Back to Journals » Journal of Pain Research » Volume 12
Ameliorative Effects Of N-Acetylcysteine As Adjunct Therapy On Symptoms Of Painful Diabetic Neuropathy
Authors Heidari N, Sajedi F ![]() , Mohammadi Y
, Mohammadi Y ![]() , Mirjalili M
, Mirjalili M ![]() , Mehrpooya M
, Mehrpooya M
Received 22 August 2019
Accepted for publication 1 November 2019
Published 19 November 2019 Volume 2019:12 Pages 3147—3159
DOI https://doi.org/10.2147/JPR.S228255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Narges Heidari,1 Firozeh Sajedi,2 Younes Mohammadi,3 Mahtabalsadat Mirjalili,4 Maryam Mehrpooya1
1Department of Clinical Pharmacy, School of Pharmacy, Hamadan University of Medical Sciences, Hamadan, Iran; 2Department of Internal Medicine, School of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran; 3Modeling of Noncommunicable Diseases Research Center, School of Public Health, Hamadan University of Medical Sciences, Hamadan, Iran; 4Department of Clinical Pharmacy, School of Pharmacy, Shiraz University of Medical Sciences, Shiraz, Iran
Correspondence: Maryam Mehrpooya
Department of Clinical Pharmacy, School of Pharmacy, Hamadan University of Medical Sciences, Shahid Fahmideh Ave, Hamadan 6517838678, Iran
Tel +98813821868
Fax +988138381591
Email [email protected]
Purpose: Painful diabetic neuropathy (PDN) is a variant of diabetic peripheral neuropathy which is highly prevalent and distressing in diabetic patients. Despite its high burden, the optimal treatment of PDN has remained a clinical challenge. To explain the emergence and maintenance of PDN, increasing attention has been focused on dimensions of inflammation and oxidative toxic stress (OTS). Accordingly, the aim of this study was to investigate the effects of oral N-acetylcysteine (NAC), an agent with known anti-oxidant and anti-inflammatory effects, as an adjunct therapy in patients suffering from PDN.
Patients and methods: 113 eligible patients with type 2 diabetes suffering from PDN were randomly assigned to either the pregabalin + placebo or pregabalin + NAC group for 8 weeks (pregabalin at a dose of 150 mg per day, NAC and matched placebo at doses of 600 mg twice a day). Mean pain score was evaluated at baseline, week 1, 2, 4, 6, and 8 of the study based on the mean 24 hr average pain score, using an 11-point numeric rating scale (NRS). As secondary efficacy measures, mean sleep interference score (SIS) resulting from PDN, responder rates, Patient Global Impression of Change (PGIC), Clinical Global Impression of Change (CGIC), and safety were also assessed. Additionally, serum levels of total antioxidant capacity (TAC), total thiol groups (TTG), catalase activity (CAT), glutathione peroxidase (GPx), superoxide dismutase (SOD), nitric oxide (NO), and malondialdehyde (MDA) were assessed at baseline and at the end of the study.
Results: Ninety patients completed the eight-week course of the study. The decrease in mean pain scores and mean sleep interference score in pregabalin + NAC group was greater in comparison with pregabalin + placebo group (p value<0.001 in both conditions). Moreover, more responders (defined as ≥50% reduction in mean pain score from baseline to end-point) were observed in the pregabalin + NAC group, in comparison with pregabalin + placebo group (72.1% vs 46.8%). More improvement in PGIC and CGIC from baseline to the end of the study was reported in pregabalin + NAC group. Oral NAC had minimal adverse effects and was well tolerated in almost all patients. Furthermore, in respect to OTS biomarkers, adjuvant NAC significantly decreased serum level of MDA and significantly increased serum levels of SOD, GPx, TAC, and TTG.
Conclusion: The pattern of results suggests that compared to placebo and over a time period of 8 weeks, adjuvant NAC is more efficacious in improving neuropathic pain associated with diabetic neuropathy than placebo. Ameliorative effects of NAC on OTS biomarkers demonstrated that NAC may alleviate painful symptoms of diabetic neuropathy, at least in part by its antioxidant effects.
Keywords: painful diabetic neuropathy, oxidative stress, N-acetylcysteine, pregabalin
Introduction
Diabetic neuropathy is the most common long-term microvascular complication of diabetes mellitus (DM), present in up to 50% of all diabetic patients with a long-standing disease. Diabetic neuropathy is a heterogeneous set of clinical or subclinical manifestations which is associated with loss of peripheral nerve fibers.1 Distal symmetric polyneuropathy is the most prevalent form of diabetic neuropathy which is characterized by a progressive loss of distal sensation. Up to 50% of patients with distal symmetric polyneuropathy may experience sensory symptoms, either negative or positive ones. Pain is one of the most frequent symptoms in diabetic neuropathy, which is called painful diabetic neuropathy (PDN).2 PDN is characterized by burning, tingling (“pins and needles” or paresthesia), shooting (electric-shock like), lancing (stabbing) or unusual sensations. The severity of pain is moderate to severe in most patients and typically worsens during the night, resulting in sleep disturbance.3 The constant and unremitting nature of the pain can be distressing and can have a major impact on quality of life of patients and it has a negative influence on their mood and their ability to perform daily activities.4
Despite its high burden, the optimal treatment of PDN, because of its unclear mechanisms, has remained a clinical challenge. Although near-normoglycemia is generally accepted as the first approach in prevention of PDN, tricyclic agents (TCAs), serotonin–norepinephrine reuptake inhibitors (SNRIs) or γ-aminobutyric acid (GABA) analogs (gabapentin or pregabalin), followed by opioids and topical treatments are frequently used for symptomatic management of PDN. However, at best they are only partially effective because they do not influence the underlying pathologies and usually have several adverse effects.5,6 An understanding of pathomechanisms responsible for PDN pathogenesis can be helpful in introducing new treatment strategies.
The exact pathophysiological mechanisms of PDN are not fully understood and it is very likely to be multifactorial. Changes in the blood vessels that supply the peripheral nerves, metabolic and autoimmune disorders accompanied by glial cell activation, changes in sodium and calcium channels' expression, and central pain mechanisms such as increased thalamic vascularity and imbalance of the facilitatory/inhibitory descending pathways, may be potential mechanisms responsible for the development of PDN.7
Findings from numerous studies have implicated that oxidative toxic stress (OTS) and neuroinflammation play a critical role in the development and progression of diabetic neuropathy.8 Hyperglycemia leads to overproduction of reactive oxygen and nitrogen species (ROS/RNS) through activation of numerous metabolic pathways such as polyol, protein kinase C (PKC), advanced glycation end products (AGE), and hexosamine pathways. Increased free-radical production, along with defective antioxidant mechanisms cause OTS and subsequent neuronal damage. Furthermore, OTS enhances the production of various proinflammatory mediators via inducing activation of transcription factors, such as nuclear factor kappa enhancer of B cells (NF-κB), leading to neuronal damage.9 In addition to these mechanisms, impaired mitochondrial function stands at a central position in the pathogenesis of diabetic neuropathy. It has been hypothesized that high glucose concentration increases production of ROS/RNS through driving excessive electron donation to the respiratory chain in mitochondria, causing mitochondrial dysfunction. On the other hand, mitochondrial dysfunction has the ability to induce OTS and aggravate progression of nerve damage via increasing ROS/RNS production.10,11 Impaired calcium homeostasis due to mitochondrial dysfunction is also associated with overproduction of ROS/RNS.12,13 Increasing cell apoptosis and defects in axon transport resulting from mitochondrial dysfunction may be other contributing factors which augment these pathological conditions.14,15 Moreover, recent studies revealed that during the hyperglycemic diabetic state, activation and expression of matrix metalloproteinase (MMPs), a group of metal-dependent endopeptidases which have an important role in extracellular matrix (ECM) remodeling, specifically MMP-2 and MMP-9, are triggered by various pathogenic mediators, including ROS/RNS and proinflammatory cytokines. MMPs' overactivity can lead to extracellular matrix (ECM) disturbances which alter neuronal structures and functions directly or indirectly.16,17
Thus, it seems that activation of various pathogenic pathways can cumulatively cause structural nerve damage. Eventually, the damage to the nerves causes hyperexcitability in the peripheral and central neurons, causing the generation of spontaneous impulses in the axons and the dorsal root ganglia of the nerves, which leads to precipitation of the neuropathic pain.18
These causative mechanisms suggest the possible role of anti-oxidant and anti-inflammatory agents in the treatment of PDN. Within the past decade, the effectiveness of several antioxidant agents, such as alpha lipoic acid, vitamin E, and acetyl-L-carnitine has been investigated in patients with PDN,19–21 among which alpha-lipoic acid was shown to have therapeutic efficacy.22 In addition, natural agents such as curcumin, resveratrol, and melatonin, as well as mitochondria targeted antioxidants have shown promising effects in the preclinical models of diabetic neuropathy.23,24
One candidate molecule known to affect all these mentioned pathological pathways is N-acetylcysteine (NAC). NAC is a cysteine prodrug and glutathione (GSH) precursor with well-known anti-oxidant and anti-inflammatory properties.25 NAC exhibits potent anti-oxidant activity in the cell through augmentation of intracellular glutathione (GSH), which is a major component of the pathways by which cells are protected from OTS, and its direct scavenging activity of free radicals by providing sulfhydryl groups.26 Additionally, NAC treatment exhibits anti-inflammatory effects via inhibition of NF-κB activation and reducing subsequent cytokine production.27 Mitochondria-protective mechanisms of NAC may also be related to its anti-oxidant and anti-inflammatory properties.28 Moreover, some recent evidence showed NAC can induce analgesia in animal models of inflammatory and neuropathic pain via suppression of MMPs and inhibition of nociceptive responses.29,30 As a result, these multiple mechanisms of action have raised the possibility that NAC might be potentially useful for managing PDN.
To the best our knowledge, there has been no clinical study exploring the influence of oral NAC on management of PDN. Hence, the aim of this study was to evaluate the effects of oral NAC as adjunct therapy on pain management in patients with type 2 diabetes suffering from PDN. We also investigated the modulatory effects of oral NAC on serum levels of OTS biomarkers, as the main mechanism of its action in management of PDN.
Materials And Methods
This was a randomized, double-blind, 8-week placebo-controlled clinical trial study which was conducted in an outpatient specialty clinic affiliated to Hamadan University of Medical Sciences, Hamadan, Iran, from August 2018 until June 2019. Eligible patients were fully informed about the study aims and the confidential and anonymous data handling. Participants signed the written informed consent, and were randomly assigned either to the intervention or the control group. The Ethics Committee of Hamadan University of Medical Sciences approved the study protocol, which was performed in accordance with the rules laid down in the Declaration of Helsinki and its later amendments. The study was registered at the Iranian Registry of Clinical Trials (IRCT20150629022965N19; www.irct.ir).
Participants
Inclusion criteria at the screening visit were as follows: males or females between the ages of 18 and 75 years old, diagnosed with type 2 DM for ≥1 year, HbA1c (A1C) <10%, stable antidiabetic treatment regimen ≥1 month and maintaining the same treatment regimen during the study, painful distal symmetrical and sensorimotor polyneuropathy attributable to DM ≥3 months, score of ≥40 mm on the visual analog scale of the short-form McGill Pain Questionnaire (SF-MPQ), and average daily pain score ≥4 based on an 11-point (0–10) numeric rating scale (NRS) during the last week before beginning study. At baseline and during the study period patients were excluded if any of the following exclusion criteria was met: creatinine clearance (CLcr) ≤ 30 mL/min calculated according to the Cockcroft & Gault formula,31 patients with acute and chronic inflammatory conditions, consuming any antioxidant supplements or anti-inflammatory medicines other than prescribed medications, pregnancy or lactation or expecting to get pregnant during the study, medical, psychological, or pharmacological factors interfering with the collection or interpretation of study data, non-adherent to the treatment (using the medication for less than 80% of study period), and presence of any adverse effects resulting in patients’ intolerance or complications.
Intervention
113 patients who met the inclusion/exclusion criteria were allocated to either the pregabalin + NAC treatment group (intervention group; n=57) or pregabalin + placebo treatment group (control group; n=58) by block randomization method using a block size of 4 in a 1:1 ratio. The randomization was provided by an independent statistician to ensure that groups were matched for age, sex, and body mass index (BMI) where possible. Also, both the study participants and the investigators were blinded to the study medication.
All subjects were taken off any pain medication for two weeks before participating in the trial. After this washout period, the study patients were assigned to receive either pregabalin + placebo or pregabalin + NAC for 8 weeks. Pregabalin tablets at a dose of 150 mg per day at bed time were administered to all patients. Additionally, subjects according to their group allocation, were instructed to take 600 mg effervescent tablet of NAC or placebo twice a day for 8 weeks. Placebo tablets were visually identical to the NAC tablets and were prepared and packaged by the manufacturer of the NAC tablets (Osveh Pharmaceutical Company, Tehran, Iran). Adherence to treatment was determined by counting drugs left in the container at the end of each visit and patients were considered adherent to treatment if at least 80% of all doses were taken.
Efficacy Measures
The primary efficacy variable was weekly mean pain score which was evaluated based on the mean 24 hr average pain score reported in the daily patient diary. In the daily pain diary, patients described their pain during the previous 24 hrs using an 11-point NRS (0 = “no pain” and 10 = “worst possible pain”).
Secondary efficacy end-points included responder rates (defined as ≥50% reduction in mean pain score from baseline to end-point) and weekly mean sleep interference score (SIS) to evaluate the degree of sleep interference resulting from PDN. Similar to the patient’s pain score, sleep score was recorded by the patients daily in their daily sleep diaries on an 11-point NRS (0=“does not interfere with sleep” and 10=“completely interferes/unable to sleep”). Additionally, Patient Global Impression of Change (PGIC) and Clinical Global Impression of Change (CGIC) were recorded at the end of the study. PGIC and CGIC are self-evaluation and physician’s evaluation of the patient’s overall change since the start of the study on a 7-point scale (1 = very much improved; 2 = much improved; 3 = minimally improved; 4 = no change; 5 = minimally worse; 6 = much worse; 7 = very much worse).32
To evaluate the adverse effects of medications, all patients were asked at each visit if they had experienced any possible adverse effects and type and severity of adverse effects were recorded.
Measurement Of Inflammatory And OTS Biomarkers
5 mL fasting blood samples were taken from all recruited patients at the baseline and at the end of 8-week treatment period. Samples were centrifuged for 10 min. Serum specimen was separated and stored at −70°C until completion of all the samples. All samples were assessed in duplicate.
TAC, as a total antioxidant capacity, was measured by the ferric-reducing ability of plasma (FRAP method) and was expressed as micromoles per liter (µmol/L).33 Total thiol groups (TTG), as a major portion of the total body antioxidants, was measured by spectrophotometric assay based on Ellman’s method and was expressed as micromoles per liter (μmol/L).34 Superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase (CAT) serum activities were determined using spectrophotometry according to commercially available kits (Zelbio co., Germany) and serum GPx and SOD levels were presented as units per milliliter serum (U/mL) and CAT levels as milliUnits per milliliter serum (mU/mL). Malondialdehyde (MDA), as an indicator of final products of lipid peroxidation was measured using the thiobarbituric acid-reactive substances method, as described by Botsoglou,35 and was expressed as nmol/mL. Nitric oxide (NO), as main reactive nitrogen species was measured by using commercially available kits from Cayman Chemical Company based on determining the total nitrate/nitrite level and results were expressed as μmol/L.
Statistical Analyses
Per protocol analysis was exploited to analyze data of all individuals who completed the study. Data were analyzed using the SPSS for windows (SPSS Inc., Chicago, IL, USA) version 16 software. Mean ± standard deviation (SD) was used to express continuous variables. Categorical variables were reported as percentages. Mean (SD) of continuous variables was compared between two groups using independent t-test and the distribution of categorical variables between two groups was compared using Chi-squared or Fisher's exact test (if more than 20% of the categories were expected to have frequencies less than 5). Moreover, to compare means of variables including mean pain score and mean sleep interference score between two groups over time, as dependent variables, and control of their baseline differences, General Linear Model (GLM) ANOVA repeated measure was used to analyze data. Because of the deviation from sphericity assumption, we used Greenhouse-Geisser correction to perform ANOVA results. Moreover, effect sizes were extracted from Partial Eta Square (PES). Accordingly, we considered effect size as Small [S] if PES was less than 0.06, Medium [M] if PES was 0.06 <PES<0.13, and Large [L] if PES was greater than 0.14. P-values less than 0.05 were considered as significant.
Results
Totally, 113 patients who met the inclusion/exclusion criteria were divided into either intervention or control group by block randomization method. Twenty-three patients were excluded from the study due to experiencing intolerable adverse effects (7 patients), loss to follow up (9 patients), and using the medication for less than 80% of study period (7 patients). Ten patients in pregabalin + placebo group and 13 patients in pregabalin + NAC group were withdrawn from the study. Consequently, 90 patients completed the trial period (47 patients in pregabalin + placebo group and 43 patients in pregabalin + NAC group). As mentioned in the statistical analysis, all of the following results were related to 90 patients who completed the 8 weeks of the study. Figure 1 shows the flow diagram of trial participants.
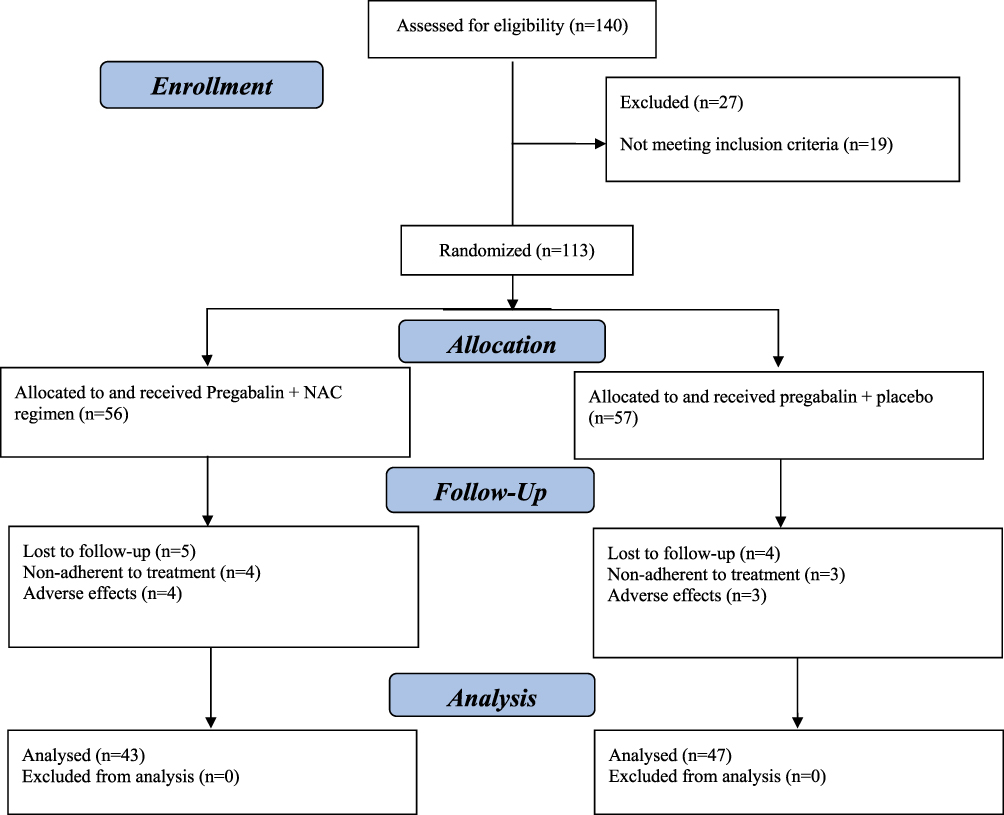 |
Figure 1 The flow diagram of the study. |
There were no significant differences between intervention group and control group regarding demographic characteristics. Of the included patients, 25.56% (23 patients) were male and 74.44% (67 patients) were female and the gender distribution was in favor of females in both groups (80.9% and 83.7% in control and intervention groups, respectively). The patients’ age ranged from 36–70 years with the mean ± SD age of 58.09 ± 7.90 years. The mean duration of diabetes and PDN in the study patients was 9.5 ± 5.9 years (ranged from 1–23 years) and 16.5 ± 12.4 months (ranged from 4–72 months), respectively. Concomitant medications for diabetes control were also similar between treatment groups (P-value = 0.31). Nearly half of the participants used insulin therapy for blood glucose control. The demographic and clinical data of the patients are shown in Table 1.
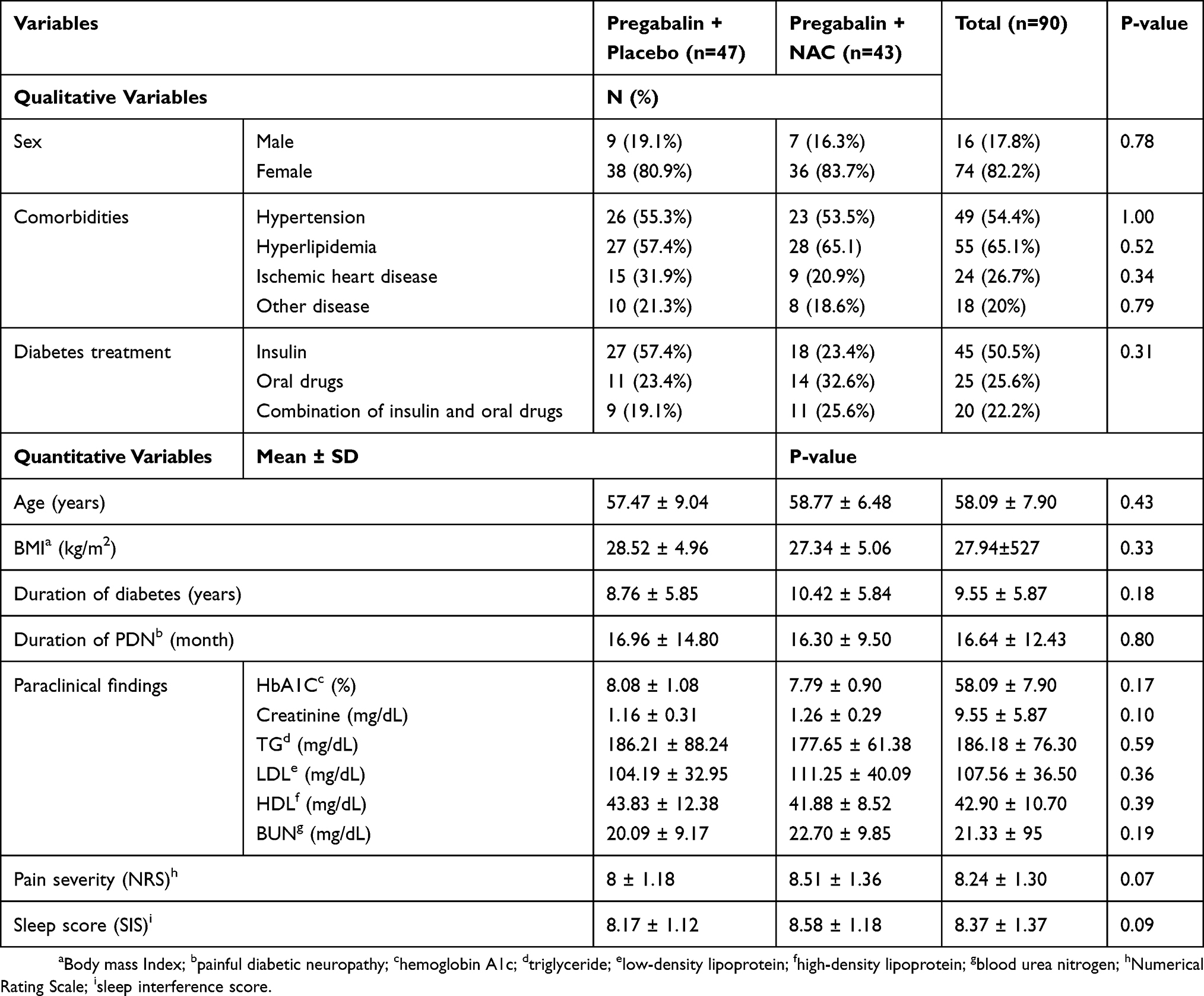 |
Table 1 Patient Demographics And Baseline Characteristics |
Primary Efficacy Measure
The study groups were similar with respect to the 24 hr NRS average pain score at baseline (8 ± 1.18 in pregabalin + placebo group and 8.51 ± 1.36 in pregabalin + NAC group; P-value=0.06) (Table 1). Although there was a statistically significant decrease in 24 hr NRS average pain score in both groups over time (P-value<0.001 in both groups), the mean change in 24 hr NRS average pain score at the end of the study period (week 8) was significantly greater in pregabalin + NAC group compared with pregabalin + placebo group (−5.20 ± 1.91 vs −3.45 ± 1.50; P-value <0.001) (Table 2). Also, the general linear model analysis demonstrated that relatively comparable decreases in mean pain scores were observed in both groups by the end of week 2, but thereafter until end of the study (week 8), the decreases in mean pain scores in pregabalin + NAC group were greater compared with pregabalin + placebo group with large treatment-effect sizes (F=14.5; df=2.9; effect size = 0.14; P-value<0.001) (Figure 2A). Therefore, combining pregabalin with NAC had significant effect on reducing the 24 hr NRS average pain score over time.
 |
Table 2 Mean Changes In NRS And SIS Scores From Baseline To Week 8 (Study End) By Treatment Group |
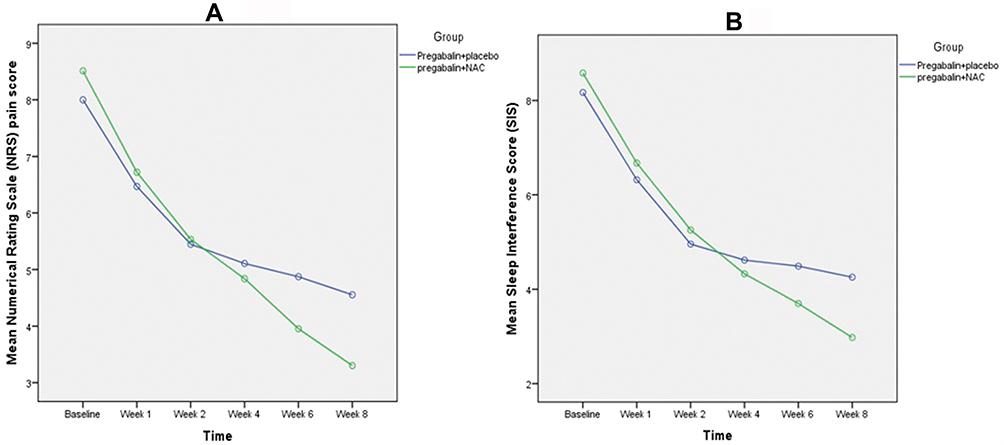 |
Figure 2 (A) Mean pain score as measured on an 11-point numerical pain rating scale from 0 (no pain) to 10 (worst possible pain) at different time points during study (P-value<0.001). (B) Mean sleep interference score as rated on an 11-point scale from 0 (did not interfere) to 10 (unable to sleep due to pain) at different time points during study (P-value<0.001). |
With respect to responder rate (defined as ≥50% reduction in mean pain score from baseline to end-point) at the end of the study period, 46.8% (22 out of 47 patients) in the pregabalin + placebo group compared to 72.1% in the pregabalin + NAC group (31 out of 43 patients) were responders, which was a statistically significant difference (P-value=0.02).
Secondary Efficacy Measures
At baseline, the mean sleep interference score resulting from PDN was comparable in the two treatment groups (Table 1). The mean sleep interference scores in both the pregabalin + NAC and pregabalin + placebo group showed an improvement from baseline to week 8, but the mean changes of sleep score from baseline to week 8 were more favorable for pregabalin + NAC treated patients compared to pregabalin + placebo treated patients (−5.60 ± 1.53 vs −3.91 ± 1.53; P-value <0.001) (Table 2). Also, as shown in Figure 2B the general linear model analysis demonstrated that between the 2 groups, treatment-effect size for mean differences of the sleep interference score was medium at week 8 (F=13.5; df =3.9; effect size = 0.13; P-value<0.001).
At the end of the study, the percentage of patients reporting “very much improved” in global impression of change (PGIC) was greater in the pregabalin + NAC group compared to the pregabalin + placebo group (39.5 vs 17%), with significant between-group difference (P-value=0.02) (Figure 3A). Also, the CGIC findings paralleled the PGIC results, with a statistically significant difference favoring pregabalin + NAC treatment modality (P-value=0.03) (Figure 3B). At the final visit, the physician reported “very much improved” and “much improved” in CGIC in 30.2% and 55.8% of the pregabalin + NAC treated patients and in 23.4% and 36.2% of the pregabalin + placebo treated patients.
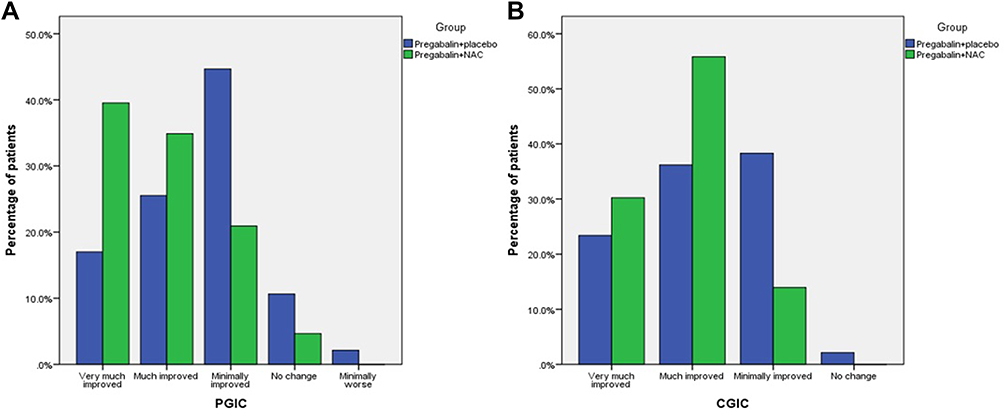 |
Figure 3 (A) Patient Global Impression of Change (PGIC), (P-value=0.03). (B) Clinical Global Impression of Change (CGIC), (P-value=0.02). |
Safety
A summary of the most common treatment-related adverse events has been shown in Table 3. The most commonly reported adverse effects by the study patients regardless of treatment group were somnolence and dizziness. All of the adverse events were mild or moderate in intensity and none of the reported adverse effects were serious or caused complication for the patients. Regardless of treatment group, patients generally tolerated the treatment well. As shown in Table 3, there were no significant differences regarding the incidence of adverse effects between the study groups. No reported adverse effects were significantly more common in the pregabalin + NAC group compared with the pregabalin + placebo group. Therefore, it seems that almost all the reported adverse effects were caused by pregabalin treatment and oral NAC was well tolerated, demonstrating comparable safety to placebo.
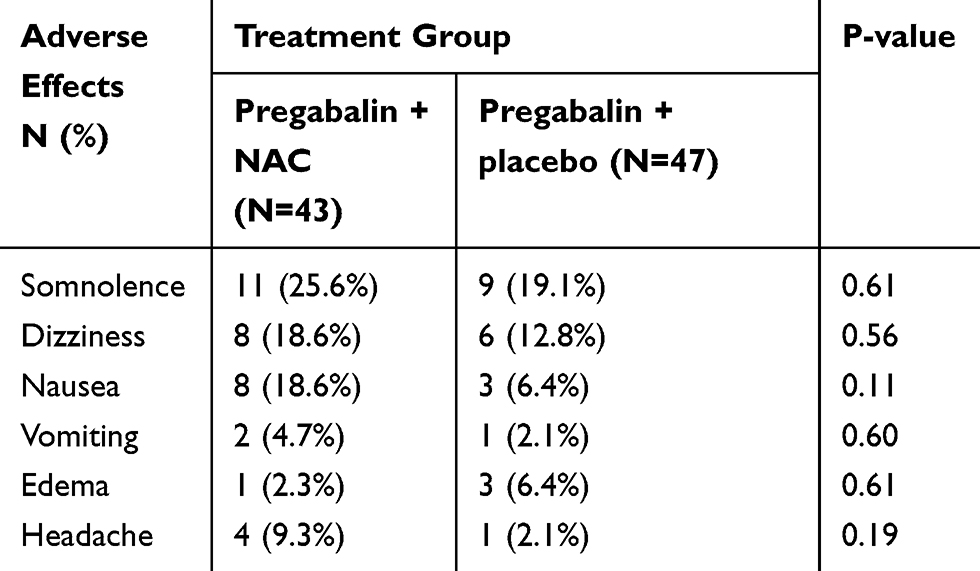 |
Table 3 Frequency Of Drug Related Adverse Effects Among Patients In Each Group |
Changes In Oxidative Stress Biomarkers; From Baseline To The End Of The Study, And Between The Groups
As shown in Table 4 at baseline, there were no significant differences in the serum levels of OTS biomarkers including TTG, TAC, GPx, CAT, SOD, NO, and MDA between the pregabalin + NAC and pregabalin + placebo groups. After 8 weeks of treatment, the serum levels of TTG, GPx, and SOD were significantly higher in the pregabalin + NAC group compared to the pregabalin + placebo group (P-value = 0.02, <0.001, and <0.001, respectively). Also, at 8th week of treatment, the MDA levels were significantly lower in the pregabalin + NAC group compared to the pregabalin + placebo group (P-value <0.001). Additionally, mean changes in the serum levels of TTG, TAC, MDA, GPx, and SOD from baseline to 8th week of the treatment showed significant difference between the two groups, in favor of the pregabalin + NAC group. Although after 8 weeks of treatment, mean changes of the serum levels of CAT and NO were more favorable in the pregabalin + NAC group compared to the pregabalin + placebo group, their changes did not display significant differences either in the inter- or the intra-group comparisons over time.
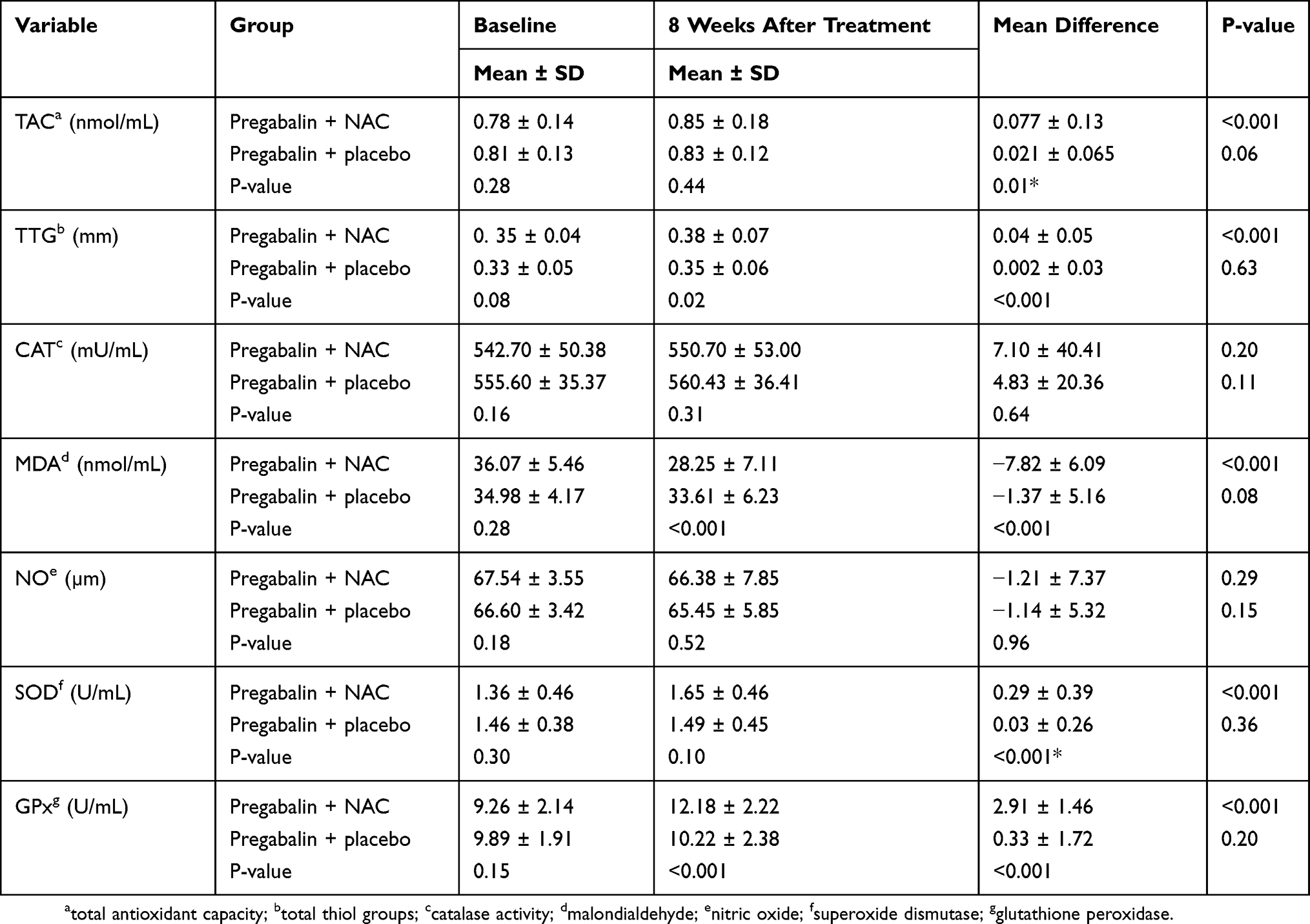 |
Table 4 The Serum Level Of OTS Biomarkers Of Two Groups, At Baseline And 8 Weeks After Treatment |
In summary, adjuvant NAC significantly decreased serum level of MDA and significantly increased serum activities of SOD and GPx, as well as serum levels of TAC and TTG.
Discussion
Our study was the first to evaluate the influence of NAC supplementation in patients with PDN in a randomized, double-blind and placebo-controlled study. The key findings of the present study were as follows: NAC as adjunct therapy is more efficacious in improving neuropathic pain associated with diabetic neuropathy compared to placebo, and proportion of patients responding to treatment was higher in the pregabalin + NAC group compared to the pregabalin + placebo group. Next, ameliorative effects of NAC on OTS biomarkers indicated that NAC may alleviate painful symptoms of diabetic neuropathy, at least in part by its antioxidant effects.
Around a fifth of people with DM develop PDN which can substantially impair quality of life in patients already burdened with chronic disease.36 Although a wide range of treatment options is available in patients with PDN, treatment outcomes are often unsatisfactory, due to partial effectiveness and associated side effects.37 On the other hand, current therapies for PDN aim to mask symptoms and do not influence the course of neuropathy; while pathogenetic treatments targeting the underlying molecular and cellular mechanisms involved in PDN, may delay, stop, or reverse the progression of neuropathy and may also alleviate pain.38 Despite the fact that the etiology of diabetic neuropathy has remained unclear, strong and growing evidence supports the important roles of mitochondrial dysfunction, OTS, and neuroinflammation in neuronal damage in diabetic patients. The dysregulation in these pathways by structural and functional changes at the level of both peripheral and central nervous systems can impair normal neuronal process, resulting in pain generation and maintenance.39–41 Considering these pathologic pathways, a large number of experimental studies have focused on the effects of antioxidant and anti-inflammatory agents in the prevention and treatment of diabetic neuropathy.42–46 These studies mostly demonstrated effectiveness of these agents in the prevention and treatment of neuropathic symptoms. However, there are limited numbers of published clinical studies regarding the use of antioxidants and anti-inflammatory agents in the treatment of diabetic neuropathy.44,47
NAC’s robust antioxidant and anti-inflammatory properties and its excellent safety profile have made it a reasonable choice in prevention and treatment of diabetic-associated complications. Previous studies have demonstrated the favorable effects of NAC supplementation on dyslipidemia and carbohydrate metabolism.48,49 Furthermore, NAC has shown protective effects against diabetes-associated cardiovascular complications and diabetic nephropathy.50,51 Recently, potential applications of NAC in prevention and treatment of peripheral neuropathy resulting from DM and other pathological conditions have been investigated in several experimental studies. In this regard, a study by Kunitomo et al showed that oral administration of NAC can alleviate thermal hyperalgesia in experimental diabetic neuropathy. They also found that supplementation with NAC can show protective effect against progression of diabetic neuropathy through attenuation of oxidative stress conditions and apoptosis.52 In another study, oral administration of NAC inhibited functional and structural abnormalities of the peripheral nerve in diabetic rats irrespective of blood glucose concentrations. This study concluded that protective effect of NAC on peripheral neurons is probably mediated through its modification effects on OTS and inflammatory biomarkers.53 Results of another study showed that overproduction of ROS plays a major role in cisplatin‐induced apoptotic neuronal cell death, and preincubation with NAC can decrease cisplatin-induced sensory neuropathy, probably via blocking the apoptosis pathways.54 Also, results of Naik et al’s study in the experimentally induced chronic constriction injury (CCI) of sciatic nerve of rats showed that endoneurial oxidative stress plays a critical role in generation of neuropathic pain in CCI model, and NAC can cause significant reduction in mechanical, thermal and cold hyperalgesia in CCI rats, probably through ROS scavenging.55 Similar to these findings, another study in an experimental model of CCI in rats revealed that the pain relieving effect of NAC may be related to its modulation effects on oxidative-stress parameters in the spinal cord.56 There are limited clinical trials about influence of NAC supplementation on treatment of neuropathic pain. In this regard, a pilot study on colorectal cancer patients receiving postoperative adjuvant oxaliplatin combined with fluorouracil chemotherapy regimen indicated that oral NAC can reduce the incidence of oxaliplatin-induced neuropathy in colon cancer patients.57 It seems that NAC can prevent oxaliplatin-induced neuropathy through increasing whole blood concentrations of glutathione and decreasing the accumulation of platinum metabolites within the peripheral nervous system.58 Also, in our previous clinical trial, we found that supplementation with NAC as adjuvant therapy to a standard medication significantly improved pain symptoms in patients suffering from rheumatoid arthritis (RA).59 Ameliorative effects of NAC on inflammatory and oxidative stress biomarkers may be the mechanism responsible for its beneficial effects on pain symptoms in RA patients.60 In accordance to these studies, the present study also provided further evidence that NAC can be efficacious in improving neuropathic pain associated with diabetic neuropathy, at least in part via ameliorative effects on oxidative stress.
Recent studies on neuropathic pain mechanisms have revealed that over-expression and activity of MMPs are also critical to the development of neuropathic pain. Increased MMPs' activity, especially MMP2 and MMP9, due to their facilitation of inflammatory cytokine maturation and induction of neural inflammation, is associated with neuronal injury, leading to precipitation of neuropathic pain.61 Therefore, MMPs inhibitors may be considered as a potential therapeutic strategy in the management of neuropathic pain. Activation of MMP-9/2 is dependent on the modification of the cysteine residue, and reaction of ROS with thiol groups can activate both MMP-2 and MMP-9.62 Furthermore, experimental studies suggest that proinflammatory cytokines, such as IL-1 and TNF-α can also stimulate the synthesis of MMPs.63 Therefore, the pharmacologic modalities that can prevent the oxidation of cysteine residue on MMP-9/2, such as NAC which contains abundant cysteine residues, may have potential ability to interfere with the activation of MMP-9/2. In this regard, results of a recent experimental study by Li et al on CCI-induced neuropathic pain in rats showed NAC significantly attenuated neuropathic pain through powerful inhibition of the activation of MMPs.29 Activation of MMP-9/2 also plays an important role in opioids-mediated nociception. Considering this fact, results of another study in a rat model of incisional pain demonstrated that NAC can attenuate the development of remifentanil-induced postoperative hyperalgesia via inhibiting MMP-9 activation in dorsal root ganglia.64
In addition to the pathological pathways mentioned previously, recent discoveries have shown that elevated glutamatergic neurotransmission in the CNS is associated with different types of pain, especially neuropathic pain.65 Therefore, it seems that ionotropic or metabotropic glutamate receptors can be considered as potential targets for treatment of neuropathic pain.66 Results of some recent experimental studies reported that an increase in expression or activation of type-2 metabotropic glutamate receptors (mGluR2) can produce antinociceptive effects in inflammatory and neuropathic pain models through reducing glutamate release from primary afferent sensory nerves.67,68 Preliminary evidence revealed that activation of the mGluR2 is another potential mechanism responsible for analgesic activity of NAC. Bernabucci et al in their recent study on mouse models of inflammatory and neuropathic pain, found that NAC could cause analgesia via reinforcing the endogenous activation of mGluR2 receptors.69 Also, some other studies reported that NAC treatment can reverse cocaine-induced metaplasticity and reduce cocaine craving in humans through activation of cystine-glutamate exchange and stimulation of extrasynaptic mGluRs.70,71
Taken together, these findings contribute to describe the clinical efficacy of NAC in the treatment of different chronic types of pain, especially neuropathic pain. The excellent safety and tolerability of the natural antioxidants such as NAC during long term treatment have made them an attractive therapeutic modality in controlling pain compared to conventional medications. Although, severe and in some instances life-threatening anaphylactoid reactions to intravenous administration of NAC have been reported,72 oral administration of NAC is relatively safe and has not caused clinically significant adverse reactions even at doses as high as 8000 mg/day. Mild gastrointestinal disturbance such as nausea, vomiting, and heartburn are the only adverse effects reported of oral administration of NAC.73 Therefore, this margin of safety and excellent efficacy make oral NAC an attractive therapeutic option in models of inflammatory and neuropathic pain.
Despite the novelty of the findings, several limitations warn against the overgeneralization of the results. The first limitation of this study is the relatively small number of subjects. More studies with larger sample sizes are warranted. A second limitation of the present study is the relatively short duration of the intervention. Similar studies are needed with longer follow-up duration to validate our findings. Third, due to structural constraints, the number of studied biomarkers was limited, and serum levels of other OTS and inflammatory biomarkers were not assessed. Further, only two samples were taken from each patient at baseline and 8 weeks after treatment. Therefore, the quality of the data does not allow a deeper inspection and understanding of the biochemical changes of diabetic neuropathy. Fourth, the statistical data analysis was performed per protocol, while an intent-to-treat analysis might have allowed observation of a modified pattern of results. Last but not least, we used a relatively modest dose of NAC in our study and we kept the NAC dosage stable throughout the entire study, perhaps higher doses of NAC would have allowed an even more favorable pattern of results.
Conclusion
In conclusion, our study adds to the limited clinical database regarding treatment of PDN. Considering the pathways in pathogenesis of diabetic neuropathy, including oxidative stress and inflammation, agents with proven antioxidant and anti-inflammatory properties such as NAC can decrease pain symptoms in patients with diabetic neuropathy, at least in part by improving oxidative balance. Further studies are needed to elucidate the exact mechanisms of NAC supplementation in PDN.
Acknowledgments
This study was supported by Vice-Chancellor of Research and Technology of Hamadan University of Medical Sciences, Hamadan, Iran (No: 9705303282). The authors thank all patients for helping and participating in the study.
Data Sharing
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request up to 2 years after publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Iqbal Z, Azmi S, Yadav R, et al. Diabetic peripheral neuropathy: epidemiology, diagnosis, and pharmacotherapy. Clin Ther. 2018;40(6):828–849. doi:10.1016/j.clinthera.2018.04.001
2. Veves A, Backonja M, Malik RA. Painful diabetic neuropathy: epidemiology, natural history, early diagnosis, and treatment options. Pain Med. 2008;9(6):660–674. doi:10.1111/j.1526-4637.2007.00347.x
3. Schreiber AK, Nones CF, Reis RC, Chichorro JG, Cunha JM. Diabetic neuropathic pain: physiopathology and treatment. World J Diabetes. 2015;6(3):432–444. doi:10.4239/wjd.v6.i3.432
4. Gore M, Brandenburg NA, Dukes E, Hoffman DL, Tai KS, Stacey B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J Pain Symptom Manage. 2005;30(4):374–385. doi:10.1016/j.jpainsymman.2005.04.009
5. Javed S, Petropoulos IN, Alam U, Malik RA. Treatment of painful diabetic neuropathy. Ther Adv Chronic Dis. 2015;6(1):15–28. doi:10.1177/2040622314552071
6. Spallone V. Management of painful diabetic neuropathy: guideline guidance or jungle? Curr Diab Rep. 2012;12(4):403–413. doi:10.1007/s11892-012-0287-2
7. Tesfaye S, Boulton AJ, Dickenson AH. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care. 2013;36(9):2456–2465. doi:10.2337/dc12-1964
8. Elmarakby AA, Sullivan JC. Relationship between oxidative stress and inflammatory cytokines in diabetic nephropathy. Cardiovasc Ther. 2012;30(1):49–59. doi:10.1111/j.1755-5922.2010.00218.x
9. Sandireddy R, Yerra VG, Areti A, Komirishetty P, Kumar A. Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets. Int J Endocrinol. 2014;2014:674987. doi:10.1155/2014/674987
10. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi:10.1038/414813a
11. Hernandez-Beltran N, Moreno CB, Gutierrez-Alvarez AM. Contribution of mitochondria to pain in diabetic neuropathy. Endocrinol Nutr. 2013;60(1):25–32. doi:10.1016/j.endonu.2012.03.005
12. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS, Calcium ATP. ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–C833. doi:10.1152/ajpcell.00139.2004
13. Camello-Almaraz C, Gomez-Pinilla PJ, Pozo MJ, Camello PJ. Mitochondrial reactive oxygen species and Ca2+ signaling. Am J Physiol Cell Physiol. 2006;291(5):C1082–C1088. doi:10.1152/ajpcell.00217.2006
14. Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49(11):1932–1938. doi:10.2337/diabetes.49.11.1932
15. Einheber S, Bhat MA, Salzer JL. Disrupted axo-glial junctions result in accumulation of abnormal mitochondria at nodes of ranvier. Neuron Glia Biol. 2006;2(3):165–174. doi:10.1017/S1740925X06000275
16. Kuhad A, Singh P, Chopra K. Matrix metalloproteinases: potential therapeutic target for diabetic neuropathic pain. Expert Opin Ther Targets. 2015;19(2):177–185. doi:10.1517/14728222.2014.960844
17. Kawasaki Y, Xu ZZ, Wang X, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med. 2008;14(3):331–336. doi:10.1038/nm1723
18. Jensen TS, Backonja MM, Hernandez Jimenez S, Tesfaye S, Valensi P, Ziegler D. New perspectives on the management of diabetic peripheral neuropathic pain. Diab Vasc Dis Res. 2006;3(2):108–119. doi:10.3132/dvdr.2006.013
19. Ziegler D, Hanefeld M, Ruhnau KJ, et al. Treatment of symptomatic diabetic polyneuropathy with the antioxidant alpha-lipoic acid: a 7-month multicenter randomized controlled trial (ALADIN III Study). ALADIN III Study Group. Alpha-Lipoic Acid in Diabetic Neuropathy. Diabetes Care. 1999;22(8):1296–1301. doi:10.2337/diacare.22.8.1296
20. Sima AA, Calvani M, Mehra M, Amato A. Acetyl-L-carnitine improves pain, nerve regeneration, and vibratory perception in patients with chronic diabetic neuropathy: an analysis of two randomized placebo-controlled trials. Diabetes Care. 2005;28(1):89–94. doi:10.2337/diacare.28.1.89
21. Tutuncu NB, Bayraktar M, Varli K. Reversal of defective nerve conduction with vitamin E supplementation in type 2 diabetes: a preliminary study. Diabetes Care. 1998;21(11):1915–1918. doi:10.2337/diacare.21.11.1915
22. Vallianou N, Evangelopoulos A, Koutalas P. Alpha-lipoic Acid and diabetic neuropathy. Rev Diabet Stud. 2009;6(4):230–236. doi:10.1900/RDS.2009.6.230
23. Ganesh Yerra V, Negi G, Sharma SS, Kumar A. Potential therapeutic effects of the simultaneous targeting of the Nrf2 and NF-kappaB pathways in diabetic neuropathy. Redox Biol. 2013;1:394–397. doi:10.1016/j.redox.2013.07.005
24. Choi J, Chandrasekaran K, Inoue T, Muragundla A, Russell JW. PGC-1alpha regulation of mitochondrial degeneration in experimental diabetic neuropathy. Neurobiol Dis. 2014;64:118–130. doi:10.1016/j.nbd.2014.01.001
25. Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005;38(1):136–145. doi:10.1016/j.freeradbiomed.2004.09.025
26. Aldini G, Altomare A, Baron G, et al. N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. 2018;52(7):751–762. doi:10.1080/10715762.2018.1468564
27. Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167–177. doi:10.1016/j.tips.2013.01.001
28. Fries GR, Kapczinski F. N-acetylcysteine as a mitochondrial enhancer: a new class of psychoactive drugs? Braz J Psychiatry. 2011;33(4):321–322. doi:10.1590/S1516-44462011000400003
29. Li J, Xu L, Deng X, et al. N-acetyl-cysteine attenuates neuropathic pain by suppressing matrix metalloproteinases. Pain. 2016;157(8):1711–1723. doi:10.1097/j.pain.0000000000000575
30. Truini A, Piroso S, Pasquale E, et al. N-acetyl-cysteine, a drug that enhances the endogenous activation of group-II metabotropic glutamate receptors, inhibits nociceptive transmission in humans. Mol Pain. 2015;11:14. doi:10.1186/s12990-015-0009-2
31. Hallynck T, Soep HH, Thomis J, et al. Prediction of creatinine clearance from serum creatinine concentration based on lean body mass. Clin Pharmacol Ther. 1981;30(3):414–421. doi:10.1038/clpt.1981.181
32. Rosenstock J, Tuchman M, LaMoreaux L, Sharma U. Pregabalin for the treatment of painful diabetic peripheral neuropathy: a double-blind, placebo-controlled trial. Pain. 2004;110(3):628–638. doi:10.1016/j.pain.2004.05.001
33. Benzie IF, Szeto YT. Total antioxidant capacity of teas by the ferric reducing/antioxidant power assay. J Agric Food Chem. 1999;47(2):633–636. doi:10.1021/jf9807768
34. Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82(1):70–77. doi:10.1016/0003-9861(59)90090-6
35. Botsoglou NA, Fletouris DJ, Papageorgiou GE, Vassilopoulos VN, Mantis AJ, Trakatellis AG. Rapid, sensitive, and specific thiobarbituric acid method for measuring lipid peroxidation in animal tissue, food, and feedstuff samples. J Agric Food Chem. 1994;42(9):1931–1937. doi:10.1021/jf00045a019
36. Alleman CJ, Westerhout KY, Hensen M, et al. Humanistic and economic burden of painful diabetic peripheral neuropathy in Europe: A review of the literature. Diabetes Res Clin Pract. 2015;109(2):215–225. doi:10.1016/j.diabres.2015.04.031
37. Gore M, Brandenburg NA, Hoffman DL, Tai KS, Stacey B. Burden of illness in painful diabetic peripheral neuropathy: the patients’ perspectives. J Pain. 2006;7(12):892–900. doi:10.1016/j.jpain.2006.04.013
38. Varkonyi T, Kempler P. Diabetic neuropathy: new strategies for treatment. Diabetes Obes Metab. 2008;10(2):99–108. doi:10.1111/j.1463-1326.2007.00741.x
39. Sloan G, Shillo P, Selvarajah D, et al. A new look at painful diabetic neuropathy. Diabetes Res Clin Pract. 2018;144:177–191. doi:10.1016/j.diabres.2018.08.020
40. Kellogg AP, Pop-Busui R. Peripheral nerve dysfunction in experimental diabetes is mediated by cyclooxygenase-2 and oxidative stress. Antioxid Redox Signal. 2005;7(11–12):1521–1529. doi:10.1089/ars.2005.7.1521
41. Skundric DS, Lisak RP. Role of neuropoietic cytokines in development and progression of diabetic polyneuropathy: from glucose metabolism to neurodegeneration. Exp Diabesity Res. 2003;4(4):303–312. doi:10.1155/EDR.2003.303
42. Cameron NE, Cotter MA, Archibald V, Dines KC, Maxfield EK. Anti-oxidant and pro-oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic and streptozotocin-diabetic rats. Diabetologia. 1994;37(5):449–459. doi:10.1007/s001250050131
43. Cameron NE, Cotter MA. Effects of antioxidants on nerve and vascular dysfunction in experimental diabetes. Diabetes Res Clin Pract. 1999;45(2–3):137–146. doi:10.1016/S0168-8227(99)00043-1
44. van Dam PS. Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev. 2002;18(3):176–184. doi:10.1002/(ISSN)1520-7560
45. Sayyed SG, Kumar A, Sharma SS. Effects of U83836E on nerve functions, hyperalgesia and oxidative stress in experimental diabetic neuropathy. Life Sci. 2006;79(8):777–783. doi:10.1016/j.lfs.2006.02.033
46. Kumar A, Kaundal RK, Iyer S, Sharma SS. Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life Sci. 2007;80(13):1236–1244. doi:10.1016/j.lfs.2006.12.036
47. Oyenihi AB, Ayeleso AO, Mukwevho E, Masola B. Antioxidant strategies in the management of diabetic neuropathy. Biomed Res Int. 2015;2015:515042. doi:10.1155/2015/515042
48. Kaga AK, Barbanera PO, Do Carmo NOL, Rosa LRO, Fernandes AAH. Effect of N-acetylcysteine on dyslipidemia and carbohydrate metabolism in STZ-induced diabetic rats. Int J Vasc Med. 2018;2018:6428630. doi:10.1155/2018/4305781
49. Fulghesu AM, Ciampelli M, Muzj G, et al. N-acetyl-cysteine treatment improves insulin sensitivity in women with polycystic ovary syndrome. Fertil Steril. 2002;77(6):1128–1135. doi:10.1016/S0015-0282(02)03133-3
50. Dludla PV, Dias SC, Obonye N, Johnson R, Louw J, Nkambule BB. A systematic review on the protective effect of N-acetyl cysteine against diabetes-associated cardiovascular complications. Am J Cardiovasc Drugs. 2018;18(4):283–298. doi:10.1007/s40256-018-0275-2
51. Lee ES, Kim HM, Kang JS, et al. Oleanolic acid and N-acetylcysteine ameliorate diabetic nephropathy through reduction of oxidative stress and endoplasmic reticulum stress in a type 2 diabetic rat model. Nephrol Dial Transplant. 2016;31(3):391–400. doi:10.1093/ndt/gfv377
52. Kamboj SS, Vasishta RK, Sandhir R. N-acetylcysteine inhibits hyperglycemia-induced oxidative stress and apoptosis markers in diabetic neuropathy. J Neurochem. 2010;112(1):77–91. doi:10.1111/jnc.2009.112.issue-1
53. Sagara M, Satoh J, Wada R, et al. Inhibition of development of peripheral neuropathy in streptozotocin-induced diabetic rats with N-acetylcysteine. Diabetologia. 1996;39(3):263–269. doi:10.1007/BF00418340
54. Park SA, Choi KS, Bang JH, Huh K, Kim SU. Cisplatin-induced apoptotic cell death in mouse hybrid neurons is blocked by antioxidants through suppression of cisplatin-mediated accumulation of p53 but not of Fas/Fas ligand. J Neurochem. 2000;75(3):946–953. doi:10.1046/j.1471-4159.2000.0750946.x
55. Naik AK, Tandan SK, Dudhgaonkar SP, et al. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur J Pain. 2006;10(7):573–579. doi:10.1016/j.ejpain.2005.08.006
56. Horst A, de Souza JA, Santos MCQ, Riffel APK, Kolberg C, Partata WA. Effects of N-acetylcysteine on spinal cord oxidative stress biomarkers in rats with neuropathic pain. Braz J Med Biol Res. 2017;50(12):e6533. doi:10.1590/1414-431x20176533
57. Lin PC, Lee MY, Wang WS, et al. N-acetylcysteine has neuroprotective effects against oxaliplatin-based adjuvant chemotherapy in colon cancer patients: preliminary data. Support Care Cancer. 2006;14(5):484–487. doi:10.1007/s00520-006-0018-9
58. Cascinu S, Catalano V, Cordella L, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. J Clin Oncol. 2002;20(16):3478–3483. doi:10.1200/JCO.2002.07.061
59. Batooei M, Tahamoli-Roudsari A, Basiri Z, et al. Evaluating the effect of Oral N-acetylcysteine as an adjuvant treatment on clinical outcomes of patients with rheumatoid arthritis: a randomized, double blind clinical trial. Rev Recent Clin Trials. 2018;13(2):132–138. doi:10.2174/1574887113666180307151937
60. Hashemi G, Mirjalili M, Basiri Z, et al. A pilot study to evaluate the effects of oral N-acetyl cysteine on inflammatory and oxidative stress biomarkers in rheumatoid arthritis. Curr Rheumatol Rev. 2019;15(3):246–253. doi:10.2174/1573403X14666180926100811
61. Ji RR, Xu ZZ, Wang X, Lo EH. Matrix metalloprotease regulation of neuropathic pain. Trends Pharmacol Sci. 2009;30(7):336–340. doi:10.1016/j.tips.2009.04.002
62. Uemura S, Matsushita H, Li W, et al. Diabetes mellitus enhances vascular matrix metalloproteinase activity: role of oxidative stress. Circ Res. 2001;88(12):1291–1298. doi:10.1161/hh1201.092042
63. Ryan ME, Ramamurthy NS, Sorsa T, Golub LM. MMP-mediated events in diabetes. Ann N Y Acad Sci. 1999;878:311–334. doi:10.1111/j.1749-6632.1999.tb07692.x
64. Liu Y, Ni Y, Zhang W, Sun YE, Ma Z, Gu X. N-acetyl-cysteine attenuates remifentanil-induced postoperative hyperalgesia via inhibiting matrix metalloproteinase-9 in dorsal root ganglia. Oncotarget. 2017;8(10):16988–17001. doi:10.18632/oncotarget.15217
65. Osikowicz M, Mika J, Przewlocka B. The glutamatergic system as a target for neuropathic pain relief. Exp Physiol. 2013;98(2):372–384. doi:10.1113/expphysiol.2012.069922
66. Bleakman D, Alt A, Nisenbaum ES. Glutamate receptors and pain. Semin Cell Dev Biol. 2006;17(5):592–604. doi:10.1016/j.semcdb.2006.10.008
67. Chiechio S, Caricasole A, Barletta E, et al. L-Acetylcarnitine induces analgesia by selectively up-regulating mGlu2 metabotropic glutamate receptors. Mol Pharmacol. 2002;61(5):989–996. doi:10.1124/mol.61.5.989
68. Zammataro M, Chiechio S, Montana MC, et al. mGlu2 metabotropic glutamate receptors restrain inflammatory pain and mediate the analgesic activity of dual mGlu2/mGlu3 receptor agonists. Mol Pain. 2011;7:6. doi:10.1186/1744-8069-7-6
69. Bernabucci M, Notartomaso S, Zappulla C, et al. N-Acetyl-cysteine causes analgesia by reinforcing the endogenous activation of type-2 metabotropic glutamate receptors. Mol Pain. 2012;8:77. doi:10.1186/1744-8069-8-77
70. Moussawi K, Pacchioni A, Moran M, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12(2):182–189. doi:10.1038/nn.2250
71. Amen SL, Piacentine LB, Ahmad ME, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36(4):871–878. doi:10.1038/npp.2010.226
72. Prescott L. Oral or intravenous N-acetylcysteine for acetaminophen poisoning? Ann Emerg Med. 2005;45(4):409–413. doi:10.1016/j.annemergmed.2004.09.028
73. De Rosa SC, Zaretsky MD, Dubs JG, et al. N-acetylcysteine replenishes glutathione in HIV infection. Eur J Clin Invest. 2000;30(10):915–929. doi:10.1046/j.1365-2362.2000.00736.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.