Back to Journals » Clinical Ophthalmology » Volume 9
An evaluation of Retaine™ ophthalmic emulsion in the management of tear film stability and ocular surface staining in patients diagnosed with dry eye
Authors Ousler G, Devries D ![]() , Karpecki P, Ciolino JB
, Karpecki P, Ciolino JB
Received 2 October 2014
Accepted for publication 26 November 2014
Published 5 February 2015 Volume 2015:9 Pages 235—243
DOI https://doi.org/10.2147/OPTH.S75297
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
George Ousler III,1 Douglas K Devries,2 Paul M Karpecki,3 Joseph B Ciolino4
1Ora, Inc, Andover, MA, USA; 2Eye Care Associates of Nevada, Sparks, NV, USA; 3Koffler Vision Group, Lexington, KY, USA; 4Massachusetts Eye and Ear, Boston, MA, USA
Abstract: A single-center, open-label study consisting of two visits over the course of approximately 2 weeks was conducted to evaluate the efficacy of Retaine™ ophthalmic emulsion in improving the signs and symptoms of dry eye. Forty-two subjects were enrolled and received 1–2 drops twice daily of Retaine™ beginning at the first visit (day 1) and ending at the second visit. Subjects were instructed to complete a symptomatology diary twice daily prior to drop instillation through the morning of the second visit. Ocular sign and symptom assessments, visual acuity procedures, and comfort assessments were conducted during both visits. A statistically significant reduction was observed in mean breakup area on the second visit between the predose time and the postdose time (P=0.026). On the second visit, subjects had significantly less corneal fluorescein staining in the superior (P=0.002), central (P=0.017), corneal sum (P=0.011), and all ocular regions combined (P=0.038) than on the first visit. On the second visit, statistically significant reductions in dryness (P<0.001), grittiness (P=0.0217), ocular discomfort (P=0.0017), and all symptoms (P<0.001) were also seen as measured by the Ora Calibra™ Ocular Discomfort and 4-Symptom Questionnaire (0–5 scale). Subjects reported a statistically significant improvement in their abilities to work with a computer at night (P=0.044). Mean drop comfort scores ranged from 1.29–1.81 on the Ora Calibra™ 0–10 Drop Comfort Scale, on which 0 is very comfortable and 10 is very uncomfortable. Retaine™ demonstrates promising results as a novel artificial tear option for individuals suffering from dry eye. The unique mechanism of action of Retaine™ provides enhanced comfort and improves the quality of life of dry eye subjects while reducing the ocular signs of dry eye.
Keywords: artificial tears, dry eye syndrome, quality of life, cationic, lipid emulsion, mean breakup area
Introduction
Dry eye is a disorder of the tear film and ocular surface. This disease causes discomfort, decreased visual function, and instability of the tear film. It has been estimated that about 4.91 million Americans (3.23 million women and 1.68 million men) who are 50 years and older suffer from a moderate to severe form of dry eye.1,2 This does not include the population of individuals who have either less severe symptoms or more episodic manifestations of the disease and only note their symptoms during contact with adverse contributing factors, such as low humidity, extended visual tasking, or contact lens wear. Affecting one in three patients who seek treatment from an eye care practitioner, dry eye is one of the most common ophthalmic diseases and can drastically impact the quality of life (QoL) of those plagued by the disease.3
Currently, artificial tears remain the first line of treatment for patients with dry eye disease.3,4 Lipid emulsions are considered to be the latest generation of artificial tears and offer dry eye patients a novel option in an effort to combat their dry eye signs and symptoms.5 Emulsions provide a high encapsulation rate and enhanced ocular penetration.5 Cationic oil-in-water emulsions offer an additional advantage in that an electrostatic interaction occurs with the negatively charged cells of the ocular surface and thus improves the residence time of the formulation on the ocular surface.5 Cationic oil-in-water emulsions extend the benefits of negatively charged oil-in-water emulsions and offer significant advantages over the existing marketed anionic emulsions.
Retaine™ ophthalmic emulsion (marketed outside the United States as Cationorm®, Santen, Osaka, Japan) is a cationic emulsion that contains mineral oil and targets all three layers of the tear film. Retaine™ contains Novasorb®, a proprietary cationic oil-in-water nanoemulsion technology with novel bioadhesive properties. The principle of the Novasorb® technology is based on electrostatic interactions between the positively charged oil nanodroplets and the negatively charged ocular surface epithelium. This electrostatic attraction aims to increase the residence time on the ocular surface and enhance ocular drug bioavailability6–8 in order to enhance protection and restoration of a healthy tear film and corneal epithelium. Electrostatic forces allow the emulsion to spread evenly over the ocular surface,7 and the nanoscale size of the oil droplets contributes to the stability of the emulsion and to ocular absorption. Tear hyperosmolarity, a key feature in dry eye, is improved by the hypo-osmotic properties of the drop. Collectively, these properties provide long-lasting and effective relief for individuals with dry eye.
The purpose of the current study was to evaluate RetaineTM after 2 weeks of treatment for the management of ocular surface staining, visual function, and tear film stability in patients diagnosed with dry eye disease.
Methods
This was a single-center, open-label study evaluating the efficacy of bilateral RetaineTM administration (1–2 drops twice daily [BID] for 14 days) in the management of ocular surface staining, visual function, and tear film stability in patients diagnosed with dry eye. The study comprised two visits over the course of approximately 2 weeks and was conducted at Andover Eye, in Andover, Massachusetts. The study was performed according to a protocol approved by an external independent review board (Alpha Independent Review Board, San Clemente, CA, USA), and in compliance with the ethical principles of Good Clinical Practices, the Declaration of Helsinki, the International Conference on Harmonization guidelines, and all applicable local, state, and federal requirements relevant to the use of investigational drugs. Written informed consent was obtained prior to initiation of any study procedures. The study is listed on http://clinicaltrials.gov as NCT02139033.
Forty-nine subjects were screened in order to enroll 42 subjects, a typical sample size for this type of clinical evaluation. No other statistical considerations were used to determined sample size, and enrolled subjects were to receive identical doses of RetaineTM. During the first visit (day 1 the study), all candidates underwent screening procedures to determine eligibility. Initial screening procedures included assessments of ocular discomfort, ocular symptoms, QoL, visual acuity (VA), and corrected VA (CVA) degradation between blinks. After this initial set of assessments, each subject underwent a review of qualification criteria before being able to continue with screening procedures. Eligible study subjects were at least 18 years of age and had a history of dry eye for at least 6 months, used eye drops for dry eye symptoms, or had the desire to use eye drops for dry eye symptoms.
For the assessment of signs and symptoms, various calibrated scales were used throughout the study by the investigators and the subjects. Investigators graded fluorescein staining and lissamine green staining in each eye with the Ora Calibra™ Corneal and Conjunctival Staining Scale9–12 (Ora Calibra, Andover, MA, USA), (0–4 scale with 0.5 increments) on which 0 is no staining and 4 is severe staining. This scale divides each eye into five staining areas: the inferior, superior, and central regions relative to the cornea and the temporal and nasal regions relative to the conjunctiva. Each area is graded separately; the sum of the three corneal regions generates a corneal sum score, and the addition of the scores for the nasal and temporal conjunctiva areas generates an all-region combined score. Investigators graded conjunctival redness in each eye by using the Ora Calibra™ Conjunctival Redness Scale,13 a 5-point scale (0–4 with 0.5 unit increments) on which 0 is none and 4 is severe. Investigators graded lid margin redness in each eye by using the Ora CalibraTM Lid Margin Redness Scale,12 a 4-point scale (0–3) on which 0 is none and 3 is severe. Subjects graded ocular discomfort by using the Ora Calibra™ Ocular Discomfort Scale,9,10 a 5-point scale (0–4) on which 0 is no discomfort and 4 is constant discomfort. Subjects also graded the severity of their dry eye symptoms by using the Ora Calibra™ Ocular Discomfort and 4-Symptom Questionnaire,9,10,12 a 6-point scale (0–5) on which 0 is no pain and 5 is the worst and in which five ocular symptoms are individually graded: overall discomfort, burning, dryness, grittiness, and stinging. Subjects scored QoL parameters by using a 5-point, Ora CalibraTM Dry Eye Quality of Life (QoL) Questionnaire14 (0–4) on which 0 is minimal, 3 is severe, and 4 is not applicable; they scored “how troubled” they had been by ocular discomfort in the previous week for the following categories: 1) eyesight issues; 2) reading at night; 3) watching television at night; 4) driving at night; and 5) working with a computer at night. Subjects graded drop comfort with two grading systems: 1) an 11-point Ora Calibra™ Drop Comfort Scale (0–10) on which 0 is very comfortable and 10 is very uncomfortable, and 2) a three-word best descriptor choice that best defines eye comfort after drop use.
At the time of the first visit, eligible subjects were required to have moderate to severe signs and symptoms to be considered for the study. Subjects were required to have sufficient symptom scores on at least one of the five symptoms of the Ora Calibra™ Ocular Discomfort and 4-Symptom Questionnaire. Subjects also were required to have sufficient dry eye signs as determined by average tear film breakup time (TFBUT), total corneal fluorescein staining, inferior corneal fluorescein staining, and total lissamine green conjunctival staining. Subjects had to meet all dry eye sign criteria in the same eye. Subjects were excluded from the study if they met any of the following conditions: 1) had a corrected VA ≥ logMAR+0.7 as assessed according to the Early Treatment of Diabetic Retinopathy Study (ETDRS) scale in both eyes; 2) had any clinically significant slit lamp biomicroscopy (SLE) findings that required therapeutic intervention and/or may have interfered with study parameters; 3) had any ongoing ocular infection or active ocular inflammation; 4) had any uncontrolled systemic disease; 5) had a history of laser-assisted in situ keratomileusis (LASIK) surgery within 12 months prior to the first visit; or 6) had used Restasis® within 30 days prior to the first visit. Subjects were required to avoid ophthalmic medications, including artificial tear substitutes, for 2 hours prior to the first study visit, and to discontinue use of any topical ophthalmic prescription or over-the-counter solutions, artificial tears, gels, or scrubs for the duration of the trial (except for medications allowed for the conduct of the study).
After these evaluations, candidates underwent a second review of the qualification criteria wherein eligible candidates who satisfied all eligibility requirements were enrolled in the study. For enrolled subjects, efficacy and safety measurements obtained during the first visit were considered baseline values, and adverse events (AE) were assessed.
After enrollment, subjects received one dose (one to two drops) of study drug administered bilaterally by a trained study technician. After this first bilateral dose, subjects assessed drop comfort. Subjects were dispensed a subject diary and a sufficient quantity of study drug for the duration of the study. During the evening of the first day, after returning home from the first visit, subjects self-administered a single dose bilaterally prior to bed. Throughout the remainder of the study, which lasted up to and including the morning of the second visit, subjects were to administer bilateral doses BID, once in the morning and once in the evening before bed. However, on the morning of the second visit, drug instillation was not to occur within 2 hours prior to the study visit. Subjects were instructed to score their symptoms in the diary prior to each self-administered instillation of study drug; subjects completed diary entries twice a day, morning and evening, except on days of the first visit and the second visit; on those days subjects completed only one diary entry.
The second visit occurred on days 13–17 of the study at different times for different patients. On this visit, prior to in-office study drug instillation, subjects underwent measurements of ocular discomfort, ocular symptoms, QoL, VA, CVA degradation between blinks, conjunctival redness, lid margin redness, interblink interval (IBI), tear film stability, TFBUT, fluorescein staining, and lissamine green staining, as well as SLE and an AE query. Subjects then received one dose of study drug bilaterally administered by a trained study technician. After instillation, subjects graded drop comfort. CVA degradation between blinks and tear film stability were assessed, and AE information was collected. Study drug vials and diaries were collected from subjects.
The primary efficacy measures were TFBUT and corneal fluorescein staining. Evaluations of TFBUT and corneal fluorescein staining were performed before the dose given on the first visit and before the dose given on the second visit. The secondary efficacy measures were ocular discomfort, ocular symptoms, QoL, conjunctival redness, lid margin redness, IBI, tear film stability, CVA degradation between blinks, conjunctival fluorescein staining scores, lissamine green staining scores, and drop comfort.
Tear film stability and IBI, both assessments of drying on the ocular surface, were measured by the Ocular Protection Index (OPI) 2.0 System, an automated measure of ocular surface protection under normal blink pattern and normal visual conditions.15,16 Video capture and analysis using the OPI 2.0 System allows for a calculation of mean corneal surface exposure, otherwise known as mean breakup area (MBA). Final endpoints were percent change in MBA and IBI in seconds. The OPI 2.0 assessment was performed in each eye before the dose given on the first visit, before the dose given on the second visit, and after the dose given on the second visit. Decay in CVA was measured by the Interblink Interval Visual Acuity Decay (IVAD) test. IVAD is a diagnostic tool that evaluates functional VA between blinks. This computer-based task involves identification of Landolt ‘c’s at individualized best-corrected VA between blinks.17 The IVAD test was performed in each eye before the dose given on the first visit, before the dose given on the second visit, and after the dose given on the second visit.
Both conjunctival fluorescein and lissamine green staining grading were performed before the dose given on the first visit and again before the dose given on the second visit. Subjects graded drop comfort immediately after the dose, 1 minute after the dose, and 2 minutes after the dose. Subjects also assessed drop comfort 3 minutes after the dose given on the first visit and again 3 minutes after the dose given on the second visit by selecting three descriptive words for eye comfort in both eyes. Before the dose given on the first visit and again before the dose given on second visit, subjects scored QoL parameters. Before the dose given on the first visit and again before the dose given on second visit, investigators graded conjunctival and lid margin redness. Ocular discomfort was graded by the subjects before the dose given on the first visit and again before the dose given on second visit. As previously mentioned, subjects also graded their symptoms and recorded their scores for each ocular symptom in their diary before taking the dose on the evening of the first visit (on day 1 of the study), prior to study drug instillation every morning and evening during the at-home BID dosing period, and before the dose taken on the morning of the second visit.
The analysis for all primary and secondary efficacy measures was based on changes from baseline values. The safety measures were VA as assessed by the ETDRS scale, SLE, and AEs (elicited and observed).
Statistical methods
Analyses were conducted on the intent-to-treat (ITT) population, which included all enrolled subjects. Primary and secondary efficacy analyses were performed on the ITT population.
Primary efficacy analyses
The primary analyses of the change from baseline in TFBUT and corneal fluorescein staining were analyzed by paired t-tests with significance defined as P≤0.05. Descriptive statistics (number, mean, and standard deviation) were summarized for each parameter by study visit.
Secondary efficacy analyses
The secondary analyses of the change from baseline in ocular discomfort, ocular symptoms, QoL, conjunctival redness, lid margin redness, IBI, tear film stability, decay in CVA between blinks, conjunctival fluorescein staining, and lissamine green staining were analyzed by paired t-tests with significance defined as P≤0.05.
Descriptive statistics (number, mean, and standard deviation) were summarized by study visit. For categorical variables, counts and percentages were used to summarize the data. Missing data were not replaced.
Results
A total of 42 subjects were enrolled in this study with no withdrawals. No subjects were discontinued or dropped out. Therefore, the ITT population comprised 42 subjects and the per protocol and safety populations were identical to the ITT population. According to the inclusion/exclusion criteria, subjects were allowed to enroll if one eye satisfied the inclusion/exclusion criteria. Consequently, among the ITT population (number[n]=42), not all eyes satisfied all inclusion/exclusion criteria. Therefore, analyses of parameters evaluated at the eye level were performed only on “evaluable eyes”, which were eyes in the ITT population that satisfied all inclusion/exclusion criteria. A total of 75 eyes were evaluable in the ITT population (n=42). In the ITT population, 13 subjects were male and 29 subjects were female; the mean subject age was 61.6 years (range 26–89 years).
Primary efficacy endpoints
TFBUT
TFBUT was measured in each eye before the dose given during the first visit and again before the dose given during the second visit. Lower TFBUT values indicated a shorter length of corneal protection by the tear-film and hence a greater severity of dry eye. Higher TFBUT values indicated increased duration of tear-film integrity and hence longer time periods of corneal protection by the tear-film. The mean TFBUT ± standard deviation in evaluable eyes of the ITT population was 1.45±0.57 seconds on the first visit and 1.40±0.50 seconds on the second visit; the difference was not significant.
Corneal fluorescein staining
Corneal fluorescein staining evaluations were performed in each eye before the dose given on the first visit and again before the dose given on the second visit. Lower scores indicated less staining and less severe dry eye. On the second visit, corneal fluorescein staining was significantly lower in evaluable eyes of the ITT population in superior, central, corneal sum, and all ocular regions combined than on the first visit. In the superior area, the mean score for fluorescein staining decreased to 1.93 from 2.2 (P=0.002). In the central area, the mean score decreased to 0.95 from 1.25 (P=0.017) (Figure 1). For the corneal sum, the mean score decreased to 4.95 from 5.55 (P=0.011) (Figure 1). For all regions combined (ie, scores of both corneal and conjunctival fluorescein staining), the mean score decreased to 8.67 from 9.36 (P=0.038). Although the mean score for the nasal area was slightly higher for the second visit than for the first visit and the mean score for the temporal inferior region for the second visit was less than for the first visit, these changes were not statistically significant.
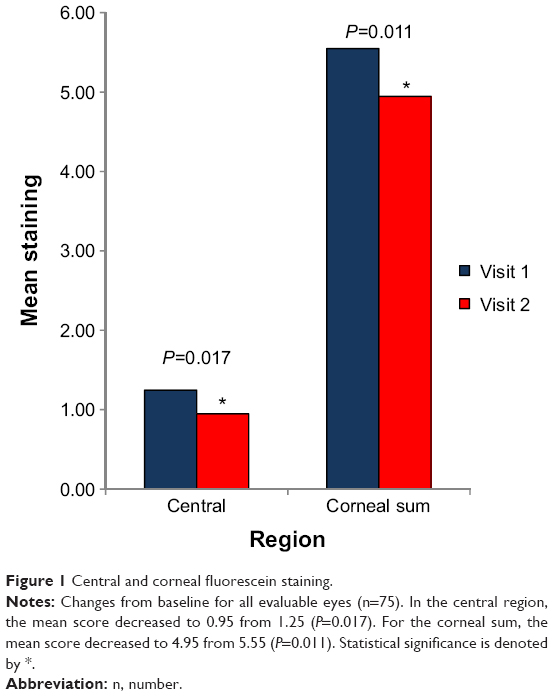 | Figure 1 Central and corneal fluorescein staining. |
Secondary efficacy endpoints
Secondary endpoints included ocular discomfort, ocular symptoms, QoL, conjunctival redness, lid margin redness, IBI, tear film stability, decay in CVA between blinks, conjunctival fluorescein staining, lissamine green staining, and drop comfort.
Ocular discomfort
On the second visit, ocular discomfort scores in evaluable eyes of the ITT population were significantly lower than on the first visit (1.91 versus 2.20; P<0.0284).
Ocular symptoms
On the second visit, the ITT population had significant reductions in three of the five ocular symptoms that were assessed on the first visit. Significant reductions were observed in discomfort (1.95 on the second visit versus 2.55 on the first visit; P=0.0017), dryness (2.02 on the second visit versus 2.88 on the first visit; P<0.001), and grittiness (1.02 at the second visit versus 1.40 at the first visit; P=0.0217). Although burning and stinging scores were lower on the second visit than they were on the first visit, these reductions were not significant. Nevertheless, the overall reduction of all ocular symptoms from baseline (the scores for all five symptoms combined) was significant (6.67 on the second visit versus 8.71 on the first visit; P<0.001). Figure 2 depicts the change from baseline for all five ocular symptoms separately and combined in the ITT population.
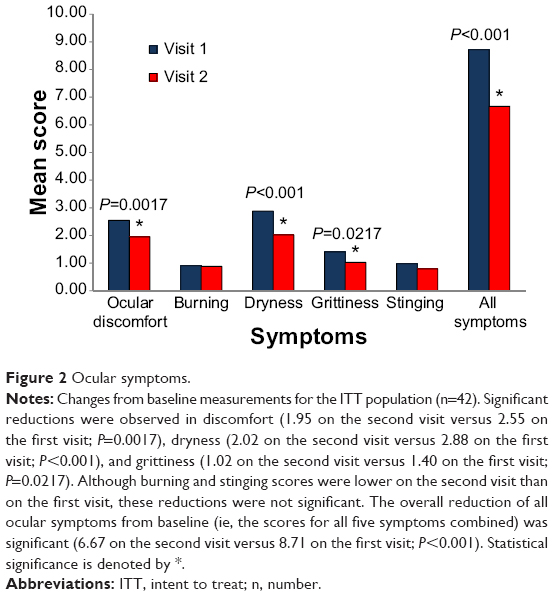 | Figure 2 Ocular symptoms. |
Dry eye QoL
On the second visit, the ITT subjects reported a significant improvement in the symptoms that occur when they work at the computer at night (1.38 on the second visit versus 1.67 on the first visit; P=0.044). Subjects also reported substantial improvement in reading at night; however, the score was not statistically significant (1.50 on the second visit versus 1.86 on the first visit; P=0.062). QoL scores also decreased for eyesight issues, watching TV at night, and driving at night, but these reductions were not significant.
Conjunctival and lid margin redness
On the second visit, conjunctival redness was significantly lower in the evaluable eyes of the ITT population than it had been on the first visit (1.54 versus 1.79; P<0.001). However, no significant reduction was observed in lid margin redness from the time of the first visit to the time of the second visit.
Tear film stability and IBI
After the dose given on the second visit, tear film stability had significantly improved in the evaluable eyes of the ITT population, as measured by percent change in MBA between the readings taken before and after the dose given on that visit (0.41% versus 0.25%; P=0.026), a reduction of approximately 40%. However, the percent change in MBA was minimal after 14 days of BID dosing (0.39% versus 0.42% respectively). Figure 3 depicts the percent change in MBA at the postdose time during the second visit from the predose time during the same visit. No significant change was observed in IBI between the predose assessment taken on the second visit and the predose assessment taken on the first visit, nor was there a change between the predose and the postdose results during the second visit.
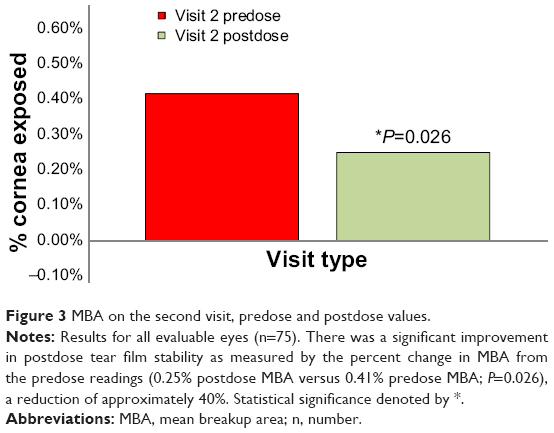 | Figure 3 MBA on the second visit, predose and postdose values. |
CVA degradation between blinks
The effects of dry eye on visual function were assessed by testing CVA degradation between blinks. The IVAD test was performed during the first visit before the dose was given, during the second visit before the dose was given, and during the second visit after the dose was given. The time for CVA was measured in seconds. Although the time at CVA (time to one-line loss of CVA) in the ITT population was 41% higher on the second visit than on the first visit, this improvement was not statistically significant (12.18 seconds versus 8.59 seconds; P=0.0697). Figure 4 depicts IVAD outcomes in evaluable subjects (n=32) of the ITT population.
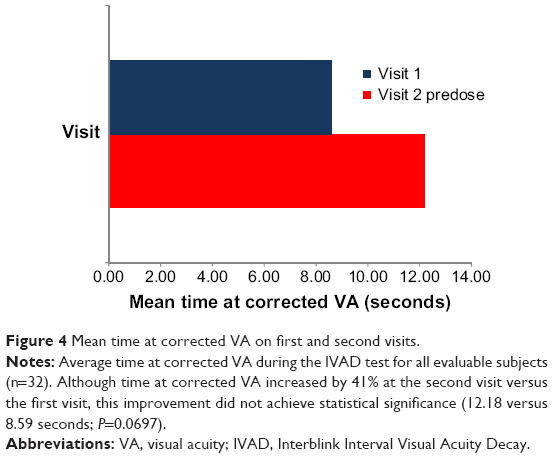 | Figure 4 Mean time at corrected VA on first and second visits. |
Drop comfort
Drop comfort scale
Mean drop comfort scores ranged from 1.29 to 1.81 across all the first visit and the second visit postdose time points for the ITT population. All drop comfort scores were consistently higher on the second visit than on the first visit; however, differences were not significant.
Drop comfort questionnaire
In the ITT population, the majority of the subjects responded to the drop comfort questionnaire with positive descriptor words during the first visit after the dose was given and again during the second visit after the dose was given. Subjects were asked to select three terms that best described the drops. The percentage of subjects that selected the positive descriptor words of cool, refreshing, smooth, soothing, and comfortable ranged from 55% to 69% (Figure 5). Smaller percentages of subjects selected the positive words thick (12%) and filmy (33%). The percentage of subjects that selected the negative descriptor words of sticky, burning, stinging, irritating, and gritty ranged from 14% to 29% (Figure 5). Only 2% of subjects selected the negative words of itchy and fuzzy. Among the ITT population, 45% of subjects selected only positive descriptor words and 2% selected only negative descriptor words.
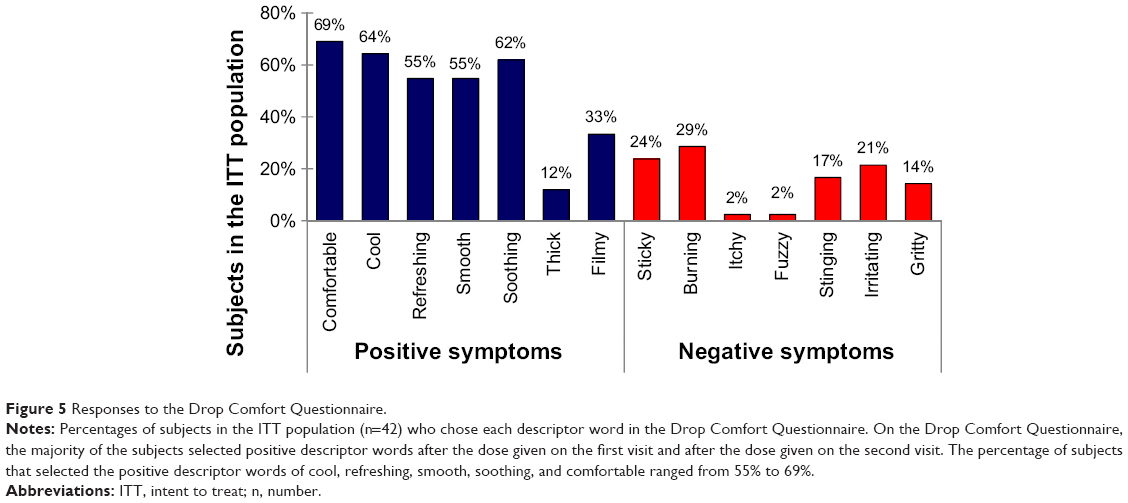 | Figure 5 Responses to the Drop Comfort Questionnaire. |
Safety measures
One AE, a decrease in VA, was reported during the study. The event was mild in severity, resolved the same day, and was deemed not to be related to the study drug. No subjects withdrew because of AEs. No serious AEs occurred during the study. No concerns were observed during any of the clinical examinations (SLE and ETDRS).
Discussion
Dry eye continues to be a challenging disease. The outcomes of this study suggest the Retaine™ has the potential to significantly reduce the symptoms of dry eye. It is extremely challenging to achieve statistically significant improvements in both signs and symptoms, as evidenced by the high number of failed clinical trials involving therapy for dry eye. This single-center, open-label study was designed to explore many potential efficacy endpoints relative to Retaine™. The finding of improvement in both a sign and symptom endpoint with Retaine™ indicates that it is an effective therapy for dry eye patients. Although no significant change from baseline was observed in TFBUT values, there were significant reductions in corneal fluorescein staining in the superior region, the central region, the sum of all three corneal regions, and all five ocular regions combined from the predose time during the first visit to the predose time during the second visit. The significant reduction in central staining is clinically relevant, as the central cornea is critical to visual function.18 Improvement in the sum of corneal regions also indicates a global treatment effect.19
Significant improvements were also observed in a number of secondary efficacy endpoints. Tear film stability, as measured by the percent change in MBA, significantly improved from the predose time of the second visit to the postdose time of the second visit. This improvement indicates that Retaine™ caused an immediate, statistically significant beneficial effect. Additionally, a second quantitative measurement, conjunctival redness, was significantly lower on the second visit than at the time of the baseline.
For most eye care professionals, an artificial tear that demonstrates an improvement in symptoms and the patient’s QoL is the optimal choice. While a practitioner will consider the impact an artificial tear has on signs of the disease, a positive response from the patient is of utmost importance in this symptom-driven disease. In this study, not only did Retaine™ achieve statistical improvement in three key symptoms (discomfort, dryness, and grittiness), but also a reduction in all symptoms combined. Prior studies have demonstrated that dry eye has a significant impact on a patient’s QoL,20,21 similar to the level of severe angina.22 Achieving a statistically significant improvement in a QoL measure (working at the computer at night) indicates how an efficacious tear substitute can benefit the patient on a global level.
While not statistically significant, study subjects saw a improvement (41%) in time at CVA from the first visit to the second visit. Generally, IVAD Test results correlated with central staining and QoL results. Of the 42 enrolled subjects, only 32 subjects were available for both evaluations, so it is possible that this smaller cohort lacked the statistical power to achieve significance.
Overall, the study drug was well tolerated and scored consistently high in drop comfort parameters. The results of the drop comfort questionnaire indicated that the majority of subjects used positive descriptor words to characterize drop comfort; of the subjects evaluated, 45% selected only positive drop comfort descriptor words and only 2% selected only negative drop comfort descriptor words.
While the open-label design of the study does not offer a comparison with a placebo or other active treatment, the breadth of examinations and procedures do provide a complete picture of the benefits of the product. Additionally, we understand that 2 weeks is a short treatment period for an artificial tear, although the findings after only 2 weeks of treatment are very encouraging and highlight the fast onset of action for this compound.
Conclusion
This study demonstrates that Retaine™, equipped with Novasorb® technology, provides effective relief from both the signs and symptoms of dry eye sufferers. The data presented herein indicate a therapeutic effect of Retaine™ on the signs and symptoms of dry eye after 2 weeks of treatment, as well as an immediate-onset transient improvement in tear film stability.
Total dry eye relief is evidenced by the reduction in MBA and corneal fluorescein staining, both of which indicate ocular surface protection, and by the promotion of global health through symptomatic relief and improvements in QoL. Further research is warranted to confirm these results and better understand how this product can fit into the dry eye treatment armamentarium as a whole.
Disclosure
George Ousler III is an employee of Ora, Inc. Douglas Devries is a consultant for OCuSOFT. Paul M Karpecki is a consultant for OCuSOFT. Joseph B Ciolino is a consultant for Ora, Inc. and is supported by a Career Development Award from Research to Prevent Blindness, Inc. and the National Eye Institute 1K08EY019686-01. The authors report no other conflicts of interest in this work.
References
Schaumberg DA, Dana R, Buring JE, Sullivan DA. Prevalence of dry eye disease among US men: estimates from the Physicians’ Health Studies. Arch Ophthalmol. 2009;127(6):763–768. | ||
Schaumberg DA, Sullivan DA, Buring JE, Dana MR. Prevalence of dry eye syndrome among US women. Am J Ophthalmol. 2003;136(2): 318–326. | ||
Gayton JL. Etiology, prevalence, and treatment of dry eye disease. Clin Ophthalmol. 2009;3:405–412. | ||
Management and therapy of dry eye disease: report of the Management and Therapy Subcommittee of the International Dry Eye WorkShop (2007). Ocul Surf. 2007;5(2):163–178. | ||
Lallemand F, Daull P, Benita S, Buggage R, Garrigue JS. Successfully improving ocular drug delivery using the cationic nanoemulsion, novasorb. J Drug Deliv. 2012;2012:604204. | ||
Daull P, Lallemand F, Garrigue JS. Benefits of cetalkonium chloride cationic oil-in-water nanoemulsions for topical ophthalmic drug delivery. The Journal of pharmacy and pharmacology. 2014;66(4):531–541. | ||
Rabinovich-Guilatt L, Couvreur P, Lambert G, Dubernet C. Cationic vectors in ocular drug delivery. J Drug Target. 2004;12(9–10):623–633. | ||
Royle L, Matthews E, Corfield A, et al. Glycan structures of ocular surface mucins in man, rabbit and dog display species differences. Glycocon J. 2008;25(8):763–773. | ||
Rodriguez JD, Ousler GW 3rd, Johnston PR, Lane K, Abelson MB. Investigation of extended blinks and interblink intervals in subjects with and without dry eye. Clin Ophthalmol. 2013;7:337–342. | ||
Gomes PJ, Ousler GW, Welch DL, Smith LM, Coderre J, Abelson MB. Exacerbation of signs and symptoms of allergic conjunctivitis by a controlled adverse environment challenge in subjects with a history of dry eye and ocular allergy. Clin Ophthalmol. 2013;7:157–165. | ||
Ousler GW 3rd, Abelson MB, Johnston PR, Rodriguez J, Lane K, Smith LM. Blink patterns and lid-contact times in dry-eye and normal subjects. Clin Ophthalmol. 2014;8:869–874. | ||
Meerovitch K, Torkildsen G, Lonsdale J, et al. Safety and efficacy of MIM-D3 ophthalmic solutions in a randomized, placebo-controlled Phase 2 clinical trial in patients with dry eye. Clin Ophthalmol. 2013;7: 1275–1285. | ||
Rodriguez JD, Johnston PR, Ousler GW 3rd, Smith LM, Abelson MB. Automated grading system for evaluation of ocular redness associated with dry eye. Clin Ophthalmol. 2013;7:1197–1204. | ||
Pollard S, Ousler GW III, Lipkin N, Abelson MB. Dry Eye Quality of Life Questionnaire (DEQLQ©) – Outcome in a Population of Normals and Patients Diagnosed with Dry Eye. Association for Research in Vision and Ophthalmology. In: Investigative Ophthalmology and Visual Science Annual Meetings Abstracts Online; April 25–29, 2004; Fort Lauderdale, FL. Abstract 82. Available from: http://abstracts.iovs.org/cgi/content/abstract/45/5/82. Accessed November 21, 2014. | ||
Abelson R, Lane KJ, Rodriguez J, et al. Validation and verification of the OPI 2.0 System. Clin Ophthalmol. 2012;6:613–622. | ||
Abelson R, Lane KJ, Rodriguez J, et al. A single-center study evaluating the effect of the controlled adverse environment (CAE(SM)) model on tear film stability. Clin Ophthalmol. 2012;6:1865–1872. | ||
Torkildsen G. The effects of lubricant eye drops on visual function as measured by the Inter-blink interval Visual Acuity Decay test. Clin Ophthalmol. 2009;3:501–506. | ||
Lemp MA. Advances in understanding and managing dry eye disease. Am J Ophthalmol. 2008;146(3):350–356. | ||
Cohen S, Martin A, Sall K. Evaluation of clinical outcomes in patients with dry eye disease using lubricant eye drops containing polyethylene glycol or carboxymethylcellulose. Clin Ophthalmol. 2014;8:157–164. | ||
Paulsen AJ, Cruickshanks KJ, Fischer ME, et al. Dry eye in the beaver dam offspring study: prevalence, risk factors, and health-related quality of life. Am J Ophthalmol. 2014;157(4):799–806. | ||
Uchino M, Schaumberg DA. Dry Eye Disease: Impact on Quality of Life and Vision. Curr Ophthalmol Rep. 2013;1(2):51–57. | ||
Buchholz P, Steeds CS, Stern LS, et al. Utility assessment to measure the impact of dry eye disease. Ocul Surf. 2006;4(3):155–161. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.