Back to Archived Journals » Research and Reports in Biology » Volume 7
AMPK in the central nervous system: physiological roles and pathological implications
Authors Rosso P, Fioramonti M, Fracassi A, Marangoni M, Taglietti V ![]() , Siteni S, Segatto M
, Siteni S, Segatto M
Received 14 October 2015
Accepted for publication 30 November 2015
Published 29 January 2016 Volume 2016:7 Pages 1—13
DOI https://doi.org/10.2147/RRB.S90858
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Zvi Kelman
Pamela Rosso,1,2,* Marco Fioramonti,3,* Anna Fracassi,2 Martina Marangoni,4 Valentina Taglietti,5 Silvia Siteni,2,6 Marco Segatto5
1Institute of Cell Biology and Neurobiology, National Research Council (CNR), 2Department of Sciences, University of Rome “Roma Tre”, 3Laboratory of Translational Oncology, University Campus Bio-Medico of Rome, 4Medical Genetics Unit, University Hospital of Rome “Tor Vergata”, Rome, 5Department of Biosciences, University of Milan, Milan, 6Department of Experimental Oncology, Regina Elena National Cancer Institute, Rome, Italy
*These authors contributed equally to this work
Abstract: 5′ AMP-activated protein kinase (AMPK) is considered the master metabolic regulator in all eukaryotes, as it maintains cellular energy homeostasis in a variety of tissues, including the brain. In humans, alterations in AMPK activity can lead to a wide spectrum of metabolic disorders. The relevance of this protein kinase in the pathogenesis of diabetes and metabolic syndrome is now well established. On the contrary, correlations between AMPK and brain physiopathology are still poorly characterized. The aim of this review is to summarize and discuss the current knowledge about the prospective involvement of AMPK in the onset and the progression of different neurological diseases.
Keywords: AMPK, brain, neurodegeneration, stroke, tumor, autophagy
Introduction
The maintenance of energy homeostasis is guaranteed by the tight regulation of molecular and functional mechanisms involved in energy intake and expenditure. The AMP-activated protein kinase (AMPK) is a sensor of AMP:ATP ratio and mediates adaptive changes as a function of low energy conditions. AMPK is considered the metabolic regulator of the whole organism, and it has become the leading factor in the study of obesity, diabetes, cachexia, and other metabolic disorders. However, during the last decade, increasing evidence sustains a pivotal role for AMPK also in the physiopathology of the central nervous system (CNS). AMPK is implicated in several neuronal aspects, such as neuronal proliferation and differentiation, synaptic connectivity, and neuroprotection. Thus, alterations in AMPK could easily lead to the onset of different neurological and neurodegenerative diseases.
AMPK: a pivotal metabolic regulator of the whole organism
Structure and regulation
In most species, AMPK exists as a heterotrimeric protein complex, containing α, β, and γ subunits in equal stoichiometry. The α subunit is responsible for the catalytic kinase activity, whereas the β and γ are regulatory subunits. In particular, mammalian γ subunits contain four cystathionine-β-synthase (CBS) domains, which occur as tandem pairs and function as nucleotide-binding sites for AMP or ATP.1 When intracellular ATP levels are low, AMP or ADP directly and mutually bind to the γ regulatory subunits of AMPK, thus inducing a conformational change that leads to AMPK activation.2,3 High AMP levels also protect AMPK from dephosphorylation events operated by the major classes of serine/threonine phosphatases (PP1, PP2A, and PP2Cα). The activatory phosphorylation of AMPK is mainly mediated by liver kinase B1 (LKB1), which phosphorylates the α catalytic subunit at Thr172.4 However, several studies have demonstrated that AMPK can be also phosphorylated on Thr172 in response to intracellular calcium levels by calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2).5–8 (For extensive review, see works by Hardie5 and Moussa and Li8).
There are two genes encoding the α catalytic subunits (α1 and α2), two β genes (β1 and β2), and three γ genes (γ1, γ2, and γ3). Some of these isoforms display a tissue-specific expression pattern, and several experimental data also highlight functional distinctions for the two catalytic α subunits, which reflect in AMP- and LKB1-responsiveness and nuclear localization. AMPKα2 isoform is particularly abundant in the nucleus, even though AMPKα1 is able to translocate to the nucleus under distinct and peculiar conditions.9,10 Moreover, the myristoylation of the β isoforms is an essential prerequisite for AMPK activation and its localization to membranes.11
Even though all the AMPK subunits are expressed in the brain, interesting findings underlie a specific distribution pattern of the various subunit isoforms among cell types in the CNS. For instance, the α2 represents the dominant catalytic subunit in neurons. On the other hand, α subunits are not generally expressed in glial cells at basal conditions, but only in activated astrocytes. The γ1 subunit is mainly expressed in neurons but absent in astrocytes, and the amount of β1 and β2 subunits also differs according to the brain cell type (for extensive review, see Turnley et al12).
AMPK orchestrates autophagy and cell proliferation
Eukaryotic cells adaptively respond to low-nutrient conditions by blocking cell growth and proliferation. These events are mediated by AMPK through the activation of p53, the inhibition of the mammalian target of rapamycin (mTOR) complex 1 (mTORC1), and the induction of autophagy. The inhibition of cell-cycle progression-induced by AMPK occurs via MDMX phosphorylation on Ser342, which leads to an increased binding of MDMX to 14-3-3. The subsequent inactivation of MDMX is responsible for the enhanced stability and activation of p53, which provokes a cell-cycle checkpoint.13
Beyond effects on p53, AMPK directly phosphorylates the tumor suppressor tuberous sclerosis complex 2 (TSC2) thus stimulating its GAP activity toward Rheb, which in turn leads to the inactivation of mTORC1 and the inhibition of cell proliferation.8,14 AMPK also blocks the positive trophic effects of mTORC1 through the direct phosphorylation of regulatory associated protein of mTOR (raptor) (Figure 1).15
 | Figure 1 AMPK modulates cell-cycle progression and cell growth. |
Besides the regulation of cell growth and proliferation, mTORC1 suppresses the autophagic flux through the inhibition of uncoordinated 51-like kinase 1 (ULK1) complex.16 Autophagy is required for the breakdown of cellular organelles and the recycling of cellular components under nutrient deprivation, as well as for the selective clearance of damaged organelles. In contrast to the inhibitory regulation operated by mTORC1, a variety of experimental evidence demonstrated that AMPK promotes autophagy by direct phosphorylation of ULK1 at several serine residues (eg, Ser317, Ser555, and Ser777).17,18 AMPK-dependent stimulation of ULK1 complex seems to appear as a universal event for the induction of autophagy, as it occurs under different conditions such as nutrient starvation, hypoxia, and drug administration. This route of activation results in the promotion of not only nonselective autophagy (known as bulk autophagy) but also selective autophagy: for instance, AMPK triggers mitophagy through the phosphorylation of ULK1 during stress conditions, and this pathway is also required for the clearance of damaged mitochondria in physiopathological states.19–24 In addition, recent data showed that AMPK phosphorylates Beclin1 at Ser91/94, and this event is essential for the induction of autophagy in nutrient stress response.25 An interesting study published by Sanchez et al sustains that AMPK also determines, at least in skeletal muscle, the FoxO3-dependent increase of autophagy-related proteins, such as LC3B, GABARAPL1, and Beclin1.26 Another research supports these findings, since AMPK directly activates FoxO3 transcription through the phosphorylation of different serine residues (eg, Ser413, Ser588).27 Collectively, these data highlight that AMPK drives autophagy at multiple levels (Figure 2).
 | Figure 2 AMPK drives autophagy through short- and long-term mechanisms. |
AMPK maintains energy homeostasis through short- and long-term regulation of metabolic targets
AMPK was originally discovered as the main protein kinase involved in the short-term modulation of key enzymes controlling fatty acid and cholesterol biosynthesis. In particular, AMPK was found to be responsible for the rapid regulation of acetyl-CoA carboxylase (ACC) and HMG-CoA reductase (HMGR), as the inhibitory phosphorylation mediated by this kinase is able to strongly suppress the activity of both the enzymes.28,29
In highly specialized cells of metabolic organs, such as skeletal muscle and adipose tissue, AMPK regulates glucose metabolism promoting the phosphorylation of AS160 and Tbc1d1 and the subsequent binding to 14-3-3. These events lead to an enhancement of GLUT4 translocation on the cell membrane.30 In addition, AMPK drives lipid mobilization by directly phosphorylating hormone-sensitive lipase (HSL) and adipocyte triglyceride lipase (ATGL).31,32 Recently, these mechanisms have been shown to be involved not only during homeostasis maintenance in physiological conditions but also in the development of metabolic disorders such as cancer-associated cachexia.33
The adaptive changes in energy homeostasis induced by AMPK are not limited to the short-term regulation of enzymes, as this kinase orchestrates a profound metabolic reprogramming through transcriptional control. For instance, AMPK phosphorylates a variety of transcriptional factors and cofactors, such as PGC1α, p300, histone deacetylase 5 (HDAC5), and sirtuin1, thus leading to the long-term regulation of genes involved in gluconeogenesis, lipogenesis, and mitochondrial biogenesis (for details, see Mihaylova and Shaw19). Of note, AMPK also suppresses the activation of SREBP1 and SREBP2. The inhibitory phosphorylation mediated by AMPK is a pivotal prerequisite to prevent the proteolytic processing and activation of SREBPs. SREBPs are crucial transcription factors involved in the regulation of lipogenic genes, such as ACC and HMGR.34 As a result, AMPK controls lipid metabolism regulating both the activity and the protein levels of enzymes implicated in fatty acid and cholesterol biosynthesis (Figure 3).
 | Figure 3 AMPK controls cell metabolism. |
Physiological roles of AMPK in the CNS
AMPK regulates brain cell metabolism, proliferation, and morphology
Even though the brain constitutes 2% of the total body weight, it utilizes 50% of the entire glucose supply. The energy consumption by brain cells is very high and, unlike other peripheral cells, neurons use only glucose as a source of energy. As neurons are not able to synthesize and store a sufficient amount of glycogen, it is indispensable to ensure themselves a constant and continuous source of glucose.35 During the last few years, several research groups highlighted a crucial role for AMPK in the regulation of energy production and consumption in neurons. Synaptic activation is followed by a number of molecular and cellular events characterized by high energy consumption, such as sodium pump activity, neurotransmitter receptor translocation/recycling, synaptic transport, cytoskeleton remodeling, and metabolic processes. In the light of these observations, it was demonstrated that the decrease in ATP levels following glutamate stimulation is able to strongly phosphorylate AMPK, and the activation of glutamate transmission enhances the translocation of the glucose transporter GLUT3 on cell surface. Notably, the increase of GLUT3 on cell membrane is mediated by AMPK, as the pharmacological inhibition or the knockdown of this kinase is able to completely prevent the glutamate-induced GLUT3 translocation.36
In addition to glucose metabolism, other studies indicated that AMPK acts as a key regulator of mitochondrial function and biogenesis in neurons. In Neuro2a cell line, AMPK activation by resveratrol determines an increase in the transcripts of the mitochondrial protein marker Mitofusin 2, and a rise in the master regulators of mitochondrial biogenesis, PGC-1α and mitochondrial transcription factor A (mtTFA). AMPK inhibition suppresses the transcription of these mitochondrial-related proteins, further sustaining that AMPK exerts a pivotal role in the regulation of mitochondrial metabolism in neurons.37,38
Besides the direct regulation of metabolism in neurons, AMPK has been shown to be involved in neurodevelopmental processes. For instance, AMPK maintains the genomic integrity during neural progenitor cell division in Drosophila, and any interference with the activity of this kinase provokes embryonic lethality.39 This study corroborates the finding that the ablation of AMPK β1 subunit leads to a severe reduction in the number of neurons and oligodendrocytes, as well as alterations in astrocyte proliferation in mice. Indeed, defects in AMPK functionality induce aberrant proliferation and increased apoptosis of neural stem cell progenitors (NPCs). The effects of AMPK on NPC proliferation and viability are exerted through direct phosphorylation of Rb protein, whose activation is required for the normal development of CNS.40
Increasing evidence also suggests that AMPK plays an important role in the control of cell polarization under energy-lacking conditions. As an instance, AMPK modulates the establishment of initial neuronal polarity by affecting the processes at the root of axogenesis.41 In addition, a recent work sustains that AMPK regulates neuronal development during dendrite outgrowth and branching. At molecular level, the effects on neuronal architecture are induced by the AMPK-dependent suppression of both mTOR and Akt signaling pathways.42
In adult brain, AMPK drives the cellular events involved in the aging process. An elegant study performed in Drosophila demonstrated that the activation of neuronal AMPK determines a delay in systemic aging and prolongs life span. These outcomes are mediated by the AMPK-dependent activation of autophagy and the synergic reduction of insulin-like peptide signaling.43
Hypothalamic AMPK controls food intake and body weight
Metabolic diseases are regarded as a major health problem in many Western countries. Generally, they are caused by the lack of physical activity and are strictly associated to the presence of food intake-related disorders. From a pathophysiological point of view, metabolic diseases are characterized by obesity, insulin resistance, and several atherogenic signs, leading to disruption in energy homeostasis.44
Whole-body energy balance is guaranteed not only by the functionality of high metabolic peripheral tissues such as liver, skeletal muscle, and adipose tissue, but also by the control of the CNS. The hypothalamus represents the main brain area involved in regulating food intake and energy expenditure, and it is organized into several structural and functional nuclei, including the arcuate nucleus (ARC), the paraventricular nucleus (PVN), the dorsomedial nucleus (DMN), the ventromedial nucleus (VMN), and the lateral hypothalamic area. The majority of these hypothalamic regions are involved in the production and secretion of orexigenic and anorectic neuropeptides that affect food intake and energy homeostasis. For instance, the orexigenic neuropeptides agouti-related protein (AgRP) and neuropeptide Y (NPY), as well as the anorectic neuropeptide α-melanocyte-stimulating hormone (MSH) are synthesized by the ARC.45,46 On the other hand, the orexigenic neuropeptides melanin-concentrating hormone (MCH) and orexins are produced by the lateral hypothalamic area.45,47 In addition, both the hypothalamic regions possess the capability to synthesize the anorexigenic neuropeptide cocaine-amphetamine regulated transcript (CART).48
During the last few years, a variety of hormones have been identified as powerful modulators of energy balance.49 In this context, AMPK exerts a pivotal role in nutritional regulation and energy homeostasis, as this kinase orchestrates the hormonal signaling pathways required for the production and the release of hypothalamic orexigenic and anorectic neuropeptides.50–52
The orexigenic hormones induce the activation of hypothalamic AMPK. Specifically, cannabinoids exert their permissive effects on food intake by activating cannabinoid receptors expressed in the VMN, whereas ghrelin, secreted by the gastric mucosa, binds to receptors expressed in both VMN and ARC.53,54 Different findings demonstrated that a rise in ghrelin levels is associated with an increase in food intake and body weight.55,56
At molecular level, the ghrelin-dependent increase of intracellular Ca2+ triggers CAMKK2 activity, which in turn enhances AMPK phosphorylation and the subsequent activity of AgRP/NPY and pro-opiomelanocortin (POMC) neurons.57,58 Similar orexigenic effects are also elicited by adiponectin. This adipocyte-secreted hormone mimics the outcomes induced by ghrelin through the activation of hypothalamic AMPK.59 However, the molecular mechanisms linking adiponectin and AMPK modulation are still not completely elucidated.
The anorexigenic hormones leptin and insulin control food intake and body weight by suppressing hypothalamic AMPK. The effects promoted by leptin are mediated through the modulation of neuropeptides in the ARC and PVN, whereas multiple hypothalamic regions are affected by insulin. An interesting study demonstrated that MC4 transduction pathway is required for AMPK inhibition by these hormones, and the expression of the constitutively active AMPKα2 isoform completely prevents the effects on food intake and body weight elicited by leptin.60 Tri-iodothyronine (T3) represents another anorexigenic hormone able to regulate the whole-body energy balance through the inhibition of AMPK in neurons located at the VMN. As for leptin, T3 determines norepinephrine/epinephrine release from sympathetic nerves, which stimulates fatty acid mobilization from white adipose tissue and heat production in brown adipose tissue.61 AMPK inhibition mediates the effects of anorectic hormones at least by two different mechanisms: the former involves the activation of ACC and the subsequent increase in fatty acid biosynthesis, which culminates in the translocation of fatty acids across the mitochondrial membrane; the latter is related to the activation of mTOR and the consequent phosphorylation of p70S6 kinase (p70S6K) and 4EBP1. The modulation of these pathways leads to the suppression of orexigenic AgRP/NPY neurons and the activation of anorectic POMC/CART neurons.60,62–65
Pathological involvement of AMPK in neurological conditions
AMPK in neurodegenerative disorders
Given the importance of AMPK in regulating stress responses, it is not surprising that dysfunctions of AMPK signaling are associated with several brain diseases including Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD). Here, we summarize the current knowledge about the pathophysiological roles of AMPK in regulating neuronal survival during neurodegenerative disorders.
AMPK in AD
AD is the most common form of dementia, characterized by progressive neurodegeneration, particularly affecting cortical and hippocampal brain regions. Histopathological features of AD are senile plaques, composed of β-amyloid (Aβ) peptide polymers, and intracellular neurofibrillary tangles, formed by hyperphosphorylated Tau protein.66
AD has been identified as a protein misfolding disease and is caused by accumulation of abnormally folded Aβ in the brain.67 Aβ peptides originate from the proteolysis of amyloid precursor protein (APP) – amyloidogenic pathway – by the sequential enzymatic actions of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE-1, or β-secretase) and a protein complex with presenilin 1 at its catalytic core (γ-secretase).68,69
AMPK has been identified to play an important role in the development of AD. This hypothesis was initially supported by different evidence highlighting the hyperphosphorylation of AMPK in the brains of both AD patients and AD mouse models.70–72 Moreover, a different AMPK subcellular localization is present in the brains of AD patients when compared with control subjects.70 It is now well established that AMPK is a key regulator of Aβ generation. Vingtdeux et al showed that the activation of AMPK by resveratrol lowers extracellular Aβ accumulation.73 The same research group demonstrated that this polyphenol inhibits the AMPK target mTOR, triggering autophagy and lysosomal degradation of Aβ.
Studies conducted on cultured rat cortical neurons revealed that Aβ production is significantly reduced after stimulation with the AMPK activator 5-aminoimidazole-4-carboxamide-1-d-ribofuranoside (AICAR). On the contrary, Aβ peptide levels are increased when AMPKα2 is knocked out, thus indicating the crucial involvement of AMPK in amyloidogenesis.74 AMPK controls Aβ generation through the modulation of neuronal cholesterol and sphingomyelin levels, which regulate APP distribution in lipid rafts.67 In addition, leptin-induced AMPK activation is associated with a reduction in Tau phosphorylation.75
Despite these findings, other evidence sustains an opposite role for AMPK in AD. Interestingly, the activation of AMPK by metformin is protective in females but increases memory dysfunction in males, suggesting a sex-divergent cognitive effect in a murine model of AD.76 Moreover, chronic treatment of AD mice with the same compound was reported to enhance the generation of Aβ via the upregulation of β-secretase at transcriptional level.77 Noteworthy AMPK inhibitor compound C (CC) is able to correct Aβ-induced inhibition of long-term potentiation (LTP) and the enhancement of long-term depression (LTD). Similarly, LTP impairments in APP/PS1 transgenic mice are improved by CC treatment.78
AMPK in HD
HD is an age-related neurodegenerative disorder, characterized by motor and cognitive impairment, and caused by a CAG trinucleotide expansion in exon 1 of the Htt gene. When the number of CAG repeats is 36 or more, the translated polyglutamine-expanded Htt protein interferes with the normal functions of cellular machinery and causes cytotoxicity. The major characteristic of HD is the selective loss of neurons in the neostriatum, nigrostriatal tract, and cortex, which leads to movement disorders, dementia, and eventual death.79
Recent research showed that mitochondria and several key molecular players in energy homeostasis are altered during HD progression. Notably, mutated Htt (mHtt) forms aggregates on mitochondrial membranes. This aberrant association impairs calcium homeostasis and triggers Ca2+-induced oxidative stress, decreases the activity of mitochondrial respiratory complexes (II and III), and alters mitochondrial fission and fusion, globally disrupting the neuronal energy homeostasis. In this scenario, AMPK could represent one of the leading actors in modulating metabolic events.80,81 Indeed, AMPK was found to be abnormally activated in the striatum of a transgenic mouse model of HD (R6/2).82 Ju et al highlighted that the activation of AMPKα1 in striatal neurons is closely associated with mHtt-induced cell death.79 The induction of neurotoxicity requires the nuclear translocation of AMPKα1 to suppress the expression of the survival gene Bcl2 in striatal neurons.79 Furthermore, AMPK activation by AICAR in HD mice induces neuronal apoptotic activation and worsens motor function.79,81 The same research groups showed that the induction of cAMP/PKA signaling reduces AMPK activity, thus preventing the detrimental effect of AMPKα1 in the nuclei of striatal neurons.
AMPK in PD
PD is the most common neurodegenerative movement disorder and the second most common neurodegenerative disease after AD. Clinically, it is characterized by tremor, rigidity, and bradykinesia, and pathologically by the loss of dopaminergic neurons in the substantia nigra. During the last few years, a plethora of studies identified different genes involved in familial forms of PD, and implicated aberrant mitochondrial homeostasis as one of the key contributors to PD.83 Among the disease-associated genes, recessive mutations in several genes such as α-synuclein, DJ-1, PINK, parkin, and dominant mutations in LRRK2 (especially the G2019S variant) are directly or indirectly associated with mitochondrial dysfunction in PD.84
In this pathology, AMPK activation is similarly a double-edged sword, promoting or aggravating neurodegeneration under different circumstances.85 Indeed, AMPK exerts a neuroprotective role as demonstrated in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which represents the most common experimental model used to investigate the pathogenesis of PD. Recent findings sustain that AMPK is activated by MPTP in mice and MPP(+) in SH-SY5Y cells, and the inhibition of AMPK by CC resulted in an increase of neuronal cell death.86 In addition, AMPK acts with parkin in a functionally converging manner to guarantee the quality control of organelles.85 Hence, the activation of AMPK may prevent neuronal cell death and play a role as a survival factor in PD.
In contrast to these findings, Kim et al described that poly (ADP-ribose) polymerase-1 (PARP-1) promotes ATP depletion and the subsequent activation of AMPK, which mediates the degeneration of dopaminergic neurons.87
Impact of AMPK on cancer cell fate and brain tumors
A variety of tumor types, including brain tumors, are characterized by uncontrolled and rapid cell growth. In order to sustain the enhanced rate of mitosis, tumor cells undergo profound changes in their metabolism, a phenomenon known as “Warburg effect”.88
It has been demonstrated that AMPK regulates cell growth arrest and cell death interacting both with mTOR and p53 in normal cells. Furthermore, the modulation of AMPK represents an important prerequisite for the induction and maintenance of cell proliferation under abnormal nutrient conditions in cancer cells. The role of AMPK in brain tumors is still poorly understood and controversial hypotheses have been proposed. Glioma represents a large group of common brain tumors that comprises glioblastoma and astrocytoma.89 Rios et al observed that AMPK is hyperactivated both in astrocytes expressing HRasV12 (common mutation in human astrocytoma) and in glioblastoma cell lines compared to control cells. High phospho-AMPK levels were also detected in samples derived from glioblastoma patients, whereas normal brain tissue was completely negative. CC treatment and the genetic deletion of AMPK lead to a strong decrease of cell proliferation and to a reduction of Rb phosphorylation, suggesting that AMPK could play a critical role in tumor growth by regulating Rb and, consequently, cell-cycle progression.90 Furthermore, it has been shown that AMPK activity is necessary to provide metabolic support during early stages of tumor growth.91 The relevance of AMPK in oncology has been demonstrated not only for cell growth but also for cell migration. Indeed, AMPK inhibition antagonizes the ghrelin-mediated migration of rat C6 and human U251 glioma cells, suggesting that AMPK activation is an obligatory event in ghrelin-induced glioma migration.92 In a retrospective clinical analysis, it has been revealed that high-grade human glioma expresses higher levels of AMPKα2 subunit if compared to low- and mid-grade gliomas. The α2 subunit is selectively induced in hypoxic conditions and significantly contributes to vascular endothelial growth factor (VEGF) expression in human glioma cells, thus improving neoangiogenesis and underlying the pivotal role of AMPK in glioma progression.93 In addition, the resistance to antiangiogenic therapies in glioblastoma may depend on the autophagic flux induced by the activation of hypoxia-inducible factor-1α (HIF-1α)/AMPK axis after hypoxia and lack of nutrients.94 Even though all these data sustain a protumor effect of AMPK, other researchers suggest that AMPK acts as an oncosuppressor. Guo et al showed that AMPK activation by AICAR blocks EGFR-activated glioma proliferation through the modulation of mTORC1 signaling and lipid biosynthesis.95 The antitumor effects of AMPK were also demonstrated by the induction of apoptosis in mouse astrocytoma cell line following AICAR administration.96 This finding corroborates the outcomes induced by metformin, which is able to inhibit tumor cell growth and to enhance the sensitivity of glioblastomas to chemotherapy.97
The use of different biological models (ie, different cell lines or mouse models, and different cancer mutations) may lead to the contrasting evidence about the prospective role of AMPK in glioma. In addition, AMPK activators or inhibitors may have AMPK-independent effects on cancer cells, making it more difficult to dissect the specific role of this kinase in brain tumors.98,99 Furthermore, AMPK could not show a unique behavior; conversely, it may play a protumoral or antitumoral role depending on the energetic and the genetic status of the tumor.
Another aggressive brain tumor is neuroblastoma, the most common extracranial solid tumor in children.100 Although only little knowledge is available about its role in neuroblastoma, AMPK is believed to act as an oncosuppressor. In particular, AMPK activity may lead to apoptosis via p38MAPK/p53 pathway in neuro2a and SH-SY5Y neuroblastoma cell lines.101,102 However, research in this field is still limited and more detailed analysis will be necessary to specifically assess the involvement of AMPK in the onset and progression of neuroblastoma.
AMPK regulation in stroke
Ischemic stroke is one of the most frequent leading causes of death worldwide.103 Recently, different studies showed that AMPK is rapidly activated in an energy-deprived status such as that which follows a stroke. Ischemic stroke promotes the activation of oxidative and cell death pathways, leading to cellular energy imbalance, that increases the phosphorylation of AMPK in an attempt to restore ATP levels through the enhancement of glycolysis and fatty acid oxidation, increment of glucose transport, and inhibition of glycogen synthesis.104–106 Thus, AMPK plays a crucial role in regulating ischemic stroke phenotype, but it is unclear whether its activation would be detrimental or beneficial. In vivo studies using the transient middle cerebral artery occlusion (MCAO) model in mice have shown that AMPK activation is damaging, because the pharmacological inhibition of AMPK with CC causes a reduction in stroke volume, whereas the treatment with AICAR leads to a more severe injury, suggesting that inactivation of AMPK during ischemia may be neuroprotective.107 The mechanisms through which acute AMPK activation exacerbates stroke injury are still unclear. It has been suggested that AMPK activation enhances astrocytic glycolysis which leads to progressive lactic acidosis, exacerbating lactate accumulation, and inhibits the ability of neurons to use lactate as an energy source, contributing to neuronal death. Moreover AMPK activation is responsible for the aberrant neuronal nitric oxide synthase activity, which in turn produces peroxynitrite, the strongest oxidizing agent. These data support the hypothesis that AMPK activation in the acute phase of stroke is associated with injury.108 To corroborate the idea that AMPK activation is harmful in ischemic stroke, preclinical studies demonstrated that mice deficient in AMPKα2 have less injury and show a reduced total infarct volume compared with wild-type littermates in an MCAO reperfusion model. AMPKα1-knockout mice have no difference in injury compared with wild-type mice, suggesting that AMPKα2 isoform plays a more significant role in the damaging response of AMPK activation in ischemic brain.109 On the contrary, recent clinical trials report that metformin significantly reduces the risk and incidence of stroke by actions that are independent of its glucose-lowering effects.110–112 The chronic treatment with metformin offers potent neuroprotective effects similar to preconditioning, a phenomenon by which exposure to sublethal ischemia stimulus leads to protection of the organism from subsequent severe ischemic insults.113,114 In particular, it was observed that chronic metformin treatment is able to significantly downregulate stroke-mediated brain injury, increase phospho-AMPK levels, reduce lactate formation, improve infarct damage, enhance angiogenesis, and alleviate inflammatory responses by a negative feedback mechanism, blunting ischemia-induced AMPK activation.115,116 Interestingly, only a chronic metformin treatment is beneficial for the prevention of ischemic disease and the change from a chronic to an acute regimen worsens ischemic injury and functional outcome in otherwise healthy animals, by enhancing AMPK activity and lactic acidosis.115 In addition, a recent study showed that acute metformin treatment in diabetic rats worsens the infarct size and caused significant neurological deficiencies if compared to untreated diabetic animals.117 Thus, metformin has negative effects on the severity of neurovascular injury when used acutely, but chronic treatment is protective against fatal cerebral ischemia. The explanation of these divergent results may be dependent on the extent of AMPK manipulation in vivo, on timing and amount of its activation, and on different animal strains and models.118 As ischemic stroke is a condition of severe energy depletion and AMPK is a master energy regulator, future investigations are needed to provide new information about the therapeutic potential of acute and/or chronic AMPK modulation in stroke.
AMPK in genetic and chromosomal disorders
Although a direct involvement is still unclear, different genetic and chromosomal aberrations strongly suggest a role for AMPK in the development of mental disabilities in genetic disorders. Mutations in the PRKAG2 gene (AMPKγ2 subunit) have been associated with hereditary cardiac arrhythmias (Wolff–Parkinson–White syndrome, OMIM 194200), familial hypertrophic cardiomyopathy (OMIM 600858), and glycogen storage disease of the heart (OMIM 261740).119–121 Interestingly, a polymorphism in the PRKAG2 gene has been also linked to cognitive impairments in elderly people.122
PRKAB2 gene (AMPKβ2 subunit), which maps on chromosome 1q21.1, is included in chromosomal rearrangements causing the chromosome 1q21.1 deletion (OMIM 612474) and duplication (OMIM 612475) syndromes. Deletions or duplications (copy number variations [CNVs]) of 1q21.1 chromosomal region have been associated with variable phenotypes, which include intellectual disability and/or autism, schizophrenia, congenital heart anomalies, dysmorphic features, or a normal phenotype.123 The crucial role of AMPK in brain function is particularly empathized in 1q21.1 CNV carriers, as they manifest some form of learning disabilities.123 Notably, AMPK inhibits the mTOR pathway, which is required for the modulation of learning and memory processes.124
The involvement of AMPK in brain functioning is further supported by recent studies demonstrating that 1q21.1 CNVs (candidate gene PRKAB2) were most frequently associated with schizophrenia.125,126
It has been shown that AMPKβ2 protein levels change in concordance with the 1q21.1 copy number state in patient-derived lymphoblastoid cell lines (LCLs).123 This imbalanced expression affects AMPK activity, as the basal levels of phospho-AMPKα in both 1q21.1 Del and Dup expressing cells are significantly different in comparison to wild-type cells.123 Furthermore, the AICAR-induced phosphorylation of the AMPK substrates ACC and raptor is suboptimal in 1q21.1 Del and Dup LCLs with respect to wild type, but it is more evident in the 1q21.1 Del-containing cells, demonstrating that the reduced availability of a regulatory β-isoform could affect AMPK activity to a greater extent than its overabundance.123
The critical role of AMPK in the CNS could also help to elucidate the mechanisms that contribute to altered neuronal function in patients affected by other genetic diseases, such as TSC (OMIM 191100 and 613254). TSC is an autosomal dominant disorder characterized by hamartomas in multiple organs and neurological manifestations such as seizures, hyperactivity and aggression, intellectual disability, or learning problems.20 TSC is caused by mutations in the TSC1 or TSC2 genes, leading to the disruption of TSC1–TSC2 intracellular protein complex and to the hyperactivation of mTORC1.127 It was reported that TSC1/2 deficiency reflects in a reduced autophagy because of the inactivation of ULK1 operated by mTORC1 in non-neuronal cells. On the contrary, a recent study demonstrated that TSC1/2 loss in neurons determines an enhanced induction of autophagy via AMPK-dependent phosphorylation of ULK1 at Ser555. Thus, despite the concomitant inhibitory effect of mTORC1 on ULK1 activity, AMPK seems to be the dominant modulator of autophagy in brain cells.20 As a consequence, the accumulation of p62 and autolysosomes could contribute to the altered neuronal homeostasis in TSC disease.
Conclusion
The fine regulation of energy intake and expenditure is of great importance for maintaining the integrity of physiological processes in almost all eukaryotic cells. AMPK is the master regulator of metabolic stress and mediates pivotal adaptive changes as a function of nutrient deprivation. For these reasons, AMPK is considered the leading actor in several pathologies such as obesity, diabetes, cachexia, and other metabolic disorders. Recently, increasing evidence sustains a key role for AMPK also in brain physiopathology. AMPK is involved in a variety of neuronal processes, such as neuronal proliferation, differentiation, and synaptic transmission.
It is possible that alterations in AMPK activity could reflect in the development of neurological disorders (Figure 4). The studies reviewed here highlight intriguing relationships between AMPK and neurological diseases such as stroke, AD, HD, PD, and brain tumors, whereas other findings show emerging roles for this protein kinase in the development of mental disabilities in genetic disorders. However, even though a great knowledge about the pathological role of AMPK in the brain has been reached, literature data is far from being convincing. AMPK does not show a unique behavior in neurodegenerative diseases, and both detrimental and protective roles have been hypothesized. Indeed, while AMPK activation mediates mHtt-induced toxicity in HD striatal neurons, it prevents neuronal cell death and acts as a survival factor in PD. The role of AMPK is ambiguous in AD pathogenesis, as some reports indicate that AMPK activation is strongly associated with a reduction in amyloidogenesis, whereas other studies demonstrate that AMPK inhibition corrects the Aβ-induced impairments in LTP.74,78 In the same way, many studies have described the divergent evidence about the involvement of AMPK in brain tumors and ischemic stroke. The presence of contradictory results indicates that further investigations are needed to better dissect the molecular mechanisms linking AMPK activity and the development of CNS diseases. A deep comprehension could provide useful information for designing novel and effective therapeutic strategies addressing a variety of neurological conditions.
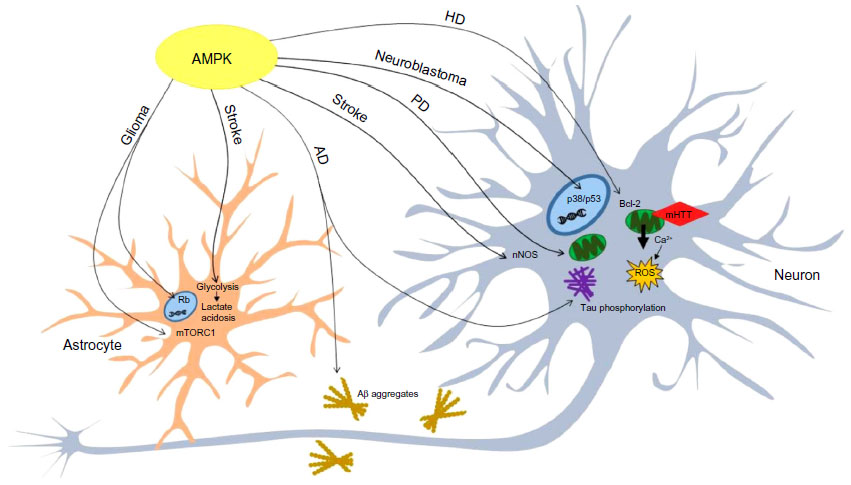 | Figure 4 AMPK is involved in the manifestation of neurological conditions. |
Disclosure
The authors report no conflicts of interest in this work.
References
Scott JW, Hawley SA, Green KA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113(2):274–284. | |
Townley R, Shapiro L. Crystal structures of the adenylate sensor from fission yeast AMP-activated protein kinase. Science. 2007;315(5819):1726–1729. | |
Xiao B, Heath R, Saiu P, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449(7161):496–500. | |
Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–2008. | |
Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895–1908. | |
Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19. | |
Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33. | |
Moussa A, Li J. AMPK in myocardial infarction and diabetes: the yin/yang effect. Acta Pharm Sin B. 2(4):368–378. | |
Salt IP, Johnson G, Ashcroft SJ, Hardie DG. AMP-activated protein kinase is activated by low glucose in cell lines derived from pancreatic beta cells, and may regulate insulin release. Biochem J. 1998;335(Pt 3):533–539. | |
Lamia KA, Sachdeva UM, DiTacchio L, et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science. 2009;326(5951):437–440. | |
Oakhill JS, Chen ZP, Scott JW, et al. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci U S A. 2010;107(45):19237–19241. | |
Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72(4):1707–1716. | |
He G, Zhang YW, Lee JH, et al. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol. 2014;34(2):148–157. | |
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. | |
Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30(2):214–226. | |
Chan EY. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci Signal. 2009;2(84):pe51. | |
Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. | |
Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J. 2011;440(2):283–291. | |
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13(9):1016–1023. | |
Di Nardo A, Wertz MH, Kwiatkowski E, et al. Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum Mol Genet. 2014;23(14):3865–3874. | |
Lee JW, Park S, Takahashi Y, Wang HG. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394. | |
Kume S, Thomas MC, Koya D. Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes. 2012;61(1):23–29. | |
Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. | |
Tian W, Li W, Chen Y, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett. 2015;589(15):1847–1854. | |
Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. | |
Sanchez AM, Csibi A, Raibon A, et al. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012;113(2):695–710. | |
Greer EL, Dowlatshahi D, Banko MR, et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17(19):1646–1656. | |
Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223(2):217–222. | |
Sato R, Goldstein JL, Brown MS. Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc Natl Acad Sci U S A. 1993;90(20):9261–9265. | |
Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89(3):1025–1078. | |
Watt MJ, Holmes AG, Pinnamaneni SK, et al. Regulation of HSL serine phosphorylation in skeletal muscle and adipose tissue. Am J Physiol Endocrinol Metab. 2006;290(3):E500–E508. | |
Ahmadian M, Abbott MJ, Tang T, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13(6):739–748. | |
Das SK, Eder S, Schauer S, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333(6039):233–238. | |
Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13(4):376–388. | |
Vilchez D, Ros S, Cifuentes D, et al. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10(11):1407–1413. | |
Weisova P, Concannon CG, Devocelle M, Prehn JH, Ward MW. Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J Neurosci. 2009;29(9):2997–3008. | |
Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104(17):7217–7222. | |
Onyango IG, Lu J, Rodova M, Lezi E, Crafter AB, Swerdlow RH. Regulation of neuron mitochondrial biogenesis and relevance to brain health. Biochim Biophys Acta. 2009;1802(1):228–234. | |
Lee JH, Koh H, Kim M, et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature. 2007;447(7147):1017–1020. | |
Dasgupta B, Milbrandt J. AMP-activated protein kinase phosphorylates retinoblastoma protein to control mammalian brain development. Dev Cell. 2009;16(2):256–270. | |
Amato S, Liu X, Zheng B, Cantley L, Rakic P, Man HY. AMP-activated protein kinase regulates neuronal polarization by interfering with PI 3-kinase localization. Science. 2011;332(6026):247–251. | |
Ramamurthy S, Chang E, Cao Y, Zhu J, Ronnett GV. AMPK activation regulates neuronal structure in developing hippocampal neurons. Neuroscience. 2014;259:13–24. | |
Ulgherait M, Rana A, Rera M, Graniel J, Walker DW. AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 2014;8(6):1767–1780. | |
Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444(7121):881–887. | |
Williams G, Harrold JA, Cutler DJ. The hypothalamus and the regulation of energy homeostasis: lifting the lid on a black box. Proc Nutr Soc. 2000;59(3):385–396. | |
Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443(7109):289–295. | |
Sakurai T. Orexins and orexin receptors: implication in feeding behavior. Regul Pept. 1999;85(1):25–30. | |
Kristensen P, Judge ME, Thim L, et al. Hypothalamic CART is a new anorectic peptide regulated by leptin. Nature. 1998;393(6680):72–76. | |
Hardie DG, Ashford ML. AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda). 2014;29(2):99–107. | |
Xue B, Kahn BB. AMPK integrates nutrient and hormonal signals to regulate food intake and energy balance through effects in the hypothalamus and peripheral tissues. J Physiol. 2006;574(Pt 1):73–83. | |
Stark R, Ashley SE, Andrews ZB. AMPK and the neuroendocrine regulation of appetite and energy expenditure. Mol Cell Endocrinol. 2013;366(2):215–223. | |
Blanco Martinez de Morentin P, Gonzalez CR, Saha AK, et al. Hypothalamic AMP-activated protein kinase as a mediator of whole body energy balance. Rev Endocr Metab Disord. 2011;12(3):127–140. | |
Jamshidi N, Taylor DA. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol. 2001;134(6):1151–1154. | |
Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol. 2006;494(3):528–548. | |
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50(8):1714–1719. | |
Cummings DE, Weigle DS, Frayo RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346(21):1623–1630. | |
Claret M, Smith MA, Batterham RL, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117(8):2325–2336. | |
Kohno D, Sone H, Minokoshi Y, Yada T. Ghrelin raises [Ca2+]i via AMPK in hypothalamic arcuate nucleus NPY neurons. Biochem Biophys Res Commun. 2008;366(2):388–392. | |
Kubota N, Yano W, Kubota T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6(1):55–68. | |
Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569–574. | |
Lopez M, Varela L, Vazquez MJ, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med. 2010;16(0):1001–1008. | |
Ropelle ER, Fernandes MF, Flores MB, et al. Central exercise action increases the AMPK and mTOR response to leptin. PLoS One. 2008;3(12):e3856. | |
Ropelle ER, Pauli JR, Fernandes MF, et al. A central role for neuronal AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) in high-protein diet-induced weight loss. Diabetes. 2008;57(3):594–605. | |
Cota D, Proulx K, Smith KA, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312(5775):927–930. | |
Blouet C, Jo YH, Li X, Schwartz GJ. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J Neurosci. 2009;29(26):8302–8311. | |
Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010; 362(4):329–344. | |
Cai Z, Yan LJ, Li K, Quazi SH, Zhao B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med. 2012;14(1):1–14. | |
Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. | |
Cole SL, Vassar R. The role of amyloid precursor protein processing by BACE1, the beta-secretase, in Alzheimer disease pathophysiology. J Biol Chem. 2008;283(44):29621–29625. | |
Vingtdeux V, Davies P, Dickson DW, Marambaud P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011; 121(3):337–349. | |
Mairet-Coello G, Courchet J, Pieraut S, Courchet V, Maximov A, Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78(1):94–108. | |
Lopez-Lopez C, Dietrich MO, Metzger F, Loetscher H, Torres-Aleman I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer’s disease. J Neurosci. 2007;27(4):824–831. | |
Vingtdeux V, Giliberto L, Zhao H, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem. 2010;285(12):9100–9113. | |
Won JS, Im YB, Kim J, Singh AK, Singh I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem Biophys Res Commun. 2010;399(4):487–491. | |
Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380(1):98–104. | |
DiTacchio KA, Heinemann SF, Dziewczapolski G. Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 2015;44(1):43–48. | |
Chen Y, Zhou K, Wang R, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci U S A. 2009;106(10):3907–3912. | |
Ma T, Chen Y, Vingtdeux V, et al. Inhibition of AMP-activated protein kinase signaling alleviates impairments in hippocampal synaptic plasticity induced by amyloid beta. J Neurosci. 2014;34(36):12230–12238. | |
Ju TC, Chen HM, Lin JT, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol. 2011;194(2):209–227. | |
Costa V, Giacomello M, Hudec R, et al. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med. 2010;2(12):490–503. | |
Liu YJ, Chern Y. AMPK-mediated regulation of neuronal metabolism and function in brain diseases. J Neurogenet. 2015;(2–3):1–9. | |
Chou SY, Lee YC, Chen HM, et al. CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem. 2005;93(2):310–320. | |
Corti O, Lesage S, Brice A. What genetics tells us about the causes and mechanisms of Parkinson’s disease. Physiol Rev. 2011;91(4):1161–1218. | |
Ng CH, Guan MS, Koh C, et al. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J Neurosci. 2012;32(41):14311–14317. | |
Hang L, Thundyil J, Lim KL. Mitochondrial dysfunction and Parkinson disease: a Parkin-AMPK alliance in neuroprotection. Ann N Y Acad Sci. 2015;1350(1):37–47. | |
Choi JS, Park C, Jeong JW. AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem Biophys Res Commun. 2010;391(1):147–151. | |
Kim TW, Cho HM, Choi SY, et al. (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis. 2013;4:e919. | |
Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8(6):519–530. | |
Appin CL, Brat DJ. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol. 2015;22(1):50–58. | |
Rios M, Foretz M, Viollet B, et al. AMPK activation by oncogenesis is required to maintain cancer cell proliferation in astrocytic tumors. Cancer Res. 2013;73(8):2628–2638. | |
Jang T, Calaoagan JM, Kwon E, Samuelsson S, Recht L, Laderoute KR. 5′-AMP-activated protein kinase activity is elevated early during primary brain tumor development in the rat. Int J Cancer. 2011;128(9):2230–2239. | |
Chen JH, Huang SM, Chen CC, et al. Ghrelin induces cell migration through GHS-R, CaMKII, AMPK, and NF-kappaB signaling pathway in glioma cells. J Cell Biochem. 2011;112(10):2931–2941. | |
Neurath KM, Keough MP, Mikkelsen T, Claffey KP. AMP-dependent protein kinase alpha 2 isoform promotes hypoxia-induced VEGF expression in human glioblastoma. Glia. 2006;53(7):733–743. | |
Hu YL, DeLay M, Jahangiri A, et al. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012;72(7):1773–1783. | |
Guo D, Cloughesy TF, Radu CG, Mischel PS. AMPK: a metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle. 2009;9(2):211–212. | |
Mukherjee P, Mulrooney TJ, Marsh J, Blair D, Chiles TC, Seyfried TN. Differential effects of energy stress on AMPK phosphorylation and apoptosis in experimental brain tumor and normal brain. Mol Cancer. 2008;7:37. | |
Sesen J, Dahan P, Scotland SJ, et al. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS One. 2015;10(4):e0123721. | |
Liu X, Chhipa RR, Pooya S, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A. 2014;111(4):E435–E444. | |
Liu X, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther. 2014;13(3):596–605. | |
Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. | |
Jung JE, Lee J, Ha J, et al. 5-Aminoimidazole-4-carboxamide-ribonucleoside enhances oxidative stress-induced apoptosis through activation of nuclear factor-kappaB in mouse Neuro 2a neuroblastoma cells. Neurosci Lett. 2004;354(3):197–200. | |
Filomeni G, Cardaci S, Da Costa Ferreira AM, Rotilio G, Ciriolo MR. Metabolic oxidative stress elicited by the copper(II) complex [Cu(isaepy)2] triggers apoptosis in SH-SY5Y cells through the induction of the AMP-activated protein kinase/p38MAPK/p53 signalling axis: evidence for a combined use with 3-bromopyruvate in neuroblastoma treatment. Biochem J. 2011;437(3):443–453. | |
Mathers CD, Boerma T, Ma Fat D. Global and regional causes of death. Br Med Bull. 2009;92:7–32. | |
Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003; 144(12):5179–5183. | |
Almeida A, Moncada S, Bolanos JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6(1):45–51. | |
Ronnett GV, Ramamurthy S, Kleman AM, Landree LE, Aja S. AMPK in the brain: its roles in energy balance and neuroprotection. J Neurochem. 2009;109 Suppl 1:17–23. | |
McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280(21):20493–20502. | |
Khan M, Dhammu TS, Matsuda F, Singh AK, Singh I. Blocking a vicious cycle nNOS/peroxynitrite/AMPK by S-nitrosoglutathione: implication for stroke therapy. BMC Neurosci. 2015;16:42. | |
Li J, Zeng Z, Viollet B, Ronnett GV, McCullough LD. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke. 2007;38(11):2992–2999. | |
Cheng YY, Leu HB, Chen TJ, et al. Metformin-inclusive therapy reduces the risk of stroke in patients with diabetes: a 4-year follow-up study. J Stroke Cerebrovasc Dis. 2014;23(2):e99–e105. | |
Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):854–865. | |
Selvin E, Bolen S, Yeh HC, et al. Cardiovascular outcomes in trials of oral diabetes medications: a systematic review. Arch Intern Med. 2008;168(19):2070–2080. | |
Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. | |
Venna VR, Li J, Benashski SE, Tarabishy S, McCullough LD. Preconditioning induces sustained neuroprotection by downregulation of adenosine 5′-monophosphate-activated protein kinase. Neuroscience. 2012;201:280–287. | |
Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J Cereb Blood Flow Metab. 2010;30(3):480–492. | |
Venna VR, Li J, Hammond MD, Mancini NS, McCullough LD. Chronic metformin treatment improves post-stroke angiogenesis and recovery after experimental stroke. Eur J Neurosci. 2014;39(12):2129–2138. | |
Li W, Qu Z, Prakash R, et al. Comparative analysis of the neurovascular injury and functional outcomes in experimental stroke models in diabetic Goto-Kakizaki rats. Brain Res. 2013;1541:106–114. | |
Nakatsu Y, Kotake Y, Hino A, Ohta S. Activation of AMP-activated protein kinase by tributyltin induces neuronal cell death. Toxicol Appl Pharmacol. 2008;230(3):358–363. | |
Gollob MH, Green MS, Tang AS, et al. Identification of a gene responsible for familial Wolff- Parkinson-White syndrome. N Engl J Med. 2001;344(24):1823–1831. | |
Blair E, Redwood C, Ashrafian H, et al. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum Mol Genet. 2001;10(11):1215–1220. | |
Burwinkel B, Scott JW, Buhrer C, et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet. 2005;76(6):1034–1049. | |
Kim E, Lee SH, Lee KS, et al. AMPK gamma2 subunit gene PRKAG2 polymorphism associated with cognitive impairment as well as diabetes in old age. Psychoneuroendocrinology. 2011;37(3):358–365. | |
Harvard C, Strong E, Mercier E, et al. Understanding the impact of 1q21.1 copy number variant. Orphanet J Rare Dis. 2011;6:54. | |
Qi S, Mizuno M, Yonezawa K, Nawa H, Takei N. Activation of mammalian target of rapamycin signaling in spatial learning. Neurosci Res. 2010;68(2):88–93. | |
Costain G, Lionel AC, Merico D, et al. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum Mol Genet. 2013;22(22):4485–4501. | |
Hosak L. New findings in the genetics of schizophrenia. World J Psychiatry. 2013;3(3):57–61. | |
Curatolo P, Moavero R, de Vries PJ. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015;14(7):733–745. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.