
Received 19 January 2026
Accepted for publication 2 May 2026
Published 20 May 2026 Volume 2026:20 597331
DOI https://doi.org/10.2147/DDDT.S597331
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Anastasios Lymperopoulos
Huanchen Li,1,* Hongkun Wu,2,* Shouhong Gao,1,* Lili Cui,1 Jingjie Xu,1 Jianguo Sun,3 Zhijun Liu,3 Zhipeng Wang,1 Xia Tao1
1Department of Pharmacy, Second Affiliated Hospital of Naval Medical University (Shanghai Changzheng Hospital), Shanghai, 200003, People’s Republic of China; 2Department of Laboratory Medicine, Second Affiliated Hospital of Naval Medical University (Shanghai Changzheng Hospital), Shanghai, 200003, People’s Republic of China; 3Research and Development Center of Chinese Medicine Resources and Biotechnology, Institute of Chinese Materia Medica, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zhipeng Wang, Department of Pharmacy, Second Affiliated Hospital of Naval Medical University (Shanghai Changzheng Hospital), No. 415, Fengyang Road, Shanghai, 200003, People’s Republic of China, Tel +86– 021– 81886202, Email [email protected] Xia Tao, Department of Pharmacy, Second Affiliated Hospital of Naval Medical University (Shanghai Changzheng Hospital), No. 415, Fengyang Road, Shanghai, 200003, People’s Republic of China, Tel +86– 021– 81886191, Email [email protected]
Objective: Atorvastatin is frequently used in combination with almonertinib in lung cancer patients who present with comorbid dyslipidemia. Both drugs are metabolized by CYP3A4, potentially leading to drug-drug interactions (DDIs) that increase the risk of adverse reactions. This study aimed to develop a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method for quantifying almonertinib and atorvastatin and evaluate the DDIs between these two drugs in rats.
Methods: An LC-MS/MS method was developed to determine almonertinib and atorvastatin in rat plasma and was fully validated in accordance with the ICH M10 bioanalytical method validation guideline and Chinese Pharmacopoeia 2020 Edition. Rats were divided into 5 groups (n=7 per group), and received the following treatments: almonertinib alone, atorvastatin alone, almonertinib combined with atorvastatin, vehicle control, or almonertinib after a long-term regimen of atorvastatin. The plasma samples were collected from each rat and analyzed using this method. The pharmacokinetic parameters were calculated and the DDIs were evaluated.
Results: An LC-MS/MS method was established and validated according to the requirements. Animal studies revealed that co-administration of almonertinib with atorvastatin significantly increased Cmax, AUC0-t, and AUC0-∞ (all p < 0.05), prolonged t1/2 (p < 0.05) and decreased CLz/F, Vz/F (both p < 0.05) of both drugs. Following a 7-day administration of atorvastatin, the DDIs between the two drugs generally weakened, but still differed significantly from those observed with single-drug administration.
Conclusion: The study successfully established an LC-MS/MS method for simultaneous determination of almonertinib and atorvastatin and assessed the DDIs of these two drugs in rat. Animal experiment demonstrated significant DDIs between two drugs no matter single dose or multiple doses were administered, which obviously increased the drug exposure and inhibited elimination. Close attention should be paid to the combination regimens of these two drugs in clinical practice. The diagram illustrates a study on drug interactions between atorvastatin and almonertinib. Atorvastatin and almonertinib are shown with their chemical structures. The study involves oral gavage administration in an animal model, represented by a rat. Blood samples are collected and plasma samples are analyzed using liquid chromatography-mass spectrometry (LC-MS). The method is established using ultra-high-performance liquid chromatography-tandem mass spectrometry (UHPLC-MS/MS). A graph depicts drug-drug interactions over time, with the x-axis labeled ’Time’ in hours and the y-axis labeled ’Concentration’ in nanograms per milliliter, showing a curve with data points from 0 to 14 hours and concentrations ranging from 0 to 30 nanograms per milliliter.Diagram of atorvastatin and almonertinib interaction study using UHPLC-MS/MS in animal model.
Keywords: almonertinib, atorvastatin, LC-MS/MS, pharmacokinetics, drug-drug interactions
Introduction
Lung cancer is the leading cause of cancer-related deaths worldwide, and non-small cell lung cancer (NSCLC) accounts for approximately 85% of all lung cancers.1,2 The common methods for treating lung cancer include surgery, chemotherapy, targeted therapy, and so on. Epidermal growth factor receptor (EGFR) is a driver of lung cancer, and cancers with EGFR mutations are usually sensitive to treatment with EGFR tyrosine kinase inhibitors (TKIs).3 The third-generation EGFR-TKI not only exhibits outstanding therapeutic efficacy but also reduces the adverse reactions associated with traditional EGFR-TKI, such as rash and diarrhea, making it a first-line and second-line treatment option for EGFR T790M mutation-positive NSCLC.4 Almonertinib is a third-generation EGFR-TKI indicated for treating advanced NSCLC with EGFR 19del, L858R, and T790M mutations. It demonstrated superior efficacy compared to other TKIs in patients with brain metastases.4–6 Due to lung cancer (particularly among middle-aged and elderly patients) often accompanied by hyperlipidemia, the concurrent use of almonertinib and statins is common in clinical practice. Atorvastatin is a common lipid-lowering drug widely employed to reduce cardiovascular disease risk.7 Moreover, cardiovascular disease has been associated with an increased risk of lung cancer incidence and mortality,8 further highlighting the potential for frequent co-administration of these two drug classes. Therefore, understanding the drug–drug interaction (DDI) between almonertinib and atorvastatin is of clinical relevance.
However, according to the Basic Principles for the Clinical Application of New Anticancer Drugs (2024 Edition),9 almonertinib should be avoided in combination with statins whenever possible. For patients with brain metastases from EGFR-mutated lung cancer, almonertinib serves as a primary treatment drug.10 Unauthorized discontinuation or substitution may lead to rapid tumor progression and pose a life-threatening risk. Meanwhile, cardiovascular disease risks require long-term management; elevated lipid levels increase the risk of myocardial infarction or stroke, which will similarly severely impact patient survival and quality of life, potentially even preclude continued anticancer therapy due to cardiovascular events. Although other EGFR-targeted agents exist, almonertinib may be the optimal choice for patients with brain metastases due to its efficacy and activity against brain metastases. Moreover, other EGFR-TKIs show similar risks of DDI. Therefore, ensuring the safety and efficacy of these two drugs when used in combination, while avoiding reduced efficacy or increased toxicity due to DDI, is a critical clinical consideration.
DDI refers to the combined effects that occur when patients take two or more drugs simultaneously or sequentially within a certain timeframe. These interactions may enhance therapeutic effects or reduce adverse reactions, but may also diminish efficacy or cause undesirable adverse reactions. The most common DDIs include multiple CYP isoenzymes and drug transporters.11 CYPs are the primary enzymes involved in drug metabolism, accounting for approximately 75% of the metabolism of clinically used drugs,12,13 and are a major source of adverse reactions. Among the various CYP enzymes, CYP3A4 is one of the most important enzymes, participating in the metabolism of numerous commonly used drugs.
Almonertinib is a substrate of CYP3A4.14 Atorvastatin is also primarily metabolized by the CYP3A4 in the liver.15 When used in combination, they may competitively bind to CYP3A4. This competition may slow the metabolic rate of both drugs, potentially leading to reduced their clearance and increased their exposure concentrations and peak concentrations in the body. Abnormally elevated atorvastatin blood concentration is a major risk factor for dose-dependent muscle toxicity (such as myalgia, myositis, or even rhabdomyolysis).15,16 P-glycoprotein (P-gp) is an efflux pump widely expressed in the gut, blood-brain barrier, and hepatobiliary mucosa, which limits drug absorption and promotes its excretion.17 Studies indicated that almonertinib was an inhibitor of P-gp,18 while atorvastatin has also been confirmed as a P-gp substrate.17 When co-administered, almonertinib may potentially reduce atorvastatin’s efflux by inhibiting P-gp function, thereby increasing its intestinal absorption and decreasing biliary excretion. Such an interaction might further contribute to atorvastatin accumulation in the body, amplifying the risk of its adverse reaction. Both almonertinib and atorvastatin can elevate blood creatine kinase (CK) levels,19,20 a biomarker of skeletal muscle injury. Nevertheless, given that both agents may individually affect muscle homeostasis, their concurrent use may theoretically increase the risk of myopathy. Therefore, close monitoring of CK levels and muscle-related symptoms may be warranted when these two drugs are co-administered.
Several studies have investigated DDIs between EGFR-TKIs and statins,21–23 but no research has yet investigated DDIs when atorvastatin is co-administered with almonertinib, as these two drug were commonly prescribed in lung cancer patients. Several studies have reported DDIs involving almonertinib,24,25 indicating that almonertinib is susceptible to the effects of CYP3A4 and P-gp metabolism or substrate drugs. Therefore, it is highly necessary to evaluate the DDI between atorvastatin and almonertinib. In addition, methods for quantifying either atorvastatin or almonertinib have been developed in the literatures,24,26 but there is currently no LC-MS/MS method that can simultaneously measure both of them, as the DDI between these two drugs may influence each other, and both the drugs may bring severe adverse reactions.
Therefore, this study developed and validated a rapid, sensitive LC-MS/MS method for simultaneous determination of almonertinib and atorvastatin. This method was applied to assess in vivo DDI between these two drugs in rats, and the findings may provide a reference for their clinical use and help to reduce adverse reactions.
Methods and Materials
Instruments
All analysis were analyzed using an Agilent 1290 series ultra-high performance liquid chromatography coupled to an Agilent 6460 triple quadrupole mass spectrometer equipped with an electrospray ionization (ESI) source (Agilent Technologies, Santa Clara, CA, USA). Data acquisition and processing were completed using Agilent Mass Hunter data processing software (Version B.07.00).
Reagents and Standards
Almonertinib (98% purity, lot No.: H2409525) was supplied by Shanghai Aladdin Bio-Chem Technology Co., Ltd (Shanghai, China). Atorvastatin (99% purity, lot No.: 25031901) was supplied by Dingrui Chemical (Shanghai) Co., Ltd (Shanghai, China). The internal standard Ceritinib-D7 (IS, 98% purity, lot No.: O23GB162867) was supplied by Yuanye Biotechnology Co., Ltd (Shanghai, China). Other reagents included acetonitrile (MS grade; Merck, Darmstadt, Germany), formic acid (MS grade; Sinopharm Group, Shanghai, China), ammonium acetate (analytical grade, Sinopharm Group), and methanol (HPLC grade; Sinopharm Group) were used in method development. Distilled water was supplied by Watsons Distilled Water Co., Ltd. (Shenzhen, China).
Chromatographic Conditions
Chromatographic separation was performed using an Atlantis™ T3 column (2.1 × 100 mm, 3 μm). The mobile phase consisted of 5 mM ammonium acetate containing 0.1% formic acid (phase A) and acetonitrile containing 0.1% formic acid (phase B). The gradient program was as follows: 0–1.0 min, 10% to 95% B; 1.0–3.5 min, held at 95% B; followed by a 1.0 min re-equilibration step at 10% B. The total run time was 3.5 min. The flow rate was maintained at 0.3 mL/min. Column temperature was maintained at 35°C, and the injection volume of 2 μL.
Mass Spectrometry Conditions
Analytes were ionized in positive ionization mode using an ESI source, with data acquired in multiple reaction monitoring (MRM) mode. MS source parameters were set as follows: capillary voltage 4000 V; nebulizer pressure 45 psi; drying and nebulizer gas were nitrogen; drying gas temperature 350°C; drying gas flow 5 L/min; sheath gas heater 300°C; sheath gas flow 12 L/min. MS parameters are shown in Table 1. The mass spectrograms of almonertinib, atorvastatin and IS are presented in Figure 1.
 |
Table 1 Mass Spectrometric Parameters for the Analytes and IS |
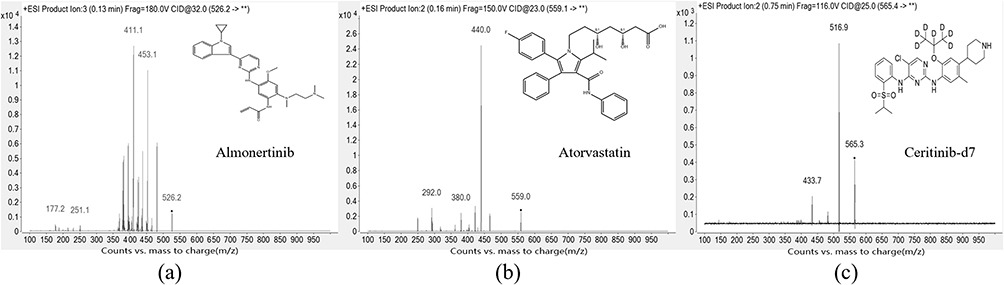 |
Figure 1 The mass spectra of (a) almonertinib, (b) atorvastatin and (c) ceritinib-d7 (IS). ** indicates the undefined product ions. |
Stock, Working Solutions and Quality Control (QC) Samples Preparation
Stock solutions of analyte standards and IS at appropriate concentrations were prepared. Stock solutions of almonertinib, atorvastatin, and ceritinib-d7 (IS) were prepared in methanol at concentrations of 1.0 mg/mL. Working solutions of almonertinib and atorvastatin were obtained by serially diluting their respective stock solutions with 10% methanol to yield concentrations of 20.0, 50.0, 100.0, 500.0, 1000.0, 2000.0, 5000.0, 7500.0, and 10000.0 ng/mL. These working solutions were added to blank rat plasma to obtain final calibration standards at concentrations of 2.0, 5.0, 10.0, 50.0, 100.0, 200.0, 500.0, 750.0, and 1000.0 ng/mL. QC samples were obtained from separately prepared stock solutions, and almonertinib and atorvastatin were prepared at four concentrations: 2.0 (lower limit of quantification, LLOQ), 5.0 (low QC, LQC), 100.0 (medium QC, MQC), and 750.0 (high QC, HQC).
The working solution of ceritinib-d7 at 100 ng/mL was prepared in acetonitrile as IS. The stock solutions, working solutions and QC samples were all stored at −80°C prior to analysis. Ceritinib-d7 was selected as the IS for the simultaneous quantification of almonertinib and atorvastatin. The choice of ceritinib-d7 was made because its retention time (with a retention time of approximately 1.99 min) was similar to that of the two analytes (the retention time of almonertinib was 1.85 min, and that of atorvastatin was 2.22 min). Moreover, as a stable isotope-labeled analogue, it can effectively compensate for the ionization efficiency and potential changes during sample processing.
Sample Pretreatment
Plasma samples were processed using a protein precipitation method. The sample preparation procedure was as follows: 100 μL of each plasma sample was mixed with 200 μL of acetonitrile solution (containing 100 ng/mL IS) in a 1.5 mL EP tube. The mixture was vortex-mixed for 3 minutes, and then centrifuged at 14,500 × g at room temperature for 10 minutes. Finally, 2 μL of the supernatant was injected into the LC-MS/MS for quantitative analysis.
During the optimization of pretreatment methods, tests were conducted using different ratios of methanol (volume ratio, V:V, 1:2, 1:3 1:4) and acetonitrile (volume ratio, V:V, 1:2, 1:3, 1:4) for protein precipitation. Analysis of retention times and peak areas indicated that acetonitrile (1:2) demonstrated superior efficacy as a protein precipitant. Additionally, we also tested various solvents for diluting the supernatant, and a mixed solvent of methanol and acetonitrile (1:1, V/V) for dilution was tested, however, this approach did not improve almonertinib’s peak shape or response. The liquid-liquid extraction and solid-phase extraction were tested too using the common solvents ethyl acetate, chloroform and the extraction column Osis HLB, MCX, and MAX cartridge, but the recovery was less than 50% for all the tests. Compared to liquid-liquid extraction and solid-phase extraction, protein precipitation is simpler, lower in cost, and less polluting. Therefore, acetonitrile (1:2) was ultimately selected for protein precipitation prior to injection.
Methodology Validation
Method validation was conducted according to the Chinese Pharmacopoeia (2020 edition) and the ICH M10, including specificity, matrix effects, extraction recovery, linearity, lower limit of quantitation (LLOQ), intra-day and inter-day precision and accuracy, carryover, and stability.27,28
Calibration Curves and LLOQ
Calibration curves for almonertinib and atorvastatin were evaluated at concentrations of 2.0, 5.0, 10.0, 50.0, 100.0, 200.0, 500.0, 750.0, and 1000.0 ng/mL. Linearity was evaluated by plotting the peak area ratio of each analyte to the IS against the nominal concentration, using a least squares linear regression model with a weighting factor of 1/x2. Deviations (RE%) between calculated and theoretical concentrations from the calibration standards were acceptable within ±15%, and the LLOQ should not exceed ±20%.
Inter-Day and Intra-Day Precision and Accuracy
Inter-day and intra-day precision and accuracy were assessed by analyzing QC samples spiked at low, medium, and high concentration levels and at the LLOQ over three consecutive days, with five replicates per concentration level per day. Precision was determined as the relative standard deviation (RSD%), while accuracy was expressed as the relative error (RE%). For QC samples at low, medium, and high concentrations, the acceptance criteria were set as RSD ≤ 15% for precision and RE within ±15% for accuracy. For the LLOQ sample, RSD ≤ 20% and RE within ±20% were considered acceptable.
Matrix Effects and Extraction Recovery
Matrix effects were assessed by comparing the peak areas of analytes in post-extraction spiked samples with those in solvent-substituted samples at equivalent concentrations. Specifically, blank plasma samples were extracted and then spiked with working solutions to obtain post-extraction spiked samples at low, medium, and high QC concentration levels (n = 3 per concentration level). Solvent-substituted samples were prepared at the same concentrations.
Extraction recovery was evaluated by comparing the peak areas of analytes in spiked samples with those in post-extraction spiked samples at the same three concentration levels (n = 3 per concentration level). Spiked samples were prepared by spiking blank plasma with working solutions prior to extraction, whereas post-extraction spiked samples were prepared by spiking blank plasma extracts after extraction.
Stability
Stability was assessed using QC samples under the following conditions, autosampler for 24 h, room temperature for 6 h, −80°C for 30 d, three freeze-thaw cycles (−80°C to room temperature), in three replicates and three concentration levels. Fresh calibration curve was constructed to calculate the measured concentrations, and analytes were deemed as stable if the deviations did not exceed ±15% of the nominal concentrations.
Carryover and Specificity
Carryover refers to the residual contamination observed in the instrument system after injecting the highest concentration sample of calibration standards followed by a blank sample. The responses of analytes in the blank sample should not exceed 20% of the LLOQ sample and should not exceed 5% of the IS. For the specificity, this study analyzed six different lots of blank matrixes, blank matrix spiked with LLOQ sample and real samples. It is acceptable that the responses of interfering substances do not exceed 20% of the LLOQ and 5% of the IS.
Evaluation of the DDI of Almonertinib and Atorvastatin in Rats
Healthy male Sprague-Dawley (SD) rats weighing 200–220 g were purchased from BiKaiKeYi biotech Co., Ltd (Shanghai, China). This study protocol was approved and supervised by the Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine (PZSHUTCM2506150003). Rats underwent acclimatization feeding for one week prior to the experiment (with ample food and water) and were maintained under appropriate conditions (12 h dark-light cycle, temperature 23°C–25°C, relative humidity 50% ± 10%). All rats were fasted for food for 12 h before the experiment, with water provided. This animal study was carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory animals, and no obvious adverse reactions were found in this study. The rats were sacrificed after the blood collection using the isoflurane and cervical dislocation.
Thirty-five healthy SD rats were randomly divided into five groups (n=7 per group). The sample size was selected based on previous pharmacokinetic interaction studies, which employed 6 animals per group.25 Almonertinib and atorvastatin were both suspended in 0.5% CMC-Na, and the dosage was converted from the clinically used dosage in humans based on body weight. Both drugs administered orally once daily. Group 1 received administration of 15 mg/kg almonertinib. Group 2 was treated with 2 mg/kg atorvastatin. Group 3 received 15 mg/kg almonertinib plus 2 mg/kg atorvastatin. Group 4 was designated as control group and only the solvent was given. The rats in Group 5 were treated with atorvastatin at 2 mg/kg qd for 7 consecutive days, followed by 15 mg/kg qd almonertinib on day 8, administered 30 min after the final dose of atorvastatin. The time interval between almonertinib and atorvastatin was set at 30 min (Group 3 and 5). The 7-day atorvastatin pretreatment period was selected based on the pharmacokinetic properties of atorvastatin in rats, which reaches steady-state plasma concentrations after approximately 5–7 days of repeated dosing.28 This regimen was designed to simulate the clinical scenario in which patients receive long-term lipid-lowering therapy prior to initiating almonertinib treatment.
Blood samples (approximately 200 μL) were collected via the orbital venous plexus into in EP tubes containing sodium heparin at the following time points relative to almonertinib administration: 0 h (pre-treatment), 0.25 h, 0.5 h, 1 h, 2 h, 3 h, 4 h, 6 h, 8 h, 12 h, 24 h, and 48 h. For Group 5, almonertinib was administered 30 min after atorvastatin on day 8. Blood samples were centrifuged at 3500×g for 10 min. The supernatant was collected and stored at −80°C until analysis.
Statistical Analysis
Pharmacokinetic parameters were calculated using non-compartmental analysis with DAS 2.1.1 software (Chinese Society of Mathematical Pharmacology, Shanghai, China). Statistical analysis of primary pharmacokinetic parameters, including AUC, Cmax, Tmax, t1/2, CLz/F, and Vz/F, was performed using SPSS 25.0 statistical software (SPSS Inc., Chicago, IL, USA). Data are presented as mean ± standard deviation (SD). For comparisons among multiple groups, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was applied for normally distributed data with equal variances; otherwise, the Kruskal–Wallis H-test followed by Dunn’s post hoc test was used. For comparisons between two groups, Student’s t test was used for normally distributed data, and the Mann–Whitney U-test was applied for non-normally distributed data. A p-value < 0.05 was considered statistically significant.
Results
Method Validation
Specificity
The retention times for atorvastatin, almonertinib, and ceritinib-d7 were 2.22, 1.85, and 1.99 minutes, respectively. The responses of the interfering compounds did not exceed 15% of the LLOQ and 5% of the IS, conforming to the criteria of Chinese Pharmacopoeia (2020 edition). Figure 2 displays representative chromatograms of almonertinib, atorvastatin, and ceritinib-d7 in different plasma samples. These include blank plasma sample (a); blank plasma sample spiked with IS (b); blank plasma sample spiked with LLOQ and IS (c); blank plasma sample spiked with two analytes and IS (d).
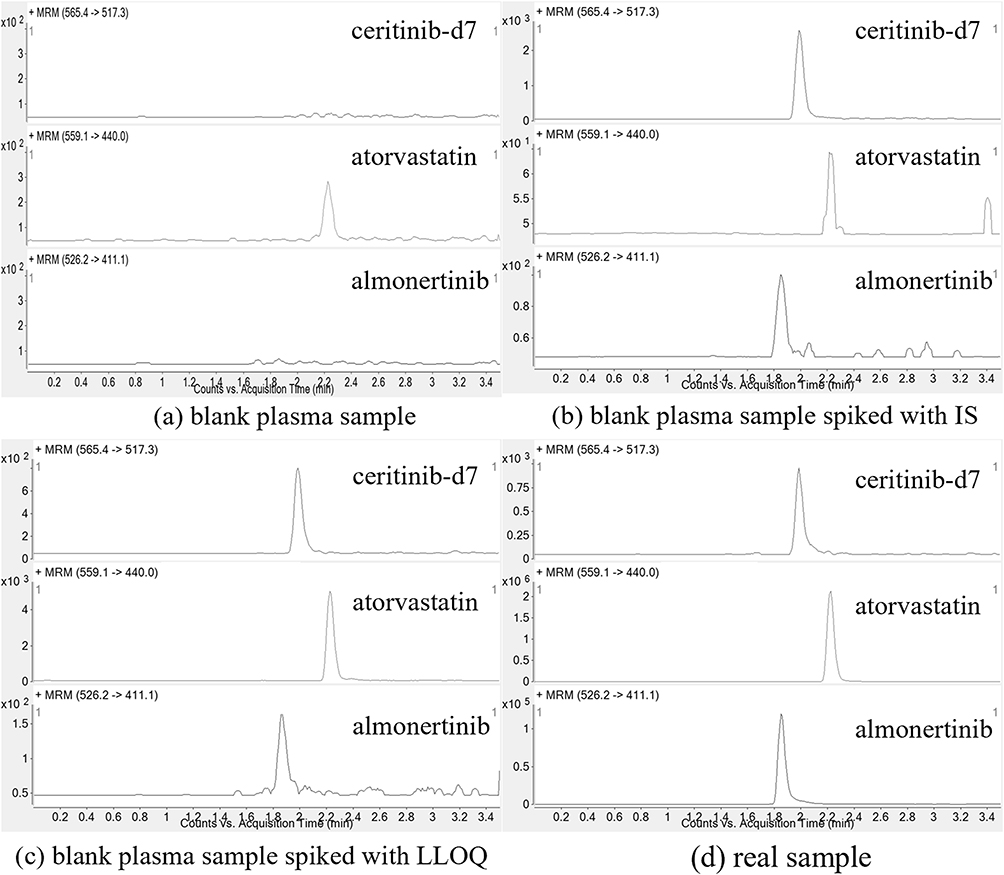 |
Figure 2 Representative MRM chromatograms of almonertinib, atorvastatin, and ceritinib-d7. (a) blank plasma sample; (b) blank plasma sample spiked with IS; (c) blank plasma sample spiked with LLOQ; (d) real sample. |
Linearity
Linear regression was performed using a 1/x2 weighted linear regression model to generate calibration curves within the concentration range of 2–1000.0 ng/mL. Table 2 presents representative calibration curves for the two drugs. The LLOQ for both almonertinib and atorvastatin is 2.0 ng/mL. The back-calculated deviation for each calibration standard was less than 9.95% for almonertinib, and less than 9.59% for atorvastatin.
 |
Table 2 Linearity Regression Parameters of Two Analytes |
Inter- and Intra-Day Precision and Accuracy
To evaluate inter- and intra-precision and accuracy, QC samples at low, medium, and high concentration levels, along with the LLOQ, were prepared freshly on each of three consecutive days. Three batches were analyzed, with five replicates per batch. As shown in Table 3, both intra-day and inter-day precision values did not exceed 10.3%, and the intra-day and inter-day accuracy ranged from −13.3% to 8.8%. Overall, the intra-day and inter-day precision and accuracy results met the requirements.
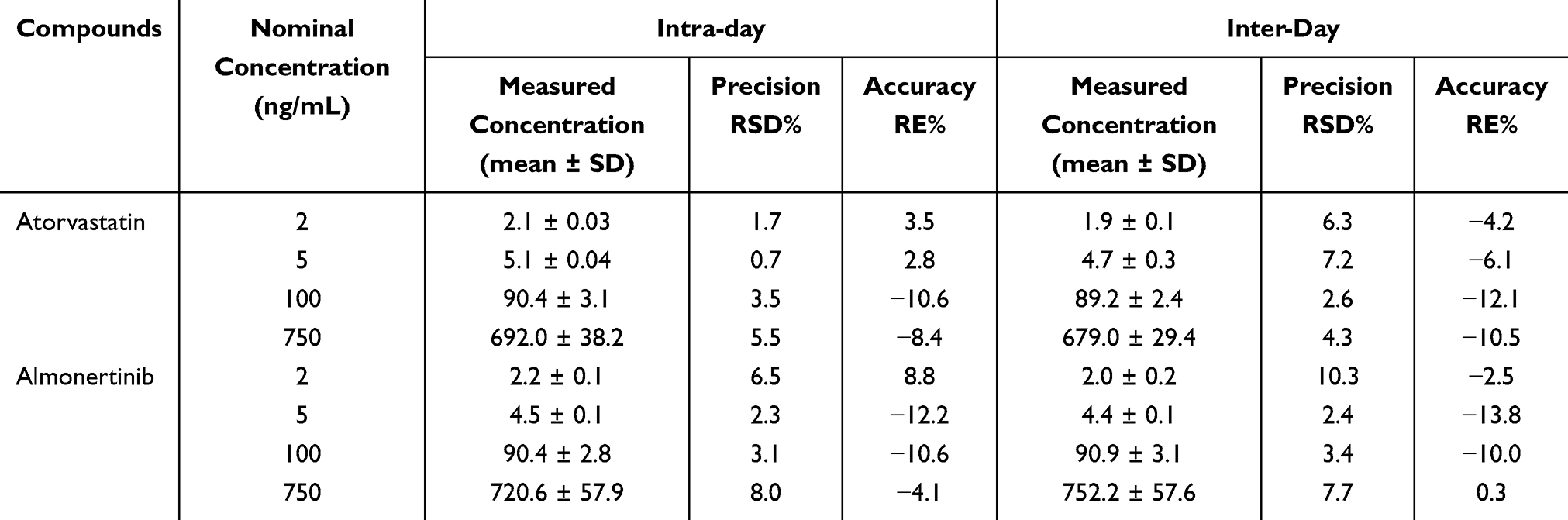 |
Table 3 Inter-Day and Intra-Day Accuracy and Precision of Two Analytes (n = 5) |
Matrix Effects and Extraction Recovery
This method employed acetonitrile as the protein precipitant in the pretreatment step. Matrix effects and extraction recovery were evaluated by analyzing samples at low, medium, and high concentration levels in triplicate. The matrix effects for almonertinib and atorvastatin ranged from 87% to 91% and 100% to 101%, respectively. Within the commonly accepted range for matrix effect evaluation (80%–120%), the interference from endogenous substances in biological samples was minimal and had no significant impact on the quantitative analysis. The extraction recoveries for almonertinib and atorvastatin in plasma samples were also acceptable, with values ranging from 88% to 93% and 92% to 96%, respectively. The RSD% for both matrix effect and recovery was less than 15%. These results indicate that protein precipitation using acetonitrile is effective and stable for extracting almonertinib and atorvastatin. The data are shown in Table 4.
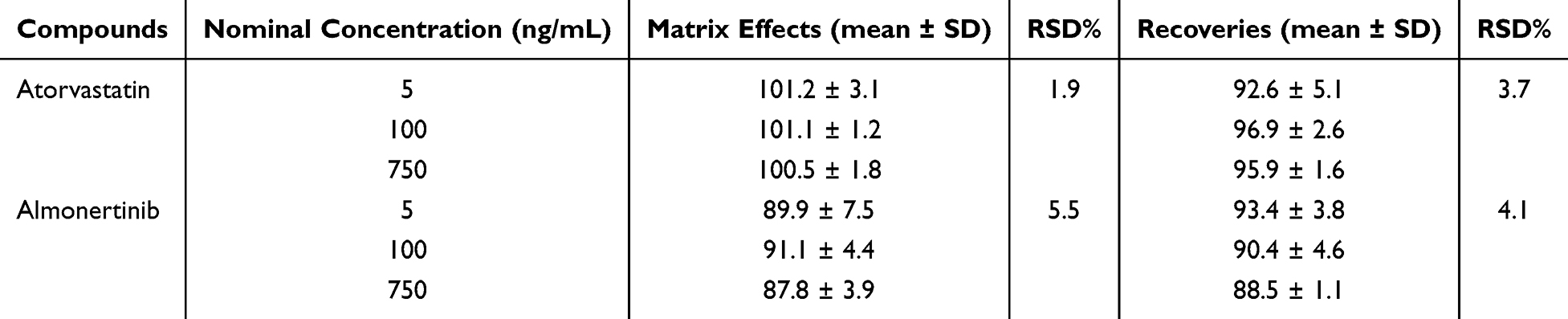 |
Table 4 Extraction Recovery and Matrix Effect of Two Analytes (n=3) |
Stability
Stability assessment was performed with three concentration levels (n = 3 per concentration level), and the results showed that samples remained stable for up to 24 h in autosampler, up to 6 h at room temperature, and up to 30 d at −80°C. Furthermore, samples remained stable after undergoing three freeze-thaw cycles (−80°C to room temperature). Both RE% and RSD% values fell within acceptable ranges. The stability of almonertinib and atorvastatin in rat plasma under various storage and processing conditions are presented in Table 5.
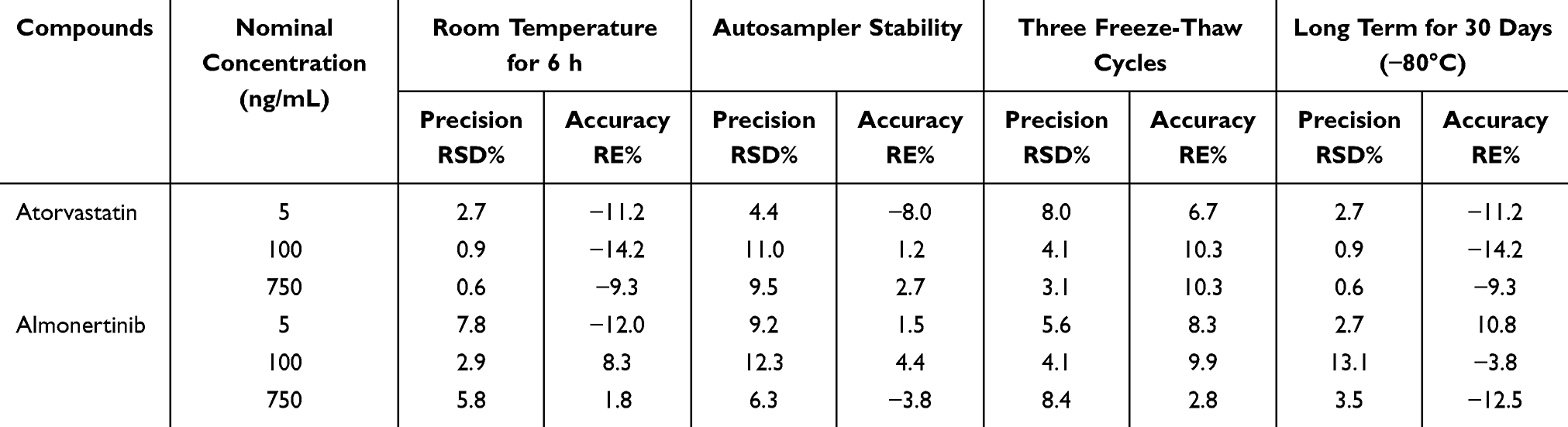 |
Table 5 Stability Results for Two Analytes (n = 3) |
Carryover
Carryover was evaluated by measuring the peak area of the blank sample after the ULOQ of the calibration curve for three cycles. The results showed that the peak areas of analytes in the blank sample after detecting the ULOQ sample of almonertinib and atorvastatin were less than 20% of the peak areas of analytes in LLOQ (2.0 ng/mL), and 5% of the IS, which met the requirements.
Animal Study for Assessing the DDIs Between Almonertinib and Atorvastatin
This newly developed method was applied to investigate the DDIs between almonertinib and atorvastatin in rats. In the Figure 3, the mean plasma concentration-time curves for almonertinib (15 mg/kg) and atorvastatin (2 mg/kg) across five groups were presented. The co-administration of almonertinib and atorvastatin significantly influenced the disposal of each other. Table 6 summarizes the pharmacokinetic parameters of almonertinib and atorvastatin. Compared with single administration of almonertinib (Group 1), co-administration with atorvastatin significantly increased almonertinib’s Cmax, AUC0-t, and AUC0-∞ by 1.21, 2.20 and 4.01-fold, respectively, and prolonged the half-life time of almonertinib (all P<0.05), while obviously decreased CLz/F by 66.3% (P < 0.05), and the Vz/F of almonertinib also showed a decreasing trend. For the atorvastatin, the co-administration with almonertinib also exerted significant effects on its pharmacokinetic parameters; its Cmax, AUC0-t, and AUC0-∞ increased significantly by 1.96, 2.49 and 3.07-fold, respectively, and the half-life time remarkably prolonged (all P < 0.05), while CLz/F decreased by 60.2% (all P < 0.05), and the Vz/F of atorvastatin displayed a decrease trend too. The Tmax of both almonertinib and atorvastatin was not influenced by each other. There is an obviously DDIs between almonertinib and atorvastatin. To further evaluate the effect of long-term administration of atorvastatin on the almonertinib, following multiple doses of atorvastatin, almonertinib was given on day 8, and its Cmax decreased significantly by 2.71-fold, while AUC0-∞ increased by 1.59-fold, additionally, the CLz/F of almonertinib decreased by 25.3% (all P<0.05), compared with the single administration of almonertinib. Compared with single administration of atorvastatin, the Cmax, AUC0-t, and AUC0-∞ in long-term group increased by 1.46, 2.19, and 2.40-fold, respectively, while CLz/F decreased by 56.15%. The DDIs of two drugs weakened in long-term group compared with combination group, especially to atorvastatin. In a word, significant pharmacokinetic DDI occurred between almonertinib and atorvastatin.
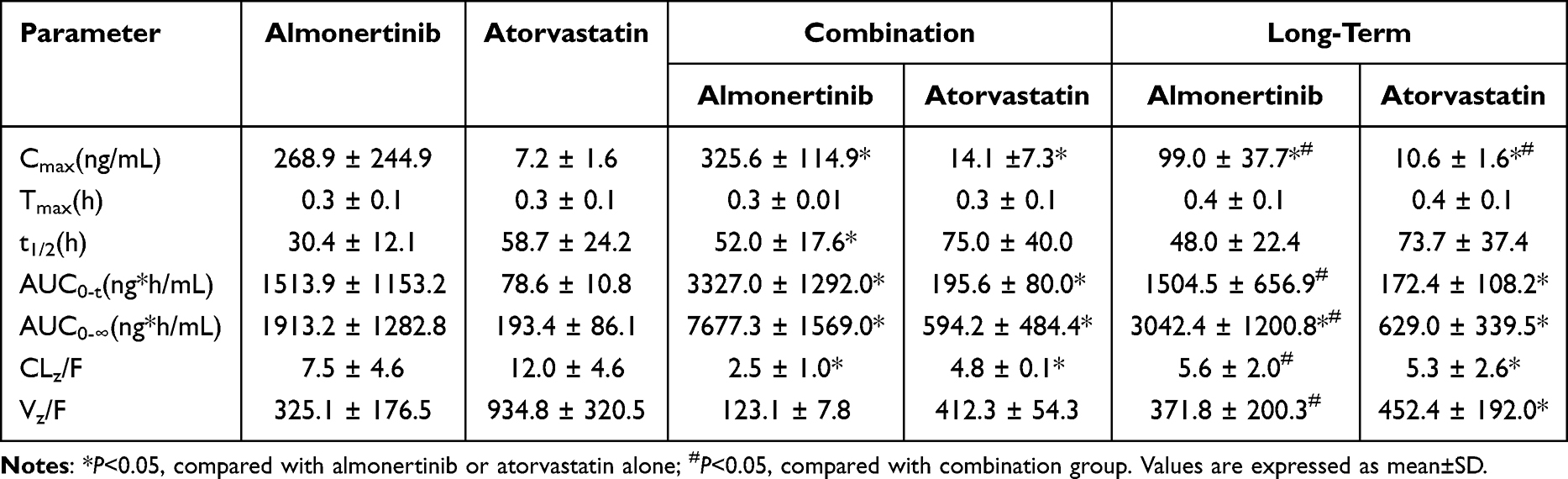 |
Table 6 Comparative Pharmacokinetic Parameters of Almonertinib and Atorvastatin |
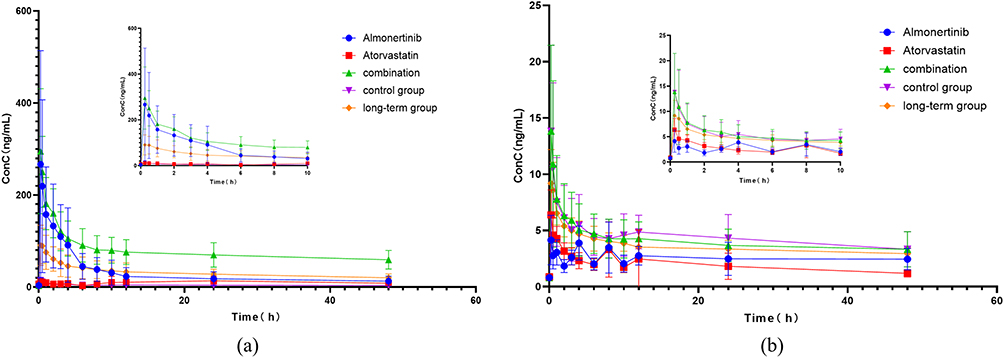 |
Figure 3 The mean plasma concentration-time profiles of almonertinib after oral atorvastatin alone and following multiple-doses almonertinib. (a) the mean plasma concentration-time curves for almonertinib; (b) the mean plasma concentration-time curves for atorvastatin. |
Discussion
The concurrent use of multiple medications can lead to DDIs, causing adverse reactions and compromising therapeutic efficacy. Therefore, identifying potential DDIs to prevent adverse reactions is crucial in combination regimens. Reports indicated that the prevalence of DDIs among elderly hospitalized patients reached as high as 45%,29 among that the incidence of DDIs induced by CYP3A4 and P-gp significantly increased, primarily involving cardiovascular-related medications.30 Given the high incidence of lung cancer in elderly patients, who often require long-term concomitant statin regimen for conditions such as cardiovascular disease and hyperlipidemia, evaluating the DDIs between almonertinib and atorvastatin is of great importance.
To simulate the recommended patient dosage in clinical practice, the final dosing regimen was determined based on clinically equivalent dose and referred to published studies on almonertinib and atorvastatin dosing.26,31 The administered doses were 15 mg/kg for almonertinib and 2 mg/kg for atorvastatin, and oral gavage was used for administration. The half-life time(t1/2) of almonertinib is approximately 30 h, similar with its t1/2 in lung cancer patients, but much longer than a study reported t1/2 in rat(2.18±1.05 h),6,31 while that of atorvastatin is about 57 h, significantly longer than that reported in human(8.8 ± 2.06 h) or in rat(2.74 ± 0.23 h),7,32 and this results may be explained by the differences in experimental conditions, sample size, strains of animals and so on; importantly, a study reported that the CMC-Na in solvent showed inhibitory effect on absorption process,33 and this study added 0.5% CMC-Na, which may detain some drugs in the intestine.
This study found that compared with monotherapy, the combined use of the two drugs increased the Cmax of almonertinib by approximately 1.21-fold and the Cmax of atorvastatin by approximately 1.96-fold, along with a marked prolongation of half-life. Concurrently, CLz/F and Vz/F were significantly reduced. Therefore, it can be hypothesized that almonertinib and atorvastatin interact with each other, may inhibit their metabolism and elimination, as the Tmax showed no obviously change after combination. Additionally, we found that the AUC0-∞ value of almonertinib observed increased approximately 4-fold. This might be due to the small sample size, which limited the reliability of the statistics, but all seven rats showed an increase in exposure, and no obvious outliers dominated the average effect. We speculate that these alterations may be attributed to both drugs being metabolized by the CYP3A4, and almonertinib and atorvastatin are substrates for P-gp and BCRP efflux transporters; inhibition of CYP3A4 and P-gp and/or BCRP can lead to increased systemic exposure of both drugs.14,17 Although no mechanistic experiments were conducted in this study to confirm these pathways, the observed pharmacokinetic changes are consistent with such a mechanism. Some studies reported that the combination of erlotinib and simvastatin caused rhabdomyolysis in patients,34 suggesting that vigilance regarding muscle-related adverse reactions may also be warranted for the almonertinib–atorvastatin combination, even though no such events have been reported to date.
Since achieving steady-state plasma concentrations requires 5–7 half-lives for a drug in vivo, this study designed a 7-day oral administration of atorvastatin to achieve steady-state concentrations in rats before co-administration, which could comprehensively investigate the effects of atorvastatin administration on the almonertinib disposal. After a long-term of atorvastatin administration, it was found that the Cmax of both drugs decreased significantly compared with the combination group. This may be attributed to prolonged exposure of atorvastatin to CYP3A4/P-gp/BCRP, which may induce upregulation of these enzymes, leading to accelerated catabolism and elimination. However, this interpretation remains speculative and requires further investigation, as the present study did not assess enzyme or transporter expression levels. The CYP3A4/P‑gp pathways exist in both rats and human. Although quantitative extrapolation requires caution, the qualitative DDI observed is relevant to clinical practice. Co‑administration may increase drug exposure and myopathy risk. Monitoring creatine kinase and muscle symptoms is advisable, and prospective clinical studies are warranted.
This study has several limitations; (1) the mechanisms mentioned are largely speculative and require further investigations; (2) no animal model of lung cancer was used to observe pharmacokinetic interactions; (3) species differences exist between rats and humans. These represent major limitations. Our findings should therefore be considered as preliminary evidence of a potential DDI. Therefore, further researches in clinical settings are needed to confirm whether similar DDIs occur in humans.
Conclusion
This study established a rapid, sensitive, and reliable LC-MS/MS method for the simultaneous quantification of almonertinib and atorvastatin in rat plasma. The method features a short run time (3.5 min) and simple, cost-effective plasma sample preparation. Co-administration significantly altered the pharmacokinetic profiles compared with monotherapy, resulting in increased exposure and prolonged half-life of both drugs. Following 7-day’s atorvastatin pretreatment, these interaction effects were attenuated, suggesting that the duration of atorvastatin exposure may influence the extent of the interaction. While the observed exposure increases may theoretically elevate the risk of dose-dependent adverse reactions, this study did not directly assess safety endpoints. The findings should be interpreted with caution, as the study was conducted in healthy rats and mechanistic experiments were not performed. Further pharmacokinetic and clinical studies are warranted to clarify the clinical significance of this DDI.
Data Sharing Statement
The datasets are available from the corresponding author, Zhipeng Wang, on reasonable request.
Ethical Approval
This study protocol was approved and supervised by the Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine (PZSHUTCM2506150003).
Consent for Publication
All authors approved the final manuscript and the submission to this journal.
Author Contributions
All authors made a significant contribution to the work in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82274173, 82272390), Shanghai Leading Talent Program of Eastern Talent Plan (SHSLJRC-TX), and the “Shenlan” talent project of Naval Medical University (SL63).
Disclosure
The authors declare no competing interests.
References
1. Siegel RL, Kratzer TB, Giaquinto AN, et al. Cancer statistics, 2025. CA Cancer J Clin. 2025;75(1):10–13. doi:10.3322/caac.21871
2. Chen Z, Fillmore CM, Hammerman PS, et al. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. doi:10.1038/nrc3775
3. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. doi:10.1002/1878-0261.12155
4. Lu S, Dong X, Jian H, et al. AENEAS: a randomized Phase III trial of aumolertinib versus gefitinib as first-line therapy for locally advanced or metastaticnon-small-cell lung cancer with EGFR Exon 19 deletion or L858R mutations. J Clin Oncol. 2022;40(27):3162–3171. doi:10.1200/JCO.21.02641
5. Nagasaka M, Zhu VW, Lim SM, et al. Beyond osimertinib: the development of third-generation EGFR tyrosine kinase inhibitors for advanced EGFR+ NSCLC. J Thorac Oncol. 2021;16(5):740–763. doi:10.1016/j.jtho.2020.11.028
6. Duan JC, Zhong J, Sun BY, et al. Aumolertinib with carboplatin-pemetrexed versus aumolertinib for nonsmall cell lung cancer with EGFR and concomitant tumor suppressor genes (ACROSS2): an open-label, multicenter, randomized Phase 3 study. CA Cancer J Clin. 2026;76(2):e70071. doi:10.3322/caac.70071
7. Choi Y, S Lee, Jang IJ, et al. Pharmacokinetic interaction between fimasartan and atorvastatin in healthy male volunteers. Drug Des Devel Ther. 2018;12:2301–2309. doi:10.2147/DDDT.S165171
8. Wang C, Lu D, Cronin-Fenton D, et al. Cardiovascular disease and risk of lung cancer incidence and mortality: a nationwide matched cohort study. Front Oncol. 2022;12:950971. doi:10.3389/fonc.2022.950971
9. CHINA. G O O N H C O T P S R O. Basic principles for the clinical application of new antineoplastic drugs (2024 edition). 2024.
10. Riely GJ, Wood DE, Ettinger DS, et al. Non-small cell lung cancer, version 4.2024, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2024;22(4):249–274. doi:10.6004/jnccn.2204.0023
11. Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360(9340):1155–1162. doi:10.1016/S0140-6736(02)11203-7
12. Christians U, Schmitz V, Haschke M. Functional interactions between P-glycoprotein and CYP3A in drug metabolism. Expert Opin Drug Metab Toxicol. 2005;1(4):641–654. doi:10.1517/17425255.1.4.641
13. Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008;21(1):70–83. doi:10.1021/tx700079z
14. L Liu, Li W, Yang L, et al. Itraconazole and rifampicin, as CYP3A modulators but not P-gp modulators, affect the pharmacokinetics of almonertinib and active metabolite HAS-719 in healthy volunteers. Acta Pharmacol Sin. 2022;43(4):1082–1090. doi:10.1038/s41401-021-00710-8
15. Kantola T, Kivistö KT, Neuvonen PJ. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther. 1998;64(1):58–65. doi:10.1016/S0009-9236(98)90023-6
16. Lund M, Petersen TS, Dalhoff KP. Clinical implications of P-glycoprotein modulation in drug-drug interactions. Drugs. 2017;77(8):859–883. doi:10.1007/s40265-017-0729-x
17. Goard CA, Mather RG, Vinepal B, et al. Differential interactions between statins and P-glycoprotein: implications for exploiting statins as anticancer agents. Int J Cancer. 2010;127(12):2936–2948. doi:10.1002/ijc.25295
18. Wu CP, Hung TH, Lusvarghi S, et al. The third-generation EGFR inhibitor almonertinib (HS-10296) resensitizes ABCB1-overexpressing multidrug-resistant cancer cells to chemotherapeutic drugs. Biochem Pharmacol. 2021;188:114516. doi:10.1016/j.bcp.2021.114516
19. Yang JC, Camidge DR, Yang CT, et al. Safety, efficacy, and pharmacokinetics of almonertinib (HS-10296) in pretreated patients with EGFR-mutated advanced NSCLC: a multicenter, open-label, Phase 1 trial. J Thorac Oncol. 2020;15(12):1907–1918. doi:10.1016/j.jtho.2020.09.001
20. Morandi L, Angelini C, Prelle A, et al. High plasma creatine kinase: review of the literature and proposal for a diagnostic algorithm. Neurol Sci. 2006;27(5):303–311. doi:10.1007/s10072-006-0701-0
21. Rysz MA, Kinzi J, Schäfer AM, et al. Simultaneous quantification of atorvastatin, erlotinib and OSI-420 in rat serum and liver microsomes using a novel liquid chromatography-mass spectrometry method. J Pharm Biomed Anal. 2023;236:115716. doi:10.1016/j.jpba.2023.115716
22. Shen X, G Fan, G Liu, et al. Severe adverse cutaneous reactions induced by gefitinib combined with antihypertensive and antihyperlipidemic drugs in lung cancer: a case report. Anticancer Drugs. 2022;33(1):e802–e7. doi:10.1097/CAD.0000000000001226
23. Occhipinti M, Brambilla M, Galli G, et al. Evaluation of drug-drug interactions in EGFR-mutated non-small-cell lung cancer patients during treatment with tyrosine-kinase inhibitors. J Pers Med. 2021;11(5). doi:10.3390/jpm11050424.
24. Chen D, Chen J, Shen Y, et al. Optimization of a sensitive and reliable UPLC-MS/MS method to simultaneously quantify almonertinib and HAS-719 and its application to study the interaction with nicardipine. Pharm Biol. 2024;62(1):874–881. doi:10.1080/13880209.2024.2425648
25. Fu Y, Li Y, Li Y, et al. Influences of magnesium isoglycyrrhizinate on the pharmacokinetics of almonertinib in rats and its potential mechanisms. Biopharm Drug Dispos. 2025;46(3):95–103. doi:10.1002/bdd.70007
26. le j, liao y, li s, et al. High-throughput LC-MS/MS method for simultaneous determination of pantoprazole and atorvastatin in rat plasma: application to a pharmacokinetic interaction study. Curr Drug Metab. 2021;22(6):481–490. doi:10.2174/1389200222666210520090632
27. Bu C, Jiang L, L Cui, et al. LC-MS/MS method for quantification of 23 TKIs in Plasma: assessing the relationship between anlotinib trough concentration and toxicities. Clin Chim Acta. 2025;566:120028. doi:10.1016/j.cca.2024.120028
28. USE I C F H O T R F P F H. ICH Harmonised Guideline: Bioanalytical Method Validation and Study Sample Analysis (M10); 2022.
29. Espinosa-Bosch M, Santos-Ramos B, Gil-Navarro MV, et al. Prevalence of drug interactions in hospital healthcare. Int J Clin Pharm. 2012;34(6):807–817. doi:10.1007/s11096-012-9697-0
30. Gallo P, De Vincentis A, Pedone C, et al. Drug-drug interactions involving CYP3A4 and p-glycoprotein in hospitalized elderly patients. Eur J Intern Med. 2019;65:51–57. doi:10.1016/j.ejim.2019.05.002
31. Wang Z, Li Y, He X, et al. In vivo evaluation of the pharmacokinetic interactions between almonertinib and rivaroxaban, almonertinib and apixaban. Front Pharmacol. 2023;14:1263975. doi:10.3389/fphar.2023.1263975
32. S Sun, Wand R, J Fan, et al. Effects of Danshen tablets on pharmacokinetics of atorvastatin calcium in rats and its potential mechanism. Pharm Biol. 2018;56(1):104–108. doi:10.1080/13880209.2018.1424209
33. Wang L, Peng J, Wang X, et al. Carboxymethylcellulose sodium improves the pharmacodynamics of 1-deoxynojirimycin by changing its absorption characteristics and pharmacokinetics in rats. Pharmazie. 2012;67(2):168–173. doi:10.31083/ph.2012.1106
34. Veeraputhiran M, Sundermeyer M. Rhabdomyolysis resulting from pharmacologic interaction between erlotinib and simvastatin. Clin Lung Cancer. 2008;9(4):232–234. doi:10.3816/CLC.2008.n.036
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Olanzapine Pharmacokinetics: A Clinical Review of Current Insights and Remaining Questions
Kolli P, Kelley G, Rosales M, Faden J, Serdenes R
Pharmacogenomics and Personalized Medicine 2023, 16:1097-1108
Published Date: 21 December 2023
Development of HPLC-MS/MS Method for Simultaneous Detection of Esketamine and Norketamine: Application to Pharmacokinetics Drug Interactions Affected by Dexmedetomidine
Zhou W, Guo Z, Zhang X, Wang W, Zheng Z, Qiu X
Drug Design, Development and Therapy 2025, 19:8589-8600
Published Date: 22 September 2025