Back to Journals » International Journal of Women's Health » Volume 18
An Integrative Genome-Wide Analysis Reveals Shared and Subtype-Specific Genetic Links Between Endometriosis and Menstrual-Cycle-Related Traits
Authors Fu F, Lin F, Yao X, Xuan Z, Gu J, Shen D, Hu S
Received 27 December 2025
Accepted for publication 27 June 2026
Published 14 July 2026 Volume 2026:18 591799
DOI https://doi.org/10.2147/IJWH.S591799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Everett Magann
Feifei Fu,1 Feng Lin,2 Xiangjun Yao,1 Zhangbiao Xuan,1 Jialu Gu,1 Dong Shen,1 Shuqin Hu1
1Department of Gynecology, Affiliated Hospital of Shaoxing University, Shaoxing, Zhejiang, 312000, People’s Republic of China; 2Department of Urology, Affiliated Hospital of Shaoxing University, Shaoxing, Zhejiang, 312000, People’s Republic of China
Correspondence: Shuqin Hu, Department of Gynecology, Affiliated Hospital of Shaoxing University, Shaoxing, Zhejiang, 312000, People’s Republic of China, Email [email protected]
Purpose: This study aimed to systematically assess the genetic comorbidity of endometriosis (EM) and its subtypes with menstrual-cycle-related traits, and to identify shared genetic mechanisms and key regulatory pathways underlying their association.
Methods: Using FinnGen-derived genome-wide association study (GWAS) data for overall EM and its lesion location-defined subtypes, including endometriosis of ovary (EO), deep endometriosis (DE), endometriosis of pelvic peritoneum (EPP), endometriosis of rectovaginal septum and vagina (ERSV), and endometriosis of intestine (EI), together with GWAS summary statistics for menstrual-cycle-related traits from European female populations, we conducted a comprehensive multi-subtype cross-trait analysis. Genetic correlations were assessed using linkage disequilibrium score regression (LDSC) and high-definition likelihood (HDL). Shared signals were prioritized using Pleiotropic Analysis under Composite Null Hypothesis (PLACO), Functional Mapping and Annotation of GWAS (FUMA), Multi-marker Analysis of GenoMic Annotation (MAGMA), and Hypothesis Prioritization in Multi-Trait Colocalization (HyPrColoc), followed by functional enrichment and protein-protein interaction (PPI) network analyses.
Results: Distinct genetic association patterns were observed in EM and across its different subtypes. Age at menarche (AAM) was negatively genetically correlated with EM and EPP, frequency of irregular menstruation (FIM) was positively correlated with EM, EO, and EPP, whereas length of menstrual cycle (LMC) was negatively correlated with EM, DE, and ERSV. Cross-trait analyses identified 96 pleiotropic loci and 136 unique genes, highlighting 11p14.1 as a key shared hotspot and 8p21.2 as a recurrent pleiotropic region. Prioritized genes and pathways implicated ovarian development, sex differentiation, chromatin remodeling, cAMP/PKA signaling, and estrogen-androgen regulation, with testosterone-related colocalization supporting androgen involvement.
Conclusion: A comprehensive multi-subtype analysis using FinnGen data revealed a complex and heterogeneous shared genetic architecture between EM and menstrual-cycle-related traits. These findings support genetic evidence for precision risk stratification and subtype-informed management of EM, but require validation in larger and more diverse populations.
Keywords: endometriosis, menstrual-cycle-related traits, genome-wide association study, GWAS, genetic correlation, hormonal signaling
Introduction
Endometriosis (EM) is a prevalent, estrogen-dependent, chronic gynecological disorder characterized by the ectopic growth of endometrial-like tissue outside the uterine cavity, often resulting in chronic pelvic pain, infertility, and ovarian dysfunction.1 Epidemiological studies indicate that EM affects approximately 6–10% of women of reproductive age, with prevalence reaching 20–50% among infertile women.2,3 Twin studies further suggest a strong genetic predisposition for EM, with an estimated heritability of 47–51%, approximately 26% of which is explained by common single nucleotide polymorphisms (SNPs).4
The pathogenesis of EM is multifactorial, involving retrograde menstruation, hormonal dysregulation, and immune-inflammatory imbalance. The retrograde menstruation hypothesis proposes that refluxed menstrual blood and exfoliated endometrial cells may seed ectopic lesions; however, its high prevalence among reproductive-aged women and the fact that only a subset develop EM indicate that this mechanism alone cannot explain disease susceptibility or clinical heterogeneity.5 Hormonal dysregulation, particularly aberrant local estrogen metabolism and progesterone resistance, may promote lesion survival, proliferation, and inflammation.6 Meanwhile, impaired immune surveillance, macrophage activation, reduced natural killer cell cytotoxicity, and elevated inflammatory cytokines may facilitate immune escape and sustain lesion adhesion, invasion, angiogenesis, and chronic inflammation.7
Genetic studies further support the multifactorial nature and subtype heterogeneity of EM. A pooled-sample genome-wide association study stratified by major clinical subtype revealed distinct genetic architectures across EM subtypes. The study found that ovarian endometrioma yielded the largest number of subtype-associated variants, including rs4703908 near ZNF366 and rs227849 in the RUNX2/SUPT3H/CDC5L region. In contrast, fewer subtype-associated variants were identified for the peritoneal subtype.8 Consistent with this, a large-scale GWAS meta-analysis identified 42 genome-wide significant loci and further demonstrated that stronger genetic effects were primarily associated with severe disease phenotypes, particularly ovarian EM.9 A multi-ancestry GWAS involving nearly 1.4 million women further expanded the map of EM-associated susceptibility loci and identified more than 50 putative causal signals.10
The association between menstrual-cycle-related traits and EM risk has also attracted considerable attention. Early menarche (≤11–12 years) modestly increases the risk of EM, suggesting that menstrual characteristics in early puberty may represent important epidemiological indicators for disease susceptibility.11 Previous studies have shown that women with EM often experience earlier menarche and shorter menstrual cycles, resulting in greater cumulative menstrual exposure, which may contribute to disease risk and is consistent with, but not specific to, the retrograde menstruation hypothesis.11,12 Mendelian randomization analyses have suggested that earlier menarche, shorter menstrual cycles, and lower anti-Müllerian hormone (AMH) levels may be associated with increased EM risk.13 At the locus level, the FSHB promoter variant rs10835638 has been associated with lower follicle-stimulating hormone (FSH) levels, longer menstrual cycles, and potentially reduced EM susceptibility.14 Additionally, key genes implicated in both EM and menstrual traits, such as WNT4, ESR1, and GREB1, regulate the synthesis and metabolism of estrogen, progesterone, and androgens, thereby supporting endometrial development and functional maintenance.15 Together, these findings suggest that menstrual-cycle-related traits are not only epidemiological correlates of EM, but may also reflect shared hormonal and genetic mechanisms underlying EM susceptibility.
Although previous studies employing genetic correlation and Mendelian randomization approaches have identified potential associations between EM and menstrual-cycle-related traits, these studies were largely limited to broad correlations and causal inferences, lacking systematic, fine-scale, and multidimensional genetic analyses. To address this gap, this study integrated LDSC and HDL analyses to obtain more accurate genetic estimates of these associations. Additionally, several analytical tools, including PLACO, FUMA, MAGMA, and HyPrColoc, were applied to systematically identify pleiotropic loci and associated genes (Figure 1). Based on these findings, functional enrichment analyses and protein-protein interaction network analyses were conducted to identify key genetic pathways mediating the interaction between menstrual traits and hormonal signaling, thus elucidating shared molecular mechanisms underlying EM and menstrual-cycle-related traits at a finer genetic level. This study provides important genetic insights for identifying high-risk populations, improving early risk prediction, and enhancing precision stratification.
 |
Figure 1 Overall study design and analytical workflow. Abbreviations: LDSC, linkage disequilibrium score regression; HDL, high-definition likelihood; PLACO, Pleiotropic Analysis under Composite Null Hypothesis; FUMA, Functional Mapping and Annotation of GWAS; Coloc.abf, colocalization analysis using approximate Bayes factor; MAGMA, Multi-marker Analysis of GenoMic Annotation; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, protein-protein interaction; HyPrColoc, Hypothesis Prioritisation in Multi-Trait Colocalization. |
Methods
Sources of Summary Statistics for EM, Its Subtypes, Menstrual-Cycle-Related Traits, and Sex Hormones
This study utilized large-scale GWAS summary statistics to systematically investigate the shared genetic architecture among EM phenotypes, menstrual-cycle-related traits, and sex hormone levels. The selected traits comprise pubertal timing, menstrual regularity, cycle length, ovarian reserve, and sex hormone regulation, all of which represent key aspects of reproductive physiology relevant to EM and ensure biological relevance, comparability, and reproducibility. Summary statistics for EM and its subtypes were obtained from the FinnGen Project (Release 12), which included European female participants. These included endometriosis (EM; n = 150,350), endometriosis of ovary (EO; n = 138,038), endometriosis of pelvic peritoneum (EPP; n = 137,777), deep endometriosis (DE; n = 275,743), endometriosis of rectovaginal septum and vagina (ERSV; n = 133,386), endometriosis of intestine (EI; n = 130,777). Statistical power for GWAS of EM subtypes was further evaluated (Supplementary Table 1). Data for menstrual-cycle-related traits were likewise obtained from European women. Age at menarche (AAM) data were mainly derived from the ReproGen Consortium (n = 179,117) and the UK Biobank (n = 73,397), encompassing a total of 252,514 women;16 Anti-Müllerian hormone (AMH) levels were derived from the GWAS conducted by Ruth et al and the Doetinchem, SWAN, and ALSPAC cohorts (n = 7,049);17 data on frequent and irregular menstruation (FIM; n = 361,194) and length of menstrual cycle (LMC; n = 43,125) were obtained from the UK Biobank. GWAS summary statistics for sex hormone-related traits were also derived exclusively from female cohorts. These included total testosterone (n = 199,569), bioavailable testosterone (n = 180,386), sex hormone-binding globulin (SHBG; n = 214,989), and oestradiol (n = 53,391), obtained from the IEU OpenGWAS database. Data for progesterone levels (n = 17,956) were retrieved from the GWAS Catalog (GCST90483485).18 Collectively, these datasets facilitated a comprehensive assessment of genetic correlations among EM, menstrual-cycle-related traits, and sex hormone levels (Supplementary Table 2).
Standardization of GWAS Summary Statistics
To ensure consistency and comparability across GWAS summary statistics, the original data were standardized before analysis. First, rare variants with a minor allele frequency (MAF) below 1% were removed to reduce potential bias caused by low-frequency alleles. Second, all genomic coordinates from the GWAS datasets were standardized to the human reference genome version GRCh37 (Genome Reference Consortium Human Build 37) to ensure comparability and consistency across datasets.
Global Genetic Correlation Analysis
To assess the overall genetic correlations of EM and its subtypes with menstrual-cycle-related traits, this study used publicly available GWAS summary statistics and applied two analytical methods: linkage disequilibrium score regression (LDSC) and high-definition likelihood (HDL). The LDSC method estimates genetic correlations between traits by regressing genome-wide SNP test statistics on their corresponding linkage disequilibrium (LD) scores. This approach effectively distinguishes true polygenic signals from potential confounding effects and has been widely applied in studies investigating genetic correlations among complex diseases and quantitative traits.19,20 In this study, LD reference data for LDSC were derived from European genotype data from Phase 3 of the 1000 Genomes Project (1000 Genomes Project Consortium).21 To improve the accuracy and robustness of genetic correlation estimates, the HDL method was additionally employed. The HDL approach, built on a likelihood-based inference framework, jointly models genome-wide effect sizes using high-dimensional LD matrices, enabling more precise estimation of genetic correlations, particularly in large samples or highly polygenic contexts with extensive pleiotropy. HDL analysis was performed using the European-ancestry UK Biobank imputed LD reference panel provided by the HDL software, and for each trait pair, only SNPs shared by both GWAS summary statistics and the HDL LD reference panel were retained for analysis.22 Compared with LDSC, HDL provides greater statistical efficiency in analyzing highly correlated complex traits.
Identification of Pleiotropic Risk Loci and Colocalization Analysis
To identify shared genetic signals of EM and its subtypes with menstrual-cycle-related traits, this study applied the PLACO (Pleiotropic Analysis under Composite Null Hypothesis) method to detect pleiotropic associations among disease–trait pairs exhibiting significant global genetic correlations. PLACO operates under a composite null hypothesis framework. The null hypothesis (H0) assumes that a SNP affects neither phenotype or only one of them, whereas the alternative hypothesis (H1) proposes that the SNP significantly influences both. By jointly computing Z statistics for both phenotypes and deriving corresponding P values, PLACO effectively distinguishes genuine pleiotropic signals from false positives. It offers improved control of false discovery rates compared with traditional approaches.23 Subsequently, significant pleiotropic SNPs were functionally annotated using the FUMA (Functional Mapping and Annotation of GWAS) platform.24 PLACO-significant SNPs (P < 5 × 10−8) were further annotated using the FUMA SNP2GENE module. Based on the 1000 Genomes Project Phase 3 European reference panel, FUMA identified independent significant SNPs and their candidate SNPs in linkage disequilibrium (r2 > 0.6) and merged nearby or overlapping LD blocks within 250 kb into genomic risk loci. This procedure allowed SNP-level pleiotropic signals to be collapsed into independent pleiotropic loci for downstream functional interpretation.
To further characterize these loci, we performed functional annotation and eQTL integration analyses. First, pleiotropic SNPs were annotated using FUMA to characterize their genomic locations, functional consequences, CADD and RegulomeDB scores, chromatin states, and SNP-to-gene mapping evidence. We then integrated GTEx v8 eQTL data to evaluate whether candidate variants were associated with gene expression in available tissues. Given the limited availability of endometrium- and ovary-specific eQTL data, whole blood was selected as a relevant tissue context. Variants with significant eQTL evidence provided support for the regulatory interpretation of candidate loci.
Bayesian colocalization analysis was performed using coloc.abf to assess whether pleiotropic loci reflected shared causal variants rather than regional overlap. Summary statistics were harmonized across overlapping SNPs, and prior probabilities were set as p1 = 1 × 10−4, p2 = 1 × 10−4, and p12 = 1 × 10−5. Strong colocalization evidence was defined as PP.H4 ≥ 0.80, with loci remaining significant under stricter thresholds prioritized.25
Identification and Functional Enrichment Analysis of Pleiotropic Genes
Building on the global genetic correlation and colocalization results, we systematically identified pleiotropic genes and performed functional enrichment analyses to further elucidate the shared genetic mechanisms linking EM and its subtypes to menstrual-cycle-related traits. Gene-level analysis was conducted using MAGMA (version 1.08) implemented on the FUMA platform.26 This method, based on a linear regression framework, maps GWAS-level SNP signals to gene regions using a ±50 kb window around gene boundaries to capture proximal regulatory variants while limiting LD noise from distant variants, and accounts for LD structure to derive gene-level association statistics. Subsequently, the identified candidate genes were functionally annotated and subjected to pathway enrichment analysis using the Metascape online platform,27 which integrates several authoritative functional databases, including Gene Ontology (GO), KEGG, and Reactome. Hypergeometric testing with Benjamini–Hochberg correction was applied to adjust for multiple comparisons. The final enrichment results were visualized as clustered networks and bar plots, highlighting the potential roles of pleiotropic genes in biological processes, molecular functions, cellular components, and key signaling pathways.
Colocalization Analysis of Sex Hormone Signals
This study applied the multi-trait colocalization method HyPrColoc (Hypothesis Prioritisation in Multi-trait Colocalization) to systematically examine hormonal signaling within the identified pleiotropic risk loci.28 HyPrColoc operates within a Bayesian model-averaging framework, using GWAS summary statistics to determine whether multiple traits are influenced by a shared causal variant and to directly estimate the posterior probability (PP) of colocalization. By employing an efficient branch-and-bound clustering algorithm, the method identifies clusters of traits that share common causal variants, thereby markedly reducing the computational complexity associated with traditional pairwise colocalization approaches while preserving both statistical power and computational efficiency. Colocalization was considered robust when the posterior probability of the colocalization result was≥0.80, providing strong evidence that endometriosis-related traits and specific sex hormone-related traits share a common causal signal within the same genomic region.29
Results
Global Genetic Correlations Between EM and Its Subtypes with Menstrual-Cycle-Related Traits
Using two complementary analytical methods, LDSC and HDL, this study systematically evaluated genome-wide genetic correlations of EM and its subtypes with multiple menstrual-cycle-related traits. After FDR multiple correction to p-values derived from both methods, trait pairs that remained significant in both analyses (FDR-adjusted p < 0.05) were retained, resulting in the identification of eight phenotype pairs with robust global genetic correlations.
For AAM, significant negative genetic correlations were detected between AAM and EM (LDSC: rg = −0.11, p = 5.51×10−6; HDL: rg = −0.12, p = 4.41×10−6), as well as between AAM and EPP (LDSC: rg = −0.11, p = 2.26×10−4; HDL: rg = −0.10, p = 2.30×10−3). This suggests that a genetic predisposition to earlier age at menarche may be associated with higher genetic susceptibility to EM and EPP. Regarding FIM, significant positive genetic correlations were identified between FIM and EM (LDSC: rg = 0.46, p = 7.68×10−9; HDL: rg = 0.54, p = 1.01×10−11), EO (LDSC: rg = 0.38, p = 1.98×10−5; HDL: rg = 0.42, p = 4.96×10−8), and EPP (LDSC: rg = 0.41, p = 2.06×10−5; HDL: rg = 0.41, p = 4.42×10−5). This suggests that a genetic background related to menstrual cycle irregularity may be associated with higher genetic susceptibility to EM, EO, and EPP. For LMC, significant negative genetic correlations were observed between LMC and DE (LDSC: rg =−0.35, p = 9.99×10−5; HDL: rg =−0.30, p = 2.13×10−2), EM (LDSC: rg =−0.24, p = 1.40×10−4; HDL: rg =−0.19, p = 2.44×10−3), and ERSV (LDSC: rg =−0.35, p = 1.05×10−4; HDL: rg =−0.31, p = 1.55×10−2). This suggests that genetically determined shorter menstrual cycles may be associated with higher genetic susceptibility to EM, DE, and ERSV (Table 1 and Figure 2).
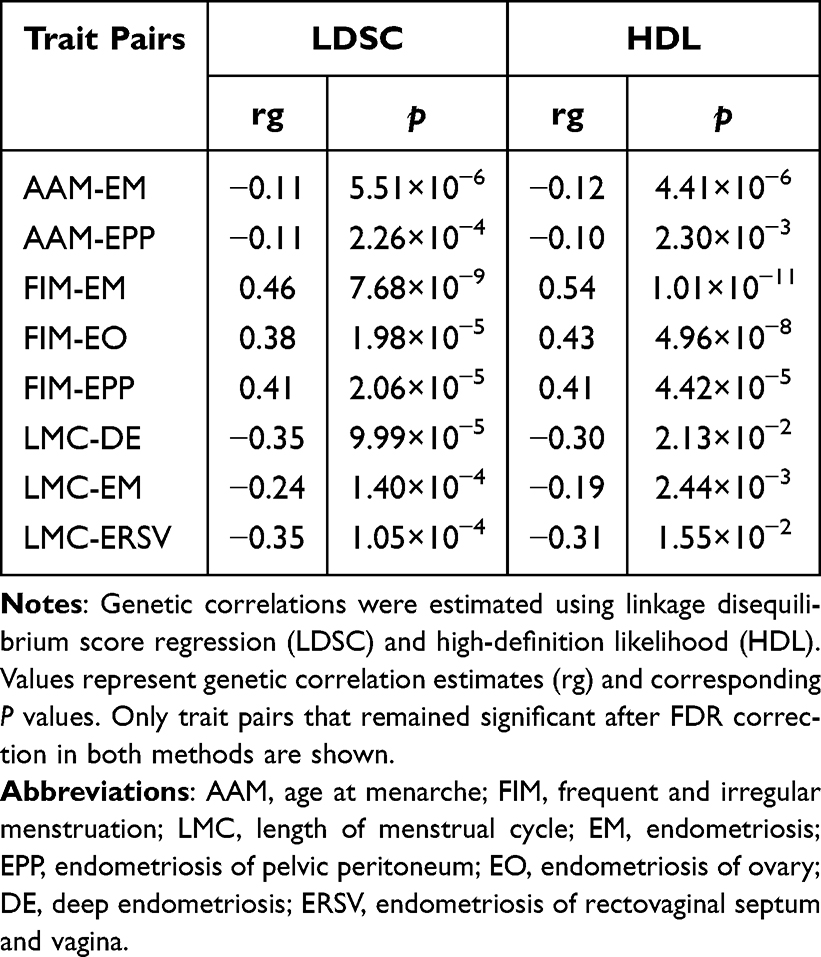 |
Table 1 Genetic Correlations of Endometriosis and Its Subtypes with Menstrual-Cycle-Related Traits |
 |
Figure 2 Genetic correlations of endometriosis and its subtypes with menstrual-cycle-related traits. (A) Heatmap of genetic correlations estimated by LDSC. (B) Heatmap of genetic correlations estimated by HDL. Abbreviations: EM, endometriosis; EO, endometriosis of ovary; DE, deep endometriosis; EPP, endometriosis of pelvic peritoneum; ERSV, endometriosis of rectovaginal septum and vagina; EI, endometriosis of intestine; AAM, age at menarche; AMH, anti-Müllerian hormone; FIM, frequent and irregular menstruation; LMC, length of menstrual cycle. |
Identification of Pleiotropic Genetic Loci Linking EM and Its Subtypes to Menstrual-Cycle-Related Traits
In the subsequent analysis of pleiotropic loci, we applied the PLACO method to investigate eight pairs of significantly correlated trait combinations identified through global genetic correlation analysis. The analysis revealed 122 to 1,523 significant pleiotropic SNPs (P < 5×10−8) across these trait pairs (Supplementary Table 3). Functional annotation of these SNPs was then conducted using the FUMA platform (Supplementary Table 4), resulting in the identification of 96 pleiotropic gene loci shared among the eight significant combinations (Supplementary Table 5). The PLACO-identified pleiotropic loci and their mapped genes formed a complex network across the eight significant trait pairs (Figure 3). This network underscores a substantial shared genetic architecture between various menstrual traits and EM, including its subtypes. In essence, a single genetic locus may exert influence over multiple phenotypes. Among these, the associations between AAM and EM were the most densely clustered. Further colocalization analysis (Figure 4A) revealed that among the 96 pleiotropic loci, 22 loci (22.92%) exhibited significant shared causal signals (PP.H4 ≥ 0.80). Of particular interest, the 11p14.1 locus showed consistent colocalization across seven trait combinations (FIM-EO, FIM-EM, FIM-EPP, AAM-EM, AAM-EPP, LMC-EM, and LMC-DE), while the 8p21.2 locus demonstrated similar pleiotropic effects across six combinations (FIM-EPP, AAM-EM, LMC-ERSV, AAM-EPP, LMC-EM, and LMC-DE). The frequency distribution (Figure 4B) further indicates that the 11p14.1 and 8p21.2 loci repeatedly appeared across multiple trait–disease combinations, highlighting their robust and wide-ranging pleiotropic effects. These loci may represent key genetic hotspots connecting EM and menstrual traits, thereby illuminating shared genetic mechanisms that may underlie both reproductive system disorders and female physiological traits.
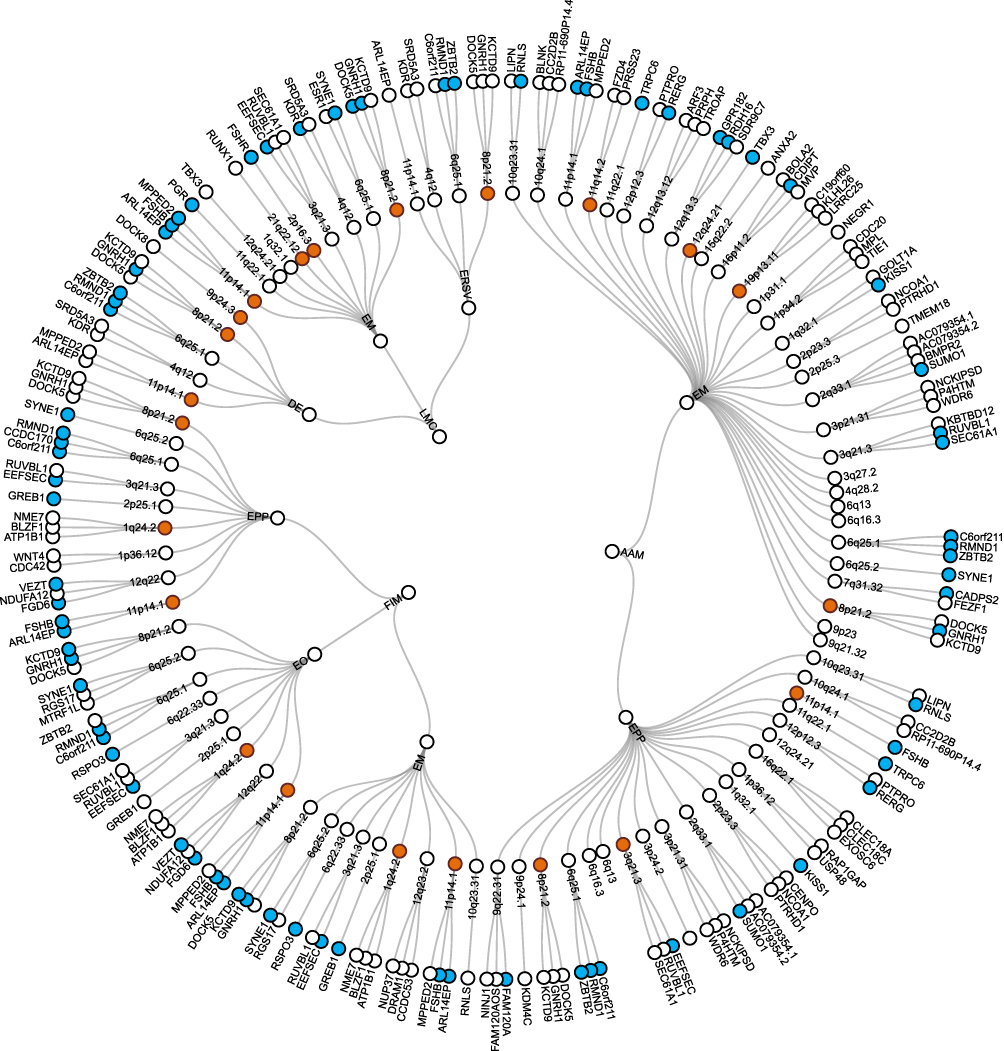 |
Figure 3 Network of pleiotropic loci and mapped genes linking endometriosis-related phenotypes to menstrual-cycle-related traits across eight trait pairs. Notes: Shared loci identified through colocalization analysis are highlighted in orange, and shared genes detected by MAGMA are highlighted in blue. |
 |
Figure 4 Colocalization evidence and recurrence of pleiotropic genomic regions across trait pairs. (A) Bubble plot of posterior probability (PP.H4) from colocalization analysis across genomic regions and trait pairs. Note: Circle size indicates the magnitude of the posterior probability of colocalization (PP.H4). Larger circles represent higher PP.H4 values. Circle color reflects PP.H4 categories: Blue: PP.H4 < 50%; Yellow: 50% ≤ PP.H4 < 80%; Orange: PP.H4 ≥ 80% (strong evidence of colocalization). Y-axis (Trait Pair): Each row corresponds to a pair of traits analyzed for colocalization. X-axis (Region): Genomic regions (cytogenetic bands) where pleiotropic or colocalized signals were detected. (B) Frequency distribution of pleiotropic genomic regions across different trait pairs. Notes: The height of each bar represents the number of trait pairs in which the corresponding genomic region was identified as a pleiotropic locus. The x-axis denotes the genomic regions (cytobands), and the y-axis indicates the number of trait pairs in which the region appeared. |
Identification of Pleiotropic Genes and Functional Enrichment Analysis
Expanding upon the identified pleiotropic loci, we further mapped significant SNP signals to nearby genomic regions using FUMA and performed gene-level pleiotropy analysis with MAGMA. Across all trait pairs, this analysis identified 227 pleiotropic gene records, corresponding to 136 unique pleiotropic genes after removing duplicated gene symbols (Supplementary Table 6). Among these unique genes, 91 were detected in two or more trait pairs and were therefore defined as recurrent pleiotropic genes. Notably, ARL14EP, C6orf211, EEFSEC, FSHB, and RMND1 were consistently significant across six trait pairs, while GNRH1 and SYNE1 were replicated in five. To further support these findings, we integrated GTEx v8 whole-blood eQTL data and found that several prioritized genes, including ARL14EP, C6orf211, EEFSEC, RMND1, and GNRH1, showed significant expression-regulatory evidence across multiple trait pairs (Supplementary Table 7). Furthermore, gene-set enrichment analysis using MAGMA demonstrated significant overrepresentation in biological pathways related to female sex differentiation, fallopian tube tissue-specific signaling, and hormone ligand-receptor interactions, implicating these processes as critical contributors to the underlying pathophysiology (Figure 5).
 |
Figure 5 Cross-trait gene-set enrichment analysis of pleiotropic genes identified by MAGMA. Abbreviations: GOBP, Gene Ontology Biological Process; GOCC, Gene Ontology Cellular Component; GOMF, Gene Ontology Molecular Function; GSEA, gene set enrichment analysis; TSEA, tissue-specific expression analysis. Notes: The upper panels summarize the number of trait pairs contributing to each enrichment category.The lower heatmap displays the significance of enrichment for each trait pair across the corresponding functional categories. |
To comprehensively characterize the functional properties of these genes, we conducted pathway enrichment analysis using the Metascape platform. The analysis revealed that, within the Gene Ontology Biological Process (GOBP) category, the candidate genes were significantly enriched in critical processes such as female gonadal development, folliculogenesis, sexual differentiation, and the ovulatory cycle, underscoring their essential roles in maintaining ovarian function and orchestrating regulation along the hypothalamic–pituitary–gonadal (HPG) axis. Under the Gene Ontology Cellular Component (GOCC) domain, significant enrichment was observed in the INO80-type complex and INO80 complex, suggesting potential involvement in transcriptional regulation and DNA repair pathways. At the Gene Ontology Molecular Function (GOMF) level, the genes were notably enriched in activities related to RNA polymerase II transcription complex binding and transcription factor complex assembly, reflecting fundamental mechanisms of transcriptional regulation. Furthermore, KEGG pathway analysis revealed robust enrichment in pathways associated with sex hormone biosynthesis and secretion, GnRH/cAMP signal transduction, and neuroendocrine regulation (Figure 6A). In parallel, a PPI network constructed via the STRING database and analyzed using the MCODE algorithm identified three highly interconnected functional modules. These modules were predominantly associated with hormone receptor signaling, neuroendocrine signaling cascades, and SUMOylation-dependent modulation of estrogen receptor activity. Notably, the Gαs signaling axis emerged as a recurrent and functionally central pathway across multiple modules, with 13 genes—including FSHB, ESR1, KISS1, and TACR3—participating in these regulatory networks (Figure 6B).
 |
Figure 6 Metascape pathway enrichment and protein-protein interaction network analyses of pleiotropic genes. (A) GO functional categories and KEGG pathway enrichment of pleiotropic genes. Note: This figure presents the enrichment results of pleiotropic genes in GO Biological Processes (GOBP), Cellular Components (GOCC), Molecular Functions (GOMF), and KEGG pathways. The length of each bar represents the enrichment significance (−log10 adjusted P value), and the size of the circles on the right indicates the number of genes involved in each pathway. (B) Protein–protein interaction (PPI) network of pleiotropic genes and MCODE-identified functional modules. Notes: Left panels show the significantly enriched pathways for the three MCODE modules, while the right panels display the corresponding PPI networks for each module. |
Identification of Shared Sex Hormone Signals Linking EM and Its Subtypes to Menstrual-Cycle-Related Traits
Preliminary analyses indicated that sex hormones may play a central role in the shared mechanisms linking EM and its subtypes to menstrual-cycle-related traits. To further substantiate this hypothesis, the HyPrColoc method was employed to perform multi-trait colocalization analyses of traits showing high colocalization posterior probabilities, with the aim of identifying potential sex hormone-associated signals within shared risk loci. Significant colocalization with bioavailable testosterone and total testosterone was observed across the FIM-EM, FIM-EO, FIM-EPP, LMC-DE, and LMC-EM combinations. Subsequent integrative analyses revealed that these colocalization signals were predominantly clustered within the 11p14.1 genomic region, where key candidate SNPs—rs11031005, rs12294104, rs10835638, and rs35078732—were consistently identified across five phenotype combinations and hormone-related traits. These findings highlight 11p14.1 as a potential genetic hotspot that may jointly regulate EM susceptibility and sex hormone levels (Supplementary Table 8).
Discussion
This study integrated GWAS summary statistics and applied multi-dimensional genetic approaches to investigate the genetic correlations between EM and various menstruation-related traits, as well as their potential shared genetic architecture. The findings not only support previous epidemiological evidence but also offer additional genomic insights into the underlying molecular pathways. We identified significant genetic correlations between EM and several menstrual traits, including AAM, FIM, and LMC. Early menarche may prolong exposure to retrograde menstrual flow, increasing the accumulation of endometrial tissue in the pelvic cavity and raising EM risk by 1.34-fold.30 Similarly, shorter cycles may lead to more frequent estrogen exposure,12 potentially promoting the proliferation and mitotic activity of ectopic endometrial cells via the IGF-1/VEGF signaling pathway, thereby accelerating disease progression.31 Dysregulation of menstrual biological processes may contribute to the development of EM by promoting the survival, migration, and ectopic implantation of shed endometrial tissue through mechanisms involving inflammation, matrix metalloproteinases (MMPs), hypoxia/angiogenesis, and impaired apoptotic regulation.32 Previous studies have linked lower AMH levels to EM.13 As a marker of ovarian reserve, AMH is influenced by ovarian lesions, inflammation, age, and surgical history.33 We found no significant genetic correlation between AMH and EM or its subtypes, suggesting that prior clinical associations may mainly reflect acquired ovarian reserve impairment or related factors. The null finding may also be due to limited sample size and statistical power. In contrast, our study further suggests that a genetic background related to menstrual rhythm may be associated with higher genetic susceptibility to EM. Notably, genetic determinants of menstrual rhythm may offer preliminary insight into increased susceptibility to EM, although their clinical utility as early indicators remains to be further validated.
In the present study, subtype-stratified LDSC and HDL analyses showed that menstrual-cycle-related traits exhibited heterogeneous genetic correlations with specific EM phenotypes rather than EM as a uniform disease entity. Specifically, AAM showed a negative genetic correlation with EPP, FIM showed positive genetic correlations with EO and EPP, and LMC showed negative genetic correlations with DE and ERSV. This view is consistent with previous evidence supporting EM heterogeneity. Menstrual cycle regularity has been reported to be associated with ovarian endometrioma and deep infiltrating EM, but not with superficial peritoneal EM.34 A narrative review further highlighted pathological, diagnostic, and therapeutic differences among these three subtypes.35 In addition, a public transcriptomic data-based study showed that ovarian endometrioma, peritoneal EM, and deep infiltrating EM share common molecular signatures while also exhibiting subtype-specific gene expression patterns.36 Moreover, susceptibility variants near WNT4, VEZT, GREB1, and FN1 have been reported to exert stronger effects in moderate-to-severe or ovarian EM.37 However, previous studies have primarily characterized EM subtype heterogeneity from clinical, pathological, transcriptomic, or severity-related genetic perspectives. In contrast, the present study links menstrual-cycle-related traits to specific EM subtypes through shared genome-wide genetic architecture.
Previous GWAS have systematically identified and consistently validated several genetic susceptibility loci associated with EM. Among the earliest and most widely recognized loci are rs7521902 at 1p36.12 (WNT4), rs13391619 at 2p25.1 (GREB1), and rs10859871 at 2q22 (VEZT).38 Building on these discoveries, Sapkota et al identified five additional loci significantly associated with EM, located at 2q35 (FN1), 6q25.1 (CCDC170, ESR1, SYNE1), 7p12.3, and 11p14.1 (FSHB).2 In the present study, we identified 11p14.1 and 8p21.2 as shared hotspot regions across multiple trait–disease combinations, indicating a stable and consistent genetic background. However, the loci identified in this study are not all entirely new reproductive signals. Among them, the 11p14.1 region contains multiple functional genes closely associated with menstrual traits, including FSHB, ARL14EP, and MPPED2. Earlier investigations have demonstrated that association signals within this region extend from the upstream of FSHB to ARL14EP within a high linkage disequilibrium block, suggesting that this region may function as an integrated genetic unit with a regulatory role in EM pathogenesis.2 This locus also influences menstrual characteristics: the FSHB promoter T allele reduces FSH expression, prolongs menstrual cycles, delays menopause, and lowers EM risk, whereas the G allele increases FSH levels, shortens cycles, increases menstrual frequency, and elevates EM risk.14 In addition to FSHB, ARL14EP and MPPED2 may provide biological context for the 11p14.1 signal. ARL14EP is involved in MHC class II vesicular trafficking and actin-cytoskeleton regulation, potentially linking this region to EM-related immune inflammation or tissue remodeling,39 whereas MPPED2 may participate in cellular differentiation and proliferation.40 Thus, although FSHB remains the most plausible effector gene, ARL14EP and MPPED2 may represent neighboring candidate genes reflecting the broader LD and regulatory architecture of 11p14.1. Another noteworthy locus, 8p21.2, harbors rs6185, a functional nonsynonymous variant within the coding region of GNRH1. Large-scale GWAS have consistently linked this variant to menstrual phenotypes, particularly shorter menstrual cycles.41,42 Additionally, the AMH region near 11q22.1 (rs10407022) is associated with ovarian follicle reserve and menstrual rhythm, while rs6933669 at 6q25.1 shows a strong association with age at menarche and regulates the expression of ESR1, RMND1, and CCDC170.43 The novelty of these loci lies in our integrated cross-subtype, menstrual/hormone-related trait, pleiotropy, colocalization, and functional annotation analyses, which further support their role as shared genetic hotspots linking menstrual-cycle regulation with EM susceptibility.
This study identified 136 pleiotropic genes, among which ARL14EP, C6orf211, EEFSEC, FSHB, and RMND1 appeared across six trait–disease combinations, making them the most frequently shared pleiotropic genes. Within the FSHB locus associated with age at menarche, several SNPs have been reported to regulate ARL14EP expression, suggesting that ARL14EP may participate in the genetic regulation of menarche timing through hormone-related pathways.44 Its expression may be influenced by genetic variation in the FSHB region and may act as a downstream effector relevant to EM biology. Previous expression-based studies reported an association between ARL14EP expression and EM risk.45,46 Genetic variants in the intronic regions of EEFSEC show significant associations with EM, menorrhagia, and irregular menstrual bleeding. These findings suggest that EEFSEC may influence the onset and progression of female reproductive disorders by regulating cell proliferation, DNA repair, and hormone-dependent tissue growth.37 The ESR1 gene encodes estrogen receptor ERα, a key regulator of estrogen signaling involved in cell proliferation, inflammation, and migration.47 Together with neighboring genes CCDC170, RMND1, and ZBTB2, ESR1 forms a regulatory network that may contribute to the pathogenesis of EM.48 CCDC170 has been implicated in cytoskeletal remodeling, cell migration, and embryo implantation, processes relevant to ectopic endometrial invasion,49 whereas RMND1 may influence mitochondrial function and hormone-dependent development of ovarian and endometrial tissues.50 The epigenetic architecture of this region further links it to menstrual physiology, as dynamically methylated CpG sites in the CCDC170 promoter vary across the menstrual cycle and may coordinately regulate ESR1, RMND1, and nearby genes, thereby affecting menarche timing and menstrual characteristics.49 Our study extends the previously reported ESR1/CCDC170/RMND1 regulatory network by identifying it as a shared pleiotropic genetic link between menstrual-cycle-related traits and EM susceptibility. Notably, C6orf211 lies immediately upstream of ESR1, and its expression in human endometrium is also positively correlated with ESR1, CCDC170, and RMND1.48
This study identified significant enrichment of candidate genes within several functional modules related to the ovarian development–ovulation–hormonal signaling axis. These results suggest that these genes may serve central roles in the shared genetic architecture of EM and menstrual-cycle-related traits by coordinating reproductive development and endocrine regulation. At the cellular component level, enrichment analysis revealed significant enrichment of the INO80 chromatin remodeling complex. This complex maintains the accessibility of estrogen response elements through ATP-dependent nucleosome sliding. Its dysfunction may disrupt cyclic gene expression and impair endometrial receptivity.51 INO80-mediated chromatin opening can also facilitate RNA polymerase II binding and transcription initiation, thereby enhancing inflammatory and proliferative signaling and potentially contributing to the sustained growth and pathological progression of ectopic lesions.52 However, because INO80 enrichment was inferred solely from gene set analysis without direct assessment of chromatin accessibility, estrogen response element accessibility, or INO80 complex activity, this finding should be considered hypothesis-generating. PPI network analysis further revealed that CALCRL, FSHR, KISS1, and TACR3 are part of the G protein-coupled receptor (GPCR) signaling pathway. Within this pathway, Gαs-mediated cAMP/PKA signaling cascades can suppress NF-κB activity, thereby modulating apoptosis and inflammatory responses and promoting immune evasion of ectopic endometrial cells.53 Furthermore, HOXA10 is essential for female reproductive tract development and has been strongly implicated in EM pathogenesis.54 Testosterone has been reported to regulate HOXA10 expression, while lower prenatal and postnatal testosterone levels have been associated with earlier menarche, shorter menstrual cycles, and endometrial thickening.55 The 11p14.1 colocalization signal indirectly supports a potential link between menstrual traits and EM susceptibility, but the testosterone-HOXA10-EM axis remains hypothetical and requires functional validation. Our colocalization analysis further showed substantial overlap between FIM- and LMC-associated genetic signals and testosterone-related traits, suggesting that androgen signaling may contribute to the development of specific EM subtypes and their shared genetic mechanisms with menstrual traits.
This study has several limitations. First, the GWAS summary statistics used in this study were primarily derived from individuals of European ancestry. Because individual-level data were unavailable, we could not further account for residual population stratification. Second, LDSC- and HDL-derived genetic correlations indicate shared genetic architecture but cannot establish causality or distinguish shared hormonal effects from subtype-specific mechanisms or residual confounding. Third, heterogeneity in EM and subtype definitions across GWAS datasets, including surgical, clinical, and self-reported diagnoses, may introduce misclassification, while overlapping subtype categories and limited case numbers for certain subtypes may reduce statistical power and compromise the robustness of subtype-specific findings. Fourth, we could not completely exclude the possibility of potential sample overlap between the UKB/ReproGen and FinnGen datasets. Furthermore, winner’s curse and the lack of an independent replication cohort may affect the robustness and generalizability of our findings. Finally, functional validation remains limited. Due to the lack of matched genotype, transcriptomic, proteomic, and disease-relevant functional experimental data, we were unable to perform allele-specific expression analysis or experimental validation. Future studies incorporating multi-ethnic cohorts, larger sample sizes, tissue-specific experimental data, and independent GWAS datasets with non-overlapping samples are warranted to further validate and extend our findings.
Conclusions
By integrating large-scale GWAS data with multiple genetic analytical approaches, this study suggests a shared yet heterogeneous genetic basis between EM and menstrual-cycle-related traits. We identified core genetic hotspots, including 11p14.1 and 8p21.2, and prioritized key pleiotropic genes. The results further showed that testosterone-related traits exhibited more stable colocalization signals, suggesting that androgen-related genetic regulation may exert differential effects across lesion types and provide clues for exploring subtype-specific therapeutic strategies. Overall, this study provides important insights into the genetic mechanisms of endometriosis and subtype-based risk stratification, and offers genetic evidence for future exploration of polygenic risk score-based risk assessment approaches.
Data Sharing Statement
The datasets analyzed during this study are publicly available. GWAS summary statistics for endometriosis and its subtypes were obtained from the FinnGen Project (Release 12; https://www.finngen.fi/en/access_results). Summary statistics for menstrual-cycle-related traits, including age at menarche, frequent and irregular menstruation, and length of menstrual cycle, were derived from publicly available GWAS conducted by the ReproGen Consortium and the UK Biobank. GWAS data for sex hormone–related traits, including total testosterone, bioavailable testosterone, sex hormone–binding globulin, and oestradiol, were obtained from the IEU OpenGWAS database (https://gwas.mrcieu.ac.uk/). Summary statistics for progesterone levels were retrieved from the GWAS Catalog (https://www.ebi.ac.uk/gwas/).
Code Availability
The corresponding software resources are available from the following repositories or official websites: LDSC (https://github.com/bulik/ldsc), HDL (https://github.com/zhenin/HDL), PLACO (https://github.com/RayDebashree/PLACO), MAGMA (https://cncr.nl/research/magma/), COLOC (https://github.com/chr1swallace/coloc), HyPrColoc (https://github.com/jrs95/hyprcoloc), and FUMA (https://fuma.ctglab.nl/; source code: https://github.com/vufuma/FUMA-webapp). Custom scripts used for GWAS summary statistics preprocessing, harmonization, quality control, format conversion, and result organization are available from the corresponding author upon reasonable request.
Ethical Approval
All data used in this study were legally obtained from publicly available datasets and were analyzed using anonymized GWAS summary statistics only. No identifiable individual-level data were accessed, and no new human participants were recruited or involved. Therefore, this study is exempt from ethical approval based on items 1 and 2 of Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects.
Acknowledgments
Our thanks should go to all participants and investigators of studies or consortiums included in this work for sharing GWAS data.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Adilbayeva A, Kunz J. Pathogenesis of Endometriosis and Endometriosis-Associated Cancers. Int J Mol Sci. 2024;25(14). doi:10.3390/ijms25147624
2. Sapkota Y, Steinthorsdottir V, Morris AP, et al. Meta-analysis identifies five novel loci associated with endometriosis highlighting key genes involved in hormone metabolism. Nat Commun. 2017;8:15539. doi:10.1038/ncomms15539
3. Treloar SA, O’Connor DT, O’Connor VM, Martin NG. Genetic influences on endometriosis in an Australian twin sample. [email protected]. Fertil Steril. 1999;71(4):701–16. doi:10.1016/s0015-0282(98)00540-8
4. Saha R, Pettersson HJ, Svedberg P, et al. Heritability of endometriosis. Fertil Steril. 2015;104(4):947–952. doi:10.1016/j.fertnstert.2015.06.035
5. Lamceva J, Uljanovs R, Strumfa I. The Main Theories on the Pathogenesis of Endometriosis. Int J Mol Sci. 2023;24(5). doi:10.3390/ijms24054254
6. Bulun SE, Cheng YH, Yin P, et al. Progesterone resistance in endometriosis: link to failure to metabolize estradiol. Mol Cell Endocrinol. 2006;248(1–2):94–103. doi:10.1016/j.mce.2005.11.041
7. Abramiuk M, Grywalska E, Malkowska P, et al. The Role of the Immune System in the Development of Endometriosis. Cells. 2022;11(13). doi:10.3390/cells11132028
8. Borghese B, Tost J, de Surville M, et al. Identification of susceptibility genes for peritoneal, ovarian, and deep infiltrating endometriosis using a pooled sample-based genome-wide association study. Biomed Res Int. 2015;2015:461024. doi:10.1155/2015/461024
9. Rahmioglu N, Mortlock S, Ghiasi M, et al. The genetic basis of endometriosis and comorbidity with other pain and inflammatory conditions. Nat Genet. 2023;55(3):423–436. doi:10.1038/s41588-023-01323-z
10. Koller D, He J, Lokhammer S, et al. Multi-ancestry genome-wide association and integrated multi-omics analyses of endometriosis and its clinical manifestations. Nat Genet. 2026;58(5):1051–1061. doi:10.1038/s41588-026-02582-2
11. Nnoaham KE, Webster P, Kumbang J, Kennedy SH, Zondervan KT. Is early age at menarche a risk factor for endometriosis? A systematic review and meta-analysis of case-control studies. Fertil Steril. 2012;98(3):702–712e706. doi:10.1016/j.fertnstert.2012.05.035
12. Wei M, Cheng Y, Bu H, Zhao Y, Zhao W. Length of Menstrual Cycle and Risk of Endometriosis: a Meta-Analysis of 11 Case-Control Studies. Medicine (Baltimore). 2016;95(9):e2922. doi:10.1097/MD.0000000000002922
13. Garitazelaia A, Rueda-Martinez A, Arauzo R, et al. A Systematic Two-Sample Mendelian Randomization Analysis Identifies Shared Genetic Origin of Endometriosis and Associated Phenotypes. Life (Basel). 2021;11(1). doi:10.3390/life11010024
14. Ruth KS, Beaumont RN, Tyrrell J, et al. Genetic evidence that lower circulating FSH levels lengthen menstrual cycle, increase age at menopause and impact female reproductive health. Hum Reprod. 2016;31(2):473–481. doi:10.1093/humrep/dev318
15. McGrath IM, Montgomery GW, Mortlock S. Insights from Mendelian randomization and genetic correlation analyses into the relationship between endometriosis and its comorbidities. Hum Reprod Update. 2023;29(5):655–674. doi:10.1093/humupd/dmad009
16. Day FR, Thompson DJ, Helgason H, et al. Genomic analyses identify hundreds of variants associated with age at menarche and support a role for puberty timing in cancer risk. Nat Genet. 2017;49(6):834–841. doi:10.1038/ng.3841
17. Verdiesen RMG, van der Schouw YT, van Gils CH, et al. Genome-wide association study meta-analysis identifies three novel loci for circulating anti-Mullerian hormone levels in women. Hum Reprod. 2022;37(5):1069–1082. doi:10.1093/humrep/deac028
18. Venkatesh SS, Wittemans LBL, Palmer DS, et al. Genome-wide analyses identify 25 infertility loci and relationships with reproductive traits across the allele frequency spectrum. Nat Genet. 2025;57(5):1107–1118. doi:10.1038/s41588-025-02156-8
19. Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. doi:10.1038/ng.3211
20. Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. doi:10.1038/ng.3406
21. Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
22. Ning Z, Pawitan Y, Shen X. High-definition likelihood inference of genetic correlations across human complex traits. Nat Genet. 2020;52(8):859–864. doi:10.1038/s41588-020-0653-y
23. Ray D, Chatterjee N. A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between Type 2 Diabetes and Prostate Cancer. PLoS Genet. 2020;16(12):e1009218. doi:10.1371/journal.pgen.1009218
24. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. doi:10.1038/s41467-017-01261-5
25. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
26. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11(4):e1004219. doi:10.1371/journal.pcbi.1004219
27. Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10(1):1523. doi:10.1038/s41467-019-09234-6
28. Foley CN, Staley JR, Breen PG, et al. A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat Commun. 2021;12(1):764. doi:10.1038/s41467-020-20885-8
29. Doostparast Torshizi A, Truong DT, Hou L, et al. Proteogenomic network analysis reveals dysregulated mechanisms and potential mediators in Parkinson’s disease. Nat Commun. 2024;15(1):6430. doi:10.1038/s41467-024-50718-x
30. Lu MY, Niu JL, Liu B. The risk of endometriosis by early menarche is recently increased: a meta-analysis of literature published from 2000 to 2020. Arch Gynecol Obstet. 2023;307(1):59–69. doi:10.1007/s00404-022-06541-0
31. Maruo T, Matsuo H, Shimomura Y, et al. Effects of progesterone on growth factor expression in human uterine leiomyoma. Steroids. 2003;68(10–13):817–824. doi:10.1016/j.steroids.2003.08.017
32. Kuan KKW, Gibson DA, Whitaker LHR, Horne AW. Menstruation Dysregulation and Endometriosis Development. Front Reprod Health. 2021;3:756704. doi:10.3389/frph.2021.756704
33. Tan Z, Gong X, Li Y, et al. Impacts of endometrioma on ovarian aging from basic science to clinical management. Front Endocrinol. 2022;13:1073261. doi:10.3389/fendo.2022.1073261
34. Chapron C, Lang JH, Leng JH, et al. Factors and Regional Differences Associated with Endometriosis: a Multi-Country, Case-Control Study. Adv Ther. 2016;33(8):1385–1407. doi:10.1007/s12325-016-0366-x
35. Imperiale L, Nisolle M, Noel JC, Fastrez M. Three Types of Endometriosis: pathogenesis, Diagnosis and Treatment. State of the Art. J Clin Med. 2023;12(3). doi:10.3390/jcm12030994
36. Jiang L, Zhang M, Wang S, Han Y, Fang X. Common and specific gene signatures among three different endometriosis subtypes. PeerJ. 2020;8:e8730. doi:10.7717/peerj.8730
37. Pathare ADS, Dzigurski J, Pujol-Gualdo N, et al. A large-scale genome-wide association study on female genital tract polyps highlights role of DNA repair, cell proliferation, and cell growth. Hum Reprod. 2025;40(4):750–763. doi:10.1093/humrep/deaf025
38. Painter JN, Anderson CA, Nyholt DR, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43(1):51–54. doi:10.1038/ng.731
39. Paul P, van den Hoorn T, Jongsma ML, et al. A Genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell. 2011;145(2):268–283. doi:10.1016/j.cell.2011.03.023
40. Liguori L, Andolfo I, de Antonellis P, et al. The metallophosphodiesterase Mpped2 impairs tumorigenesis in neuroblastoma. Cell Cycle. 2012;11(3):569–581. doi:10.4161/cc.11.3.19063
41. Mbarek H, Gordon SD, Duffy DL, et al. Genome-wide association study meta-analysis of dizygotic twinning illuminates genetic regulation of female fecundity. Hum Reprod. 2024;39(1):240–257. doi:10.1093/humrep/dead247
42. Laisk T, Kukuskina V, Palmer D, et al. Large-scale meta-analysis highlights the hypothalamic-pituitary-gonadal axis in the genetic regulation of menstrual cycle length. Hum Mol Genet. 2018;27(24):4323–4332. doi:10.1093/hmg/ddy317
43. Dunning AM, Michailidou K, Kuchenbaecker KB, et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat Genet. 2016;48(4):374–386. doi:10.1038/ng.3521
44. Ponomarenko I, Reshetnikov E, Polonikov A, et al. Candidate Genes for Age at Menarche Are Associated With Uterine Leiomyoma. Front Genet. 2020;11:512940. doi:10.3389/fgene.2020.512940
45. Gallagher CS, Makinen N, Harris HR, et al. Genome-wide association and epidemiological analyses reveal common genetic origins between uterine leiomyomata and endometriosis. Nat Commun. 2019;10(1):4857. doi:10.1038/s41467-019-12536-4
46. Koller D, He J, Lokhammer S, et al. Multi-ancestry genome-wide association study of endometriosis and its clinical manifestations in ~1.4 million women: translating gene discovery into pathogenic mechanisms and therapeutic targets. medRxiv. 2025. doi:10.1101/2025.09.03.25335012
47. Tang ZR, Zhang R, Lian ZX, Deng SL, Yu K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells. 2019;8(10). doi:10.3390/cells8101123
48. Marla S, Mortlock S, Houshdaran S, et al. Genetic risk factors for endometriosis near estrogen receptor 1 and coexpression of genes in this region in endometrium. Mol Hum Reprod. 2021;27(1). doi:10.1093/molehr/gaaa082
49. Mortlock S, Houshdaran S, Kosti I, et al. Global endometrial DNA methylation analysis reveals insights into mQTL regulation and associated endometriosis disease risk and endometrial function. Commun Biol. 2023;6(1):780. doi:10.1038/s42003-023-05070-z
50. Federici S, Rossetti R, Moleri S, et al. Primary ovarian insufficiency: update on clinical and genetic findings. Front Endocrinol. 2024;15:1464803. doi:10.3389/fendo.2024.1464803
51. Retis-Resendiz AM, Gonzalez-Garcia IN, Leon-Juarez M, et al. The role of epigenetic mechanisms in the regulation of gene expression in the cyclical endometrium. Clin Clin Epigenet. 2021;13(1):116. doi:10.1186/s13148-021-01103-8
52. Talbi S, Hamilton AE, Vo KC, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006;147(3):1097–1121. doi:10.1210/en.2005-1076
53. He SZ, Li J, Bao HC, et al. G protein‑coupled estrogen receptor/miR‑148a/human leukocyte antigen‑G signaling pathway mediates cell apoptosis of ovarian endometriosis. Mol Med Rep. 2018;18(1):1141–1148. doi:10.3892/mmr.2018.9039
54. Zanatta A, Rocha AM, Carvalho FM, et al. The role of the Hoxa10/HOXA10 gene in the etiology of endometriosis and its related infertility: a review. J Assist Reprod Genet. 2010;27(12):701–710. doi:10.1007/s10815-010-9471-y
55. Dinsdale NL, Crespi BJ. Endometriosis and polycystic ovary syndrome are diametric disorders. Evol Appl. 2021;14(7):1693–1715. doi:10.1111/eva.13244
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.